

Abstract
Aim
The aim of the study was to compare the functional and structural properties of the motor protein, myosin, and isolated myocyte contractility in heart muscle excised from hypertrophic cardiomyopathy patients by surgical myectomy with explanted failing heart and non-failing donor heart muscle.
Methods
Myosin was isolated and studied using an in vitro motility assay. The distribution of myosin light chain-1 isoforms was measured by two-dimensional electrophoresis. Myosin light chain-2 phosphorylation was measured by sodium dodecyl sulphate–polyacrylamide gel electrophoresis using Pro-Q Diamond phosphoprotein stain.
Results
The fraction of actin filaments moving when powered by myectomy myosin was 21% less than with donor myosin (P = 0.006), whereas the sliding speed was not different (0.310 ± 0.034 for myectomy myosin vs. 0.305 ± 0.019 µm/s for donor myosin in six paired experiments). Failing heart myosin showed 18% reduced motility. One myectomy myosin sample produced a consistently higher sliding speed than donor heart myosin and was identified with a disease-causing heavy chain mutation (V606M). In myectomy myosin, the level of atrial light chain-1 relative to ventricular light chain-1 was 20 ± 5% compared with 11 ± 5% in donor heart myosin and the level of myosin light chain-2 phosphorylation was decreased by 30–45%. Isolated cardiomyocytes showed reduced contraction amplitude (1.61 ± 0.25 vs. 3.58 ± 0.40%) and reduced relaxation rates compared with donor myocytes (TT50% = 0.32 ± 0.09 vs. 0.17 ± 0.02 s).
Conclusion
Contractility in myectomy samples resembles the hypocontractile phenotype found in end-stage failing heart muscle irrespective of the primary stimulus, and this phenotype is not a direct effect of the hypertrophy-inducing mutation. The presence of a myosin heavy chain mutation causing hypertrophic cardiomyopathy can be predicted from a simple functional assay.
Keywords: Contractile apparatus, Contractile function, Hypertrophy, Protein phosphorylation, Myosin, Myectomy, Hypertrophic cardiomyopathy
1. Introduction
Hypertrophic cardiomyopathy is a primary disease of cardiac muscle and is clinically defined as a hypertrophied, non-dilated left ventricle in the absence of any other aetiology. It is a common cardiac disorder affecting one in 500 of the population and is the leading cause of sudden death in the young. It has been known for some time that hypertrophic cardiomyopathy is usually familial and has been linked to mutations in the contractile apparatus of cardiac muscle. Although mutations have been identified in every component of the muscle sarcomere, the most common contractile protein mutations that cause hypertrophic cardiomyopathy are in β myosin heavy chain (MYH7 gene) and myosin-binding protein-C (MYBPC3 gene) which make up 25 and 26% of all hypertrophic cardiomyopathy cases, respectively.1–4
Many mutations identified as causing hypertrophic cardiomyopathy have been investigated with recombinant proteins incorporating the mutation or in transgenic mouse models. In vitro the mutations usually cause a gain in function, with increased rate of crossbridge turnover and increased Ca2+ sensitivity1,5–8 but this does not necessarily translate into enhanced contractility in animal models.9,10 The mechanism relating the initial mutation to the development of pathological hypertrophy is still not understood,11 nor do we know whether transgenic models of hypertrophic cardiomyopathy are directly relevant to hypertrophic cardiomyopathy as observed in human hearts.
Very few investigations have been made with heart muscle from patients with hypertrophic cardiomyopathy.8,12,13 The main reason for this has been the unavailability of suitable material. Heart punch biopsies are rarely justified and yield insufficient material for extensive functional analysis. However, a subset of hypertrophic cardiomyopathy patients (about 25%) have left ventricular outflow tract obstruction (LVOTO) caused by the contact between the mitral valve leaflet and the hypertrophied interventricular septum in systole. LVOTO is associated with an increased risk of sudden death and therefore justifies intervention.14 The surgical septal myectomy operation removes 3–7 g of myocardium from the interventricular septum in the region of the obstruction15,16 and the excised muscle is a unique source of human heart muscle with hypertrophic cardiomyopathy.
We have compared the functional and structural properties of myectomy muscle with end-stage failing and non-failing donor heart muscle to determine the molecular phenotype of hypertrophic cardiomyopathy muscle and ultimately to link this to the disease-causing mutation. Our initial studies, reported here, investigated the functional properties of isolated myocytes and of the motor protein, myosin, extracted from myectomy, failing and donor heart muscle measured by in vitro motility assay. We have found that myocytes are hypocontractile, and the fraction of actin filaments moving when powered by myectomy myosin is significantly lower than the control myosin, indicating dysfunction at the molecular level. In addition, the expression of atrial light chain-1 (ALC-1) relative to ventricular light chain-1 (VLC-1) in myectomy myosin is higher and the level of myosin light chain-2 (MLC-2) phosphorylation is lower. This pattern of results was found in every myectomy sample independent of the mutation that is presumed to initiate hypertrophy. One myosin sample showed a higher sliding speed than donor myosin which we predicted was due to a disease-causing mutation in myosin heavy chain (MHC) and this was confirmed by genotyping the tissue.
2. Methods
2.1. Collection and storage of human myocardium
2.1.1. Hypertrophic cardiomyopathy
Human myocardial samples were obtained from patients with hypertrophic cardiomyopathy undergoing surgical septal myectomy for relief of LVOTO. For in vitro motility assay, the samples were snap-frozen in liquid nitrogen and stored for later use. Local ethical approval was obtained from University College London Hospitals and the Brompton, Harefield and NHLI ethics committees for collection and use of tissue samples. All patients had cardiac investigations, including 12-lead ECG, chest X-ray, Holter monitor, cardiopulmonary exercise test, two-dimensional transthoracic echocardiography, transoesophageal echocardiography, cardiac catheterization, and coronary angiography (Table 1).
Table 1.
Patient details
| Biopsy sample | M1 | M3 | M4 | M5 | M7 | M8 |
|---|---|---|---|---|---|---|
| Diagnosis | HCM | HCM | HCM | HCM | HCM | HCM |
| Age | 24 | 42 | 49 | 31 | 54 | 42 |
| Myectomy date | 23/06/04 | 04/08/04 | 04/08/04 | 11/08/04 | 22/09/04 | 17/11/04 |
| Gender | F | M | M | M | F | M |
| Diagnosed | 2001 | 2002 | 1994 | 1996 | 1992 | 1999 |
| Family history | No HCM; no SCD | No HCM; no SCD | Father died 58 years | No HCM; no SCD; nine siblings; negative screen | No SCD; one sister w/possible HCM | Mother SCD; 47 years HCM at post-mortem |
| Current treatment | Verapamil, amiodarone | Atenolol, disopyramide | Atenolol, aspirin, disopyramide, clopidogrel, quinine | Propanolol | Atenolol, aminodarone, frumil | Disopyramide, timolol |
| ECHO appearance | ASH | ASH | Concentric LVH | Mild ASH | ASH | ASH |
| MaxLVWT (mm) | 27.5 | 23 | 16 | 20 | 22 | 23 |
| Max. ST (mm) | 27.5 | 23 | 16 | 20 | 22 | 23 |
| LVEDD (mm) | 43.6 | 42.0 | 39.0 | 44.0 | 34.0 | |
| LVESD (mm) | 23.9 | 28.0 | 21.0 | 23.0 | 12.0 | |
| LA (mm) | 45 | 41 | 48 | 41 | 43 | 44 |
| FS (%) | 45 | 33 | 48 | 46 | 64 | |
| SAM | Complete | Incomplete | Complete | Complete | Incomplete | Complete |
| MR | Mild | Mild | Mild to moderate | Trivial | Mild | Trivial |
| Resting LVOT gradient (mmHg) | 86 | 22 (133 mmHg with GTN) | 104 | 110 | 20 (stress, 190 mmHg) | 93 |
| VT on holter monitor | VT (non- sustained) | No | Isolated VEs only | No | VT (non-sustained) | No |
| ETT MVO2 (ml/min/kg) | 58% predicted; no arrhythmias; flat BP | 20.3; 68% predicted; no arrhythmias BP rise | 11.7; 35% predicted; no arrhythmias; flat BP | 62% predicted; no arrhythmia; flat BP | 15.8; 77% predicted; no arrhythmia; BP rise | 15.0; 42% predicted; no arrhythmias; BP rise |
| ECG | SR, LVH, RA | SR, LVH, RA | SR, LVH | SR, LVH | SR, LVH, RA | SR, LVH, RA |
| NYHA class | II | II–III | II | III | III | III |
2.1.2. Non-failing and failing heart muscle
Tissue samples were supplied by Prof. C. Dos Remedios, University of Sydney, Australia. Ethical approval was obtained from the Brompton, Harefield and NHLI Research Ethics Committee, London, and St Vincent's Hospital, Sydney. The investigation conformed with the principles outlined in the Declaration of Helsinki. We studied tissue from nine explanted hearts with end-stage heart failure (ejection fraction <20%) and eight non-failing donor hearts. The donor heart tissue was obtained from hearts where no suitable transplant recipient was found. The patients had no history of cardiac disease, a normal cardiac examination, normal ECG, and normal ventricular function on echocardiography within 24 h of heart explantation. Myocardium was immediately frozen in liquid nitrogen and stored for later analysis or stored in cardioplegic solution for cell isolation. Clinical and functional characteristics of these donor and failing heart samples have been previously reported.17
2.2. Myosin extraction from human heart muscle
Fifty milligrams of human cardiac tissue were crushed using a liquid-nitrogen-cooled percussion mortar and homogenized in 0.25 mL ice-cold high salt extraction buffer (0.3 mol/L KCl, 0.15 mol/L K2HPO4, 10 mmol/L Na4P2O7, 1 mmol/L MgCl2, 1 mmol/L MgATP, 10 mmol/L DTT, 3 µg/mL each of protease inhibitors, chymostatin, pepstatin A, leupeptin, and E64, pH 6.8). The homogenate was extracted at 4°C for 1 h on a rotating mixer and then centrifuged for 20 min at 337 000 g. The supernatant was collected and diluted ×20 with ice-cold 5 mmol/L DTT and allowed to precipitate on ice for 1 h. It was then spun for 20 min at 26 000 g at 4°C to collect the precipitated myosin.8 The pellet obtained was dissolved in 250 µL of resuspension buffer (0.3 mol/L KCl, 25 mmol/L imidazole, pH 7.4, 1 mmol/L EGTA, 4 mmol/L MgCl2, 10 mmol/L DTT, 1.4 µL each of 2.5 mg/mL chymostatin, pepstatin, leupeptin, and E64). The concentration of myosin was determined using the Bradford assay and the purity was assessed using sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE; Figure 2). Using this method, 1 mg of myosin can be obtained from 50 mg of tissue. Skeletal muscle actin and human cardiac muscle tropomyosin and troponin were prepared as previously described.17 The preparation of myofibrils from human heart samples is described in the Supplementary material.
Figure 2.
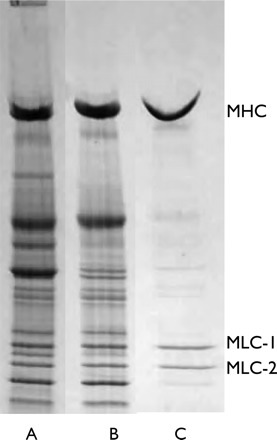
Isolation of myosin from human heart muscle. SDS–PAGE of (A) high salt extract of muscle myofibrils, (B) supernatant containing myosin and troponin–tropomyosin after ultracentrifugation, and (C) pure myosin sedimented at low salt concentration.
2.3. In vitro motility assay
The in vitro motility assay technique was used to study TRITC-phalloidin-labelled actin filaments moving over immobilized myosin.18 Detailed protocols are provided in Supplementary material.
2.4. Assay of myosin light chains
Light chain isoforms were analysed by two-dimensional electrophoresis. Thirty micrograms of muscle homogenate were separated by isoelectric focussing on 18-cm long, pI 4.5–5.5, Immobiline strips (GE Biosciences), and by SDS–PAGE in the second dimension.19 The gels were stained with SYPRO Ruby (Invitrogen).
One-dimensional SDS–PAGE gels were used to analyse the levels of phosphorylation using the Pro-Q Diamond stain developed by Molecular Probes (Invitrogen).20 Myofibrils or myosin (10–20 µg) was separated on 10 × 12 cm gels (8–18% Polyacrylamide gradient, 1% SDS: Exelgel, GE Biosciences). The gel was stained with Pro-Q Diamond according to the manufacturer's instructions and the phosphoprotein bands were visualized by UV transillumination and recorded with a digital camera (Gene Genius/CHEMI-HR16 system, Synoptics). The gel was then re-stained with SYPRO Ruby or Coomassie Blue total protein stains. The Pro-Q Diamond band volume from densitometric analysis was normalized to the corresponding total protein band volume.
2.5. Isolation and characterization of cardiomyocytes
Myocytes were isolated from fresh myectomy or transplant tissue and analysed as described previously.21 Detailed protocols are provided in the Supplementary material.
2.6. Genotyping
Screening for mutations was performed by high-resolution melting curve analysis on a Lightscanner (Idaho Technology, Inc.) with LCGreen Plus dye. Genomic DNA samples were quantitated with Nanodrop, quality-checked by gel electrophoresis, and each exon was then PCR-amplified. To promote heteroduplex formation, samples were subsequently denatured by heating to 95°C for 30 s and cooled to 20°C. Plates were centrifuged and immediately run on the Lightscanner instrument according to the manufacturer's recommendations. Amplimers with aberrant melting curves were then investigated by bidirectional sequencing.
3. Results
3.1. Properties of myocytes extracted from myectomy muscle
Ventricular myocytes from myectomy samples had a varied morphology, with some cells showing a clear rod-shaped morphology, such as those in samples from non-failing or failing ventricles, but others with a highly branched appearance (Figure 1A). Surprisingly, contraction amplitudes in myocytes from myectomy samples were low, or lower than in those from failing hearts studied during the same period, and relaxation was more impaired. Figure 1B compares myocytes at 2 mmol/L Ca2+, where amplitude was significantly lower in myectomy samples than in failing. Relaxation had a strong tendency to be slower (Figure 1) and was certainly more variable (comparison of variances P < 0.001, comparison of means with Welch's correction for unequal variances, P = 0.058). Relaxation times for myocytes from failing heart shown here were similar to those previously described from this laboratory, where relaxation times were significantly longer than those for non-failing hearts.24 At 4 mmol/L Ca2+, the amplitude difference between myectomy and failing myocytes was reduced (% shortening: myectomy 2.83 ± 0.41%, 34 cells/17 patients, failing 3.28 ± 0.21% 110 cells/40 patients), but the relaxation deficit and high variability was maintained (R90: myectomy, 0.76 ± 0.11 s, failing, 0.51 ± 0.02 s, comparison of variances P < 0.001, comparison of means with Welch's correction for unequal variances, P < 0.05).
Figure 1.
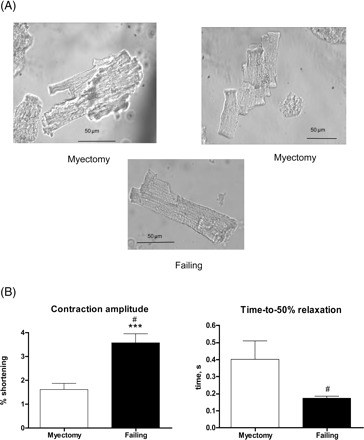
Contraction of isolated myocytes from myectomy muscle. (A) Two ventricular myocytes from myectomy samples, showing branched appearance, compared with the more usual rod-shaped appearance of a ventricular myocyte from failing heart. (B) Contraction amplitude (% shortening) and time-to-50% relaxation of ventricular myocytes from myectomy (21 myocytes/10 hearts) and failing (41 myocytes/16 hearts) samples. #P < 0.001, variances significantly different; ***P < 0.001, means significantly different.
3.2. Altered motility function of myectomy myosin
We studied myosin in six heart muscle samples from patients with hypertrophic obstructive cardiomyopathy (HOCM) undergoing septal myectomy (M) and compared these with myosin from donor heart left ventricle muscle (N). Table 1 shows the clinical details of the HOCM patients. For functional studies, myosin was extracted from 50 mg samples of frozen heart muscle by a method based on Palmiter et al.8 (Figure 2). In vitro motility assay using freshly prepared myectomy myosin showed that TRITC-phalloidin-labelled actin filaments moved at a mean speed of 0.310 ± 0.034 µm/s at 30°C. Myectomy muscle sliding speed was not significantly different from the donor heart controls (0.305 ± 0.019 µm/s). Comparison of the fraction of actin filaments that were motile showed that all of the myectomy myosin muscle samples produced a 21% lower fraction motile than the donor control heart myosin [0.54 ± 0.03 for myectomy myosin compared with 0.69 ± 0.4 for control, P < 0.001, n = 20 (four measurements each from five paired myosin preparations); see Figure 3A and B].
Figure 3.
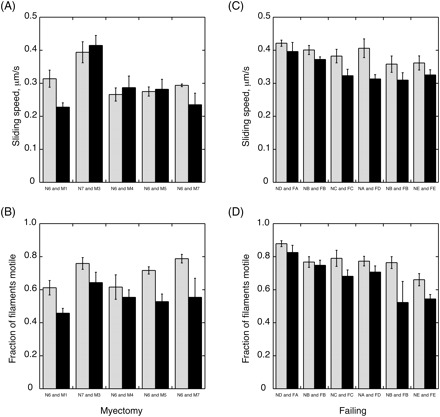
In vitro motility of human cardiac myosin with rabbit skeletal muscle actin. Actin filament motility was measured at 30°C in dual-chamber motility cells containing immobilized donor and myectomy or failing heart myosin pairs as indicated. (A, B) Myosin from five myectomy samples (M1, M3, M4, M5, M7, black bars) was compared with myosin from donor muscle samples (N6 or N7, grey bars) in paired assays. Movement was analysed to yield the sliding speed (A) and the fraction of filaments that were moving (B). There was no systematic difference in sliding speed (mean sliding speed myectomy myosin was 94 ± 0.07% of donor P = 0.44), but the fraction of filaments motile was always lower with myectomy myosin (mean fraction myectomy myosin motile was 79 ± 0.03% of donor, P = 0.004). (C, D) Myosin from failing heart muscle (FA, FB, FC, FD, FE, black bars) was compared with myosin from donor muscle (NA, NB, NC, ND, NE, grey bars) in paired assays. Movement was analysed to yield the sliding speed (C) and the fraction of filaments that were moving (D). Sliding speed was slightly reduced in failing heart myosin (mean sliding speed failing myosin was 89 ± 0.03% of donor, P = 0.004) and the fraction of filaments motile was also lower (mean fraction failing myosin motile was 82 ± 0.04% of donor, P = 0.026).
We also compared the donor myosin (N) with myosin from end-stage failing heart muscle (F) using the same preparation method. The fraction of filaments moving was 15% lower in the failing heart muscle (donor, 0.73 ± 0.02; failing, 0.63 ± 0.03, P = 0.0002) and the sliding speed was 11% lower and this difference was also significant (Figure 3 C and D).
The addition of 50 mmol/L human cardiac tropomyosin had very little effect on the fraction of filaments motile or filament sliding speed. In the presence of 40 nmol/L human cardiac troponin, the motility of thin filaments reconstituted with 10 nmol/L TRITC-F-actin and 50 nmol/L tropomyosin was regulated by Ca2+ (Figure 4). At activating Ca2+ concentrations, the fraction of filaments motile was slightly higher for pure actin, 0.76 ± 0.04 for donor control myosin, and 0.61 ± 0.09 for myectomy myosin. The lower fraction motile of myectomy myosin is thus still observed in fully reconstituted thin filaments. As previously observed,22,23 the sliding speed of Ca2+-activated thin filaments was greater than that of pure actin filaments, by 10% for donor myosin and by 36% for myectomy myosin. In relaxing conditions (pCa9), thin filament motility was switched off with both myectomy and donor heart myosin. An exception to this pattern was myosin from myectomy sample M5 which showed no relaxation in this experiment: in fact, the fraction of thin filaments motile was greater at pCa9 than pCa5. The myosin preparations were too unstable to permit determination of Ca2+ dose–response curves.
Figure 4.
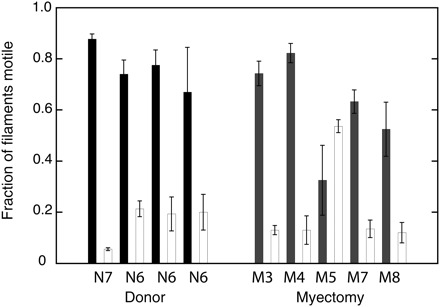
Ca2+ regulation of motility of reconstituted thin filaments. Thin filaments were reconstituted with 10 nmol/L TRITC-phalloidin-actin, 50 nmol/L human cardiac tropomyosin, and 40 nmol/L human cardiac troponin. In vitro motility of reconstituted thin filaments was measured at pCa5 (black bars) and pCa9 (grey bars) in dual-chamber cells and the fraction of filaments motile is plotted. Ca2+ regulation of motility was measured with myosin from two donor (N) heart and five myectomy (M) muscle samples. Only M5 showed abnormal regulation.
As impaired relaxation in in vitro motility assays is a feature of many mutations that are associated with hypertrophic cardiomyopathy, we examined M5 myosin further to determine whether it exhibited other characteristics in common with hypertrophic cardiomyopathy-inducing mutations (Table 2). In three series of paired comparisons with N6, M5 showed a consistently higher sliding speed ranging from 18 to 60% greater than N6 and observable with pure actin, actin–tropomyosin, and in fully reconstituted thin filaments at pCa5. Relaxation was also less complete in M5 compared with N6: at pCa9 sliding speed was faster and the fraction of filaments motile was greater for M5 myosin. All these measurements indicate a hypercontractile phenotype for M5 myosin.
Table 2.
M5 compared with N6
| Actin sliding speed, M5/N6 | Actin–tropomyosin sliding speed, M5/N6 | Actin–tropomyosin–troponin sliding speed at pCa5, M5/N6 | Actin–tropomyosin–troponin sliding speed at pCa9, M5/N6 | Actin–tropomyosin–troponin fraction motile at pCa9, M5/N6 | |
|---|---|---|---|---|---|
| Preparation 1 | 1.28 | 1.21 | 1.46 | 1.52 | 2.78 |
| Preparation 2 | 1.44 | 1.18 | 1.43 | 1.32 | 1.01 |
| Preparation 3 | 1.60 | 1.24 | 1.22 | 1.12 | 1.77 |
Comparison of motility parameters of non-failing heart myosin (N6) and HOCM myosin, M5. M5 has a consistently hypercontractile molecular phenotype.
3.3. Modifications to myosin light chains
We investigated the myosin light chains in our myectomy samples. It has been reported that ALC-1 is re-expressed in the ventricles of patients with heart failure or hypertrophy related to pressure overload. Myosin light chains were detected in muscle myofibrils by two-dimensional electrophoresis (2DE) and quantified. Compared with VLC-1, human ALC-1 has a calculated pI 0.05 units less and a molecular weight 368 Da more and thus should appear above and to the left of VLC-1. ALC-1 was found to be present as a small proportion of VLC-1 in all samples tested (Figure 5). On an average, the myectomy samples contained twice as much ALC-1 as the donor control group.
Figure 5.
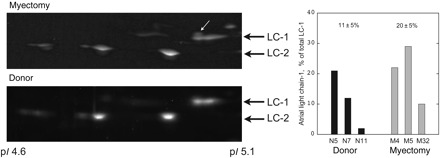
Myosin light chain detection by two-dimensional electrophoresis. Thirty micrograms of myofibrils were separated by 2D electrophoresis and stained with SYPRO Ruby. The region of the gels showing VLC-1, ALC-1, and MLC-2 are shown. VLC-1 (MYL3-HUMAN) has a pI of 5.03 and Mr of 21 800 Da, ALC-1 (MYL4-HUMAN) has a pI of 4.98 and Mr of 21 433 Da. ALC-1 was identified in a sample of mouse heart atrium located above and to the left of VLC-1 and was found to be present in all samples tested but more prominent in the myectomy samples (arrow). Densitometry of ALC-1 and VLC-1 spots was performed manually and the ratio of the two isoforms is plotted on the left. Values were quite variable with a trend towards higher ALC-1 content in the myectomy samples.
Multiple spots corresponding to phosphorylated and unphosphorylated MLC-2 isoforms were observed by 2DE. However, the Pro-Q Diamond phosphoprotein stain was not sensitive enough to identify which spots were phosphorylated, therefore phosphorylation of MLC-2 was measured by one-dimensional SDS–PAGE. In muscle myofibrils, the main phosphorylated proteins were myosin-binding protein C (MyBP-C), troponin T, troponin I, and MLC-2 (Figure 6A). The level of phosphorylation, based on the Pro-Q Diamond:Coomassie blue ratio (Q/C), of all these proteins was less in the myectomy samples. The average Q/C of MLC-2 in myectomy samples was 56% of the level in the donor heart myofibrils (Figure 6B). We can quantify this difference on the basis of a linear relationship between phosphorylation and Q/C as previously demonstrated for troponin I phosphorylation,17 taking into account the non-specific labelling of unphosphorylated protein by Pro-Q Diamond (10% of the non-failing signal for MLC-2). On this basis, the level of MLC-2 phosphorylation in myectomy muscle is 51% of phosphorylation in donor heart. In the purified myosin samples that were used for in vitro motility measurements, the level of MLC-2 phosphorylation was similarly reduced, by an average of 34% (Figure 7).
Figure 6.
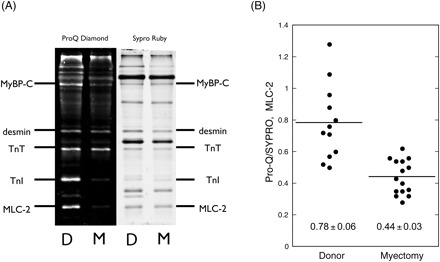
Phosphorylation of human cardiac muscle myofibrils. (A) SDS–PAGE of 10 µg myofibrils extracted from donor and myectomy muscle samples, successively stained with Pro-Q Diamond to label phosphoproteins and SYPRO Ruby total protein stain. High levels of phosphorylation are detected in MyBP-C, troponin T, troponin I, and MLC-2. (B) The level of MLC-2 phosphorylation is expressed as the ratio of Pro-Q Diamond band volume:SYPRO Ruby band volume for 12 measurements from 6 donor samples and 15 measurements from 8 myectomy samples. The mean ratios are 0.78 ± 0.06 for donor and 0.44 ± 0.03 for myectomy samples (P < 0.0001).
Figure 7.
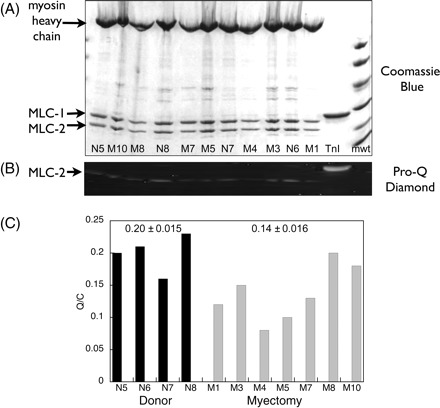
Measurement of myosin light chain-2 phosphorylation in purified myosin. (A) SDS–PAGE of purified myosin, Coomassie Blue staining. (B) Pro-Q Diamond phosphoprotein staining of the same gel showing MLC-2 bands only. (C) Relative MLC-2 phosphorylation levels. Method as described in Figure 6. The mean Pro-Q Diamond band volume/Coomassie Blue band volume ratios (Q/C) are 0.20 ± 0.015 (n = 4) for donor and 0.14 ± 0.016 (n = 7) for myectomy samples (P = 0.029).
An approximate level of phosphorylation could be calculated from the myofibril gels by comparison with troponin I in donor control, whose phosphorylation level was previously measured as 2.1 mol Pi/mol;17 we estimated that the MLC-2 phosphorylation level was 0.32 mol Pi/mol in donor heart myofibrils.
3.4. Genotyping of myectomy samples
The full MYH7 coding sequence for each patient was screened for mutations by amplification and analysis of all exons from paired genomic DNA samples; amplimers with aberrant melting curves were then investigated by bidirectional sequencing. Genomic DNA samples from M3, M4, M7, M8, and M10 revealed no MYH7 mutations, but mutations were detected in M1 and M5. Patient M1 was heterozygous for a G>A transition in exon 19, encoding the known hypertrophic cardiomyopathy-causing missense mutation Arg719Gln. Patient M5 was heterozygous for a G>A transition in exon 16, encoding the known hypertrophic cardiomyopathy-causing missense mutation Val606Met.
4. Discussion
4.1. Myocytes from myectomy samples show functional impairment
Although myectomy patients in the cohort studied were not exhibiting symptoms of end-stage failure, myocytes isolated from these hearts showed functional deficits similar to, and in some conditions greater than, those from patients explanted for end-stage idiopathic or dilated cardiomyopathy. Depression of contraction at physiological Ca2+ concentrations was more pronounced in myectomy-derived myocytes than in failing, and became equivalent to (but not better than) failing as Ca2+ concentration was raised. Similarly, slow relaxation was more evident in myectomy-derived myocytes than failing, which in turn were slower than those from non-failing donor heart. Here we described characteristics of a large population of both myectomy and transplant subjects, and it is notable that variances were significantly higher in the myectomy population. This is likely due to different effects of various mutations, which will require detailed analysis when cohorts are sufficient. However, it is clear that impairment of myocyte contraction and relaxation is a general finding in HOCM subjects.
4.2. Myosin from myectomy muscle is functionally abnormal
We then compared the functional and structural properties of myosin extracted from donor heart control, end-stage failing, and hypertrophic human heart muscle obtained from surgical myectomy operations. Myosin could be extracted in high yield and good purity within 3 h (Figure 2) and was stable for around 2 h in the in vitro motility assay. The sliding speed of 0.31–0.38 µm/s at 30°C was somewhat lower than previous measurements with human heart myosin,8,24 however we made measurements in 50 mmol/L KCl and used silica-coated glass rather than the more usual nitrocelluose surface.25,26 We found a pattern of abnormality in all the myectomy myosin samples; the fraction of filaments moving measured in the in vitro motility assay was consistently 19% lower than donor heart myosin, whereas the sliding speed, which is considered to be a measure of crossbridge turnover rate,24,26 was not significantly different (Figure 3). In order to eliminate rigor crossbridges, which would reduce motility, we used an affinity purification step and also blocked damaged myosin in the motility cell with unlabelled actin in the presence of ATP. The persistence of the lower fraction of filaments motile compared with donor muscle myosin when prepared and assayed simultaneously suggests that there is a stable difference in the myectomy myosin.
We examined the level of MLC-2 phosphorylation and found that it was significantly less in the myectomy myosin samples compared with donor heart myosin: 49% less in myofibrils and 34% less in purified myosin (Figures 6 and 7). We also measured the relative content of ALC-1 and VLC-1 in our myosin samples by 2D electrophoresis and observed an increase from 11 ± 5% ALC-1 in donor control to 20 ± 5% in the myectomy samples (Figure 5).
As these differences were observed in all myectomy samples, but only two (M1, M5) carried an MYH7 mutation, it is evident that they are common consequences of HOCM and not directly related to disease-causing mutations in myosin. It is unlikely that the observed changes in motility are related to the changes seen in myosin light chains since an increased proportion of ALC-1 should increase sliding speed27 but no change was observed. The only recorded effect of MLC-2 phosphorylation indicated a decrease in Ca2+ sensitivity28 which was not measured here. Thus the defect in myosin is likely to be due to factors that are not related to protein isoform or phosphorylation levels.
Some fresh myectomy muscle samples from the same source were investigated for mechanical performance. In isolated myocytes, the contraction amplitude was reduced and the rate of relaxation was lower than in non-failing heart samples (Figure 1) and in muscle strips there was a negative relationship between isometric force and stimulation frequency.29 It is interesting to note that these contractility parameters are similar to those observed with end-stage failing heart or pressure overload21,30 rather than the hypercontractile phenotype predicted from molecular studies of hypertrophic cardiomyopathy-causing mutations.6,8
4.3. Myosin abnormalities are similar to failing heart myosin
Many of the differences in myectomy muscle myosin resemble those observed in failing heart muscle. Comparison of myectomy myosin and failing heart myosin with donor heart by in vitro motility assay shows a similar decrease in the fraction of filaments moving (Figure 3). The lower MLC-2 phosphorylation level (17% in myectomy muscle compared with 32% in non-failing) is about the same as that reported for failing heart: 18%, compared with 40% in donor heart.31 The level of MyBP-C phosphorylation is reduced about 50% in myectomy muscle32 and a similarly reduced level of phosphorylation has been observed in failing heart.33 We have also observed a low level of MyBP-C and troponin I phosphorylation in both failing heart17,31 and myectomy muscle samples.34 An increase in ALC-1 content in ventricular myosin from less than 5% in donor heart to 2–27% in hypertrophic and failing heart muscle has been reported,27,35 comparable with our finding of 11% in donor and 20% in myectomy muscle. We did not examine the amount of α and βMHC isoforms present, however an investigation by Noguchi et al.36 showed that the in vitro motility sliding speed was not different in failing and donor heart myosin despite a variation of the proportion of αMHC from 0 to 13%.
As the combination of increased expression of ALC-1 and decreased phosphorylation of MLC-2, troponin I, and MyBP-C is commonly observed in failing heart muscle,17,33,35 it appears that the contractile proteins in myectomy muscle are modified in a similar manner to failing heart. This is compatible with our observations that in both myocytes and in vitro motility assays contractility is similarly dysfunctional in myectomy and failing heart samples (Figures 1 and 3). On the other hand, some functional changes may be different in HOCM and failing heart; for instance, troponin from myectomy muscle shows the same Ca2+ sensitivity as donor heart34 rather than the increased Ca2+ sensitivity characteristic of failing heart muscle that is observed both by in vitro motility assay and also in intact skinned muscle.17,31 The origin of the reduced fraction motile in myectomy myosin cannot be attributed to changes in isoforms or phosphorylation which would influence the rate of crossbridge turnover. The fraction motile parameter is a measure of the population of active myosins. Inactivation may be due to a specific reason such as a mutation, however in the case of myosin motility we observe it in all samples, so the inactivation is probably a result of protein damage. Damage to myosin may be due to protein oxidation because increased oxidative stress is known to be a feature of hypertrophy.37 In support of this idea, it has been shown that motility and ATPase is reduced when skeletal or smooth muscle myosin is oxidized in vitro.38,39
Clinically, all the HOCM patients had diastolic dysfunction, but ejection fraction was usually not compromised. This seems at odds with the molecular phenotype of the myectomy samples, i.e. at least as severe as that observed in end-stage failing heart. It is possible that the dysfunction in the interventricular septum, from which our myectomy samples were taken, is not representative of muscle in the rest of the heart.40 The defining feature of HOCM is a high pressure difference between the aorta and ventricle due to LVOTO caused by septal hypertrophy, therefore it is possible that local dysfunction may occur as a consequence of pressure overload secondary to the hypertrophy triggered by contractile protein mutations.
4.4. M5 has a disease-causing MYH7 mutation
As mutations in MYH7 (βMHC gene) account for up to 25% of cases of hypertrophic cardiomyopathy, it was likely from the outset that at least one of our samples would have a disease-causing mutation in the βMHC. M5 stood out from the other five myectomy samples assayed as being functionally different (Table 2). In the in vitro motility assay, filament sliding speed was greater than the other myosin samples and relaxation at pCa9 was less complete. Both these properties are characteristic of mutations that cause hypertrophic cardiomyopathy in a variety of contractile proteins that have been studied at the single filament level.5,6 A higher rate of crossbridge turnover has been recognized as a feature of the few known MHC mutations that have been studied in muscle biopsies.8 The prediction that M5 carries a disease-causing hypertrophic cardiomyopathy mutation was confirmed by the subsequent mutation screening, showing that M3, M4, M7, M8, M10 are all wild-type for the whole of MYH7 but M5 is heterozygous for Val606Met. This mutation is one of the best documented hypertrophic cardiomyopathy-causing alleles. We did not detect a specific functional abnormality in M1, which also has an MYH7 mutation (Arg719Gln), however this sample was only tested for crossbridge turnover rate using pure actin and not for Ca2+ regulation of thin-filament motility which may have shown changes characteristic of HCM mutations. Previous studies by in vitro motility assay indicate HCM mutations often cause incomplete relaxation and/or increased Ca2+ sensitivity.5–7,27 It appears that faster crossbridge turnover rate is one of several diagnostic characteristics of hypertrophic cardiomyopathy mutations.
It should be noted that the decrease in the fraction of filaments motile and changes in myosin light chain expression and phosphorylation are exhibited by all samples including M5 (Figures 3, 5, and 7). This suggests that the hypocontractile state in the interventricular septum of patients with HOCM is not a direct effect of a hypertrophy-inducing mutation. The connection between an altered molecular phenotype such as that observed in M5 and the development of pathological hypertrophy remain unknown. It is anticipated that further functional and structural investigation of myectomy muscle samples, in conjunction with genotyping, will provide data that can start to explain the molecular mechanism of hypertrophic cardiomyopathy.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by grants from the British Heart Foundation. The genotyping was supported by the Oxford Partnership Comprehensive Biomedical Research Centre with funding from the Department of Health's NIHR Biomedical Research Centres funding scheme.
Supplementary Material
Acknowledgements
The donor heart samples were provided by Prof. C. Dos Remedios, Sydney, Australia. Technical assistance was provided by O'Neal Copeland, Peter O'Gara, Max Mioulane, Joanne Franklin, Trupti Patel, Allan Zheng and Will Johnson.
Conflict of interest: There are no conflicts of interest.
References
- 1.Redwood CS, Moolman-Smook JC, Watkins H. Properties of mutant contractile proteins that cause hypertrophic cardiomyopathy. Cardiovasc Res. 1999;44:20–36. doi: 10.1016/s0008-6363(99)00213-8. [DOI] [PubMed] [Google Scholar]
- 2.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 3.Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, et al. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:602–610. doi: 10.1016/j.jacc.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 4.Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, et al. Myosin-binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. doi: 10.1016/j.jacc.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 5.Knollmann BC, Potter JD. Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. Trends Cardiovasc Med. 2001;11:206–212. doi: 10.1016/s1050-1738(01)00115-3. [DOI] [PubMed] [Google Scholar]
- 6.Robinson PJR, Mirza M, Knott A, Abdulrazzak H, Marston S, Watkins H, et al. Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. J Biol Chem. 2002;277:40710–40716. doi: 10.1074/jbc.M203446200. [DOI] [PubMed] [Google Scholar]
- 7.Redwood C, Lohmann K, Bing W, Esoposito G, Elliott K, Abdulrazzak H, et al. Investigation of a truncated troponin T that causes familial hypertrophic cardiomyopathy: Ca2+ regulatory properties of reconstituted thin filaments depend on the ratio of mutant to wild-type peptide. Circ Res. 2000;86:1146–1152. doi: 10.1161/01.res.86.11.1146. [DOI] [PubMed] [Google Scholar]
- 8.Palmiter KA, Tyska MJ, Haeberle JR, Alpert NR, Fananapazir L, Warshaw DM. R403Q and L908V mutant beta-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J Musc Res Cell Motil. 2000;21:609–620. doi: 10.1023/a:1005678905119. [DOI] [PubMed] [Google Scholar]
- 9.James J, Zhang Y, Osonska H, Sanbe A, Klevitsky R, Hewett TE, et al. Transgenic modelling of a cardiac troponin I mutation linked to familial hypertrophic cardiomyopathy. Circ Res. 2000;87:805–811. doi: 10.1161/01.res.87.9.805. [DOI] [PubMed] [Google Scholar]
- 10.Tardiff JC, Factor SM, Tompkins BD, Hewett TE, Palmer BM, Moore RL, et al. A truncated troponin T molecule in transgenic mice suggests multiple cellular mechanisms for familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:2800–2811. doi: 10.1172/JCI2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashrafian H, Watkins H. Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications cardiomyopathies—therapeutics based on molecular phenotype. J Am Coll Cardiol. 2007;49:1251–1264. doi: 10.1016/j.jacc.2006.10.073. [DOI] [PubMed] [Google Scholar]
- 12.Kohler J, Winkler G, Schulte T, McKenna W, Brenner B, Kraft T. Mutation of the myosin converter domain alters cross-bridge elasticity. Proc Natl Acad Sci USA. 2002;99:3557–3562. doi: 10.1073/pnas.062415899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirschner SE, Becker E, Antognozzi M, Kubis HP, Francino A, Navarro-Lopez F, et al. Hypertrophic cardiomyopathy-related beta-myosin mutations cause highly variable calcium sensitivity with functional imbalances among individual muscle cells. Am J Physiol Heart Circ Physiol. 2005;288:H1242–H1251. doi: 10.1152/ajpheart.00686.2004. [DOI] [PubMed] [Google Scholar]
- 14.Elliott PM, Gimeno JR, Tome MT, Shah J, Ward D, Thaman R, et al. Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. Eur Heart J. 2006;27:1933–1941. doi: 10.1093/eurheartj/ehl041. [DOI] [PubMed] [Google Scholar]
- 15.Firoozi S, Elliott PM, Sharma S, Murday A, Brecker SJ, Hamid MS, et al. Septal myotomy–myectomy and transcoronary septal alcohol ablation in hypertrophic obstructive cardiomyopathy: a comparison of clinical, haemodynamic and exercise outcomes. Eur Heart J. 2002;23:1617–1624. doi: 10.1053/euhj.2002.3285. [DOI] [PubMed] [Google Scholar]
- 16.Morrow AG, Reitz BA, Epstein SE, Henry WL, Conkle DM, Itscoitz SB, et al. Operative treatment in hypertrophic subaortic stenosis: techniques and the results of pre- and postoperative assessments in 83 patients. Circulation. 1975;52:88–102. doi: 10.1161/01.cir.52.1.88. [DOI] [PubMed] [Google Scholar]
- 17.Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol. 2007;42:247–259. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 18.Fraser IDC, Marston SB. In vitro motility analysis of actin–tropomyosin regulation by troponin and Ca2+: the thin filament is switched as a single cooperative unit. J Biol Chem. 1995;270:7836–7841. doi: 10.1074/jbc.270.14.7836. [DOI] [PubMed] [Google Scholar]
- 19.Morano I, Hadicke K, Haase H, Bohm M, Erdmann E, Schaub MC. Changes in essential myosin light chain isoform expression provide a molecular basis for isometric force regulation in the failing human heart. J Mol Cell Cardiol. 1997;29:1177–1187. doi: 10.1006/jmcc.1996.0353. [DOI] [PubMed] [Google Scholar]
- 20.Steinberg TH, Agnew BJ, Gee KR, Leung WY, Goodman T, Schulenberg B, et al. Global quantitative phosphoprotein analysis using multiplexed proteomics technology. Proteomics. 2003;3:1128–1144. doi: 10.1002/pmic.200300434. [DOI] [PubMed] [Google Scholar]
- 21.Davies CH, Davia K, Bennett JG, Pepper JR, Poole-Wilson PA, Harding SE. Reduced contraction and altered frequency response of isolated ventricular myocytes from patients with heart failure. Circulation. 1995;92:2540–2549. doi: 10.1161/01.cir.92.9.2540. [DOI] [PubMed] [Google Scholar]
- 22.Knott A, Purcell IF, Marston S. In vitro motility analysis of thin filaments from failing and non-failing human hearts induces slower filament sliding and higher Ca2+ sensitivity. J Mol Cell Cardiol. 2002;34:469–482. doi: 10.1006/jmcc.2002.1528. [DOI] [PubMed] [Google Scholar]
- 23.Bing W, Fraser IDC, Marston SB. Troponin I and troponin T interact with troponin C to produce different Ca2+-dependant effects on actin–tropomyosin filament motility. Biochem J. 1997;327:335–340. doi: 10.1042/bj3270335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malmqvist UP, Aronshtam A, Lowey S. Cardiac myosin isoforms from different species have unique enzymatic and mechanical properties. Biochemistry. 2004;43:15058–15065. doi: 10.1021/bi0495329. [DOI] [PubMed] [Google Scholar]
- 25.Kron SJ, Toyoshima YY, Uyeda TQP, Spudich JA. Assays for actin sliding movement over myosin coated surfaces. Methods Enzymol. 1991;196:399–416. doi: 10.1016/0076-6879(91)96035-p. [DOI] [PubMed] [Google Scholar]
- 26.Marston S. Random walks with thin filaments: application of in vitro motility assay to the study of actomyosin regulation. J Musc Res Cell Motil. 2003;24:149–156. doi: 10.1023/a:1026097313020. [DOI] [PubMed] [Google Scholar]
- 27.Morano M, Zacharzowski U, Maier M, Lange PE, Alexi-Meskishvili V, Haase H, et al. Regulation of human heart contractility by essential myosin light chain isoforms. J Clin Invest. 1996;98:467–473. doi: 10.1172/JCI118813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Velden J, Papp Z, Boontje NM, Zaremba R, de Jong JW, Janssen PM, et al. The effect of myosin light chain 2 dephosphorylation on Ca2+ sensitivity of force is enhanced in failing human hearts. Cardiovas Res. 2003;57:505–514. doi: 10.1016/s0008-6363(02)00662-4. [DOI] [PubMed] [Google Scholar]
- 29.Gray R, Jacques A, Peters N, McKenna W, Walker J, McIntyre H, et al. Electromechanical properties of isolated myocardium from patients with hypertrophic obstructive cardiomyopathy (HOCM) Circulation. 2005;112:U149. [Google Scholar]
- 30.McIntyre H, Fry CH. Abnormal action potential conduction in isolated human hypertrophied left ventricular myocardium. J Cardiovasc Electrophysiol. 1997;8:887–894. doi: 10.1111/j.1540-8167.1997.tb00850.x. [DOI] [PubMed] [Google Scholar]
- 31.van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, et al. Increased Ca2+ sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 32.Gallon CE, Messer AE, Copeland O, Jacques A, Tsang V, McKenna WJ, et al. Post-translational modification of TnT, TnI, MyBP-C and MLC-2 in HOCM human heart muscle. J Muscle Res Cell Motil. 2007 doi:10.1007/S10974-008-9127-Z abstract#3:9. [Google Scholar]
- 33.El-Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Yeh YH, Nattel S, et al. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J Mol Cell Cardiol. 2007;43:223–229. doi: 10.1016/j.yjmcc.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 34.Jacques A, Messer A, Tsang V, McKenna W, Marston S. Evidence for reduced troponin I phosphorylation and altered troponin function in patients with hypertrophic obstructive cardiomyopathy. J Mol Cell Cardiol. 2006;40:939. [Google Scholar]
- 35.Palmer BM. Thick filament proteins and performance in human heart failure. Heart Fail Rev. 2005;10:187–197. doi: 10.1007/s10741-005-5249-1. [DOI] [PubMed] [Google Scholar]
- 36.Noguchi T, Camp P, Jr, Alix SL, Gorga JA, Begin KJ, Leavitt BJ, et al. Myosin from failing and non-failing human ventricles exhibit similar contractile properties. J Mol Cell Cardiol. 2003;35:91–97. doi: 10.1016/s0022-2828(02)00282-1. [DOI] [PubMed] [Google Scholar]
- 37.Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93:903–907. doi: 10.1136/hrt.2005.068270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coirault C, Guellich A, Barbry T, Samuel JL, Riou B, Lecarpentier Y. Oxidative stress of myosin contributes to skeletal muscle dysfunction in rats with chronic heart failure. Am J Physiol Heart Circ Physiol. 2006;292:H1009–H1017. doi: 10.1152/ajpheart.00438.2006. [DOI] [PubMed] [Google Scholar]
- 39.Penheiter AR, Bogoger M, Ellison PA, Oswald B, Perkins WJ, Jones KA, et al. H2O2-induced kinetic and chemical modifications of smooth muscle myosin: correlation to effects of H2O2 on airway smooth muscle. J Biol Chem. 2007;282:4336–4344. doi: 10.1074/jbc.M609499200. [DOI] [PubMed] [Google Scholar]
- 40.Yamazaki T, Suzuki J, Shimamoto R, Tsuji T, Ohmoto Y, Toyo-Oka T, et al. Focalized contractile impairment at hypertrophied myocardium proven in consideration of wall stress in patients with hypertrophic cardiomyopathy. Int Heart J. 2006;47:247–258. doi: 10.1536/ihj.47.247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.