

Abstract
Oxidative stress contributes to the arrhythmogenic substrate created by myocardial ischemia-reperfusion partly through a shift in cell redox state, a key modulator of protein function. The activity of many oxidation-sensitive proteins is controlled by oxidoreductase systems that regulate the redox state of cysteine thiol groups, but the impact of these systems on ion channel function is not well defined. Thus, we examined the roles of the thioredoxin and glutaredoxin systems in controlling K+ channels in the ventricle. An oxidative shift in redox state was elicited in isolated rat ventricular myocytes by brief exposure to diamide, a thiol-specific, membrane-permeable oxidant. Voltage-clamp studies showed that diamide decreased peak outward K+ current (Ipeak) evoked by depolarizing test pulses by 41% (+60 mV; p<0.05) while steady-state outward current (Iss) measured at the end of the test pulse was decreased by 45% (p<0.05). These electrophysiological effects were not prevented by protein kinase C blockers, but the tyrosine kinase inhibitors genistein or lavendustin A blocked the suppression of both K+ currents by diamide. Moreover, inhibition of Ipeak and Iss by diamide was reversed by dichloroacetate and an insulin-mimetic. The effect of dichloroacetate to normalize Ipeak after diamide was blocked by the thioredoxin system inhibitors auranofin or 13-cis-retinoic acid, but Iss was not affected by either compound. A pan-specific inhibitor of glutaredoxin and thioredoxin systems, 1,3-bis-(2-chloroethyl)-1-nitrosourea, also blocked the dichloroacetate effect on Ipeak but only partially inhibited the recovery of Iss. These data suggest that acute regulation of cardiac K+ channels by oxidoreductase systems is mediated by redox-sensitive tyrosine kinase/phosphatase pathways. The pathways controlling Ipeak channels are targets of the thioredoxin system whereas those regulating Iss channels are likely controlled by the glutaredoxin system. Thus, cardiac oxidoreductase systems may be important regulators of ion channels affected by pathogenic oxidative stress.
Keywords: Redox, K+ channels, Glutaredoxin, Thioredoxin
INTRODUCTION
Ventricular dysfunction and arrhythmias induced by myocardial ischemia-reperfusion are closely linked to increased production of reactive oxygen species (ROS) and oxidative damage to cellular proteins. Although most amino acid residues can be oxidized, the reversible oxidative modification of proteins mainly involves the reactivity of the free thiol (-SH) group of cysteine residues [1, 2]. This side chain can exist in a number of different molecular states that can mediate activation or inactivation of the target protein [3]. Thus, thiol biochemistry plays an important role in cell biology since the redox status of critical cysteine thiols controls the structure and activity of many enzymes, receptors, transcription factors and transport proteins required for normal cell function. The thioether side chain of methionine residues also undergoes reversible oxidative modification that can profoundly affect protein function [4].
Control of the redox state of ROS-sensitive protein thiols involves the activities of endogenous enzymes that belong to the oxidoreductase superfamily [5]. Thioredoxin-1 (Trx) and glutaredoxin-1 (Grx) are members of this superfamily that function within the cytoplasm to detoxify hydroperoxides and to maintain protein thiols in a normally reduced state [2, 6]. A different pair of functionally related enzymes is also localized to the mitochondria [7]. Trx and Grx are each part of an integrated system that controls the transfer of electrons from NADPH to oxidized proteins. In the thioredoxin system, the function of Trx depends on the upstream activity of thioredoxin reductase (TrxR), which uses NADPH as cofactor to convert the oxidized, inactive form of Trx to its reduced active form [8, 9, 10]. For the glutaredoxin system, electrons are transferred from NADPH to glutathione reductase (GR), then to glutathione (GSH) which converts oxidized Grx to its active form [1, 2, 11]. While both Trx and Grx reduce oxidized thiols, they display unique substrate specificities [1, 2, 10]. In general, reduced Trx catalyzes the reduction of intra- or intermolecular protein disulfides [8, 9, 12] and possibly other thiol intermediates such as sulfenic acid and S-nitrosothiols [10]. By comparison, Grx mainly catalyzes the reduction of protein-mixed disulfides [1, 2, 11, 13]. Since a major function of Trx and Grx is to convert oxidized proteins to their reduced form, it is proposed that oxidoreductase systems act as a thiol repair mechanism that regulates the physiological function of proteins susceptible to oxidation.
Many cardiac ion channels have been shown to be sensitive to ROS and related thiol-reactive molecules in a reversible manner that suggests the channel protein itself or a modulator of the channel is controlled by cellular redox state [14–17]. In most cases, the reversibility or prevention of oxidative changes to ion channels has been examined using exogenous reductants such as dithiothreitol. While this approach helps to identify redox-mediated mechanisms of regulation, little is known about endogenous redox systems that control ion channel function under oxidative conditions. Thus, the present study was undertaken to assess the functional role of endogenous oxidoreductase systems in regulating ion channel activity. Using the thiol-specific oxidant diamide to elicit acute oxidative conditions, our electrophysiological studies suggest that voltage-gated K+ channels in rat ventricular myocytes are differentially regulated by the thioredoxin and glutaredoxin systems but that this regulation is mediated by redox-sensitive changes in tyrosine kinase/phosphatase pathways. Thus, endogenous redox systems may be important regulators of ion channel activity under pathogenic conditions associated with oxidative stress.
MATERIALS and METHODS
Isolation of ventricular myocytes. Patch-clamp technique
All animal procedures were carried out in accordance with guidelines approved by the Animal Care and Use Committee, University of Nebraska Medical Center and conformed with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). Male Sprague Dawley rats (150–180 g) were given an overdose of pentobarbital sodium (150 mg/kg, i.p.) and single ventricular myocytes were dissociated from excised, perfused hearts by a collagenase digestion procedure described previously [16, 18, 19]. Dissociated myocytes from left ventricle and septum were suspended in Dulbecco’s modified Eagle’s medium and stored in an incubator at 35° C until used, usually within 6 h of isolation. Aliquots of myocytes were transferred to a cell chamber mounted on the stage of an inverted microscope and superfused with an external solution containing (in mmol/l): 138 NaCl, 4.0 KCl, 1.2 MgCl2, 1.8 CaCl2, 18 glucose, 5 HEPES, pH 7.4. This solution also contained 0.5 mmol/l Cd2+ to block Ca2+ channels.
Ionic currents were recorded using the whole-cell configuration of the patch-clamp technique. Briefly, borosilicate glass pipettes with tip diameter of ~2 µm were filled with a solution containing (in mmol/l): 135 KCl, 3 MgCl2, 10 HEPES, 3 Na2-ATP, 10 EGTA, 0.5 Na-GTP, pH 7.2. Pipettes with resistances of 2–4 MΩ were coupled to a patch-clamp amplifier (Axopatch 200B, Axon Instruments, Union City, CA) and the liquid junction potential corrected. After formation of a gigaohm seal the membrane within the pipette was ruptured and series resistance was compensated. A computer program (pClamp, Axon Instruments) controlled command potentials and acquired current signals that were filtered at 2 kHz. Currents were sampled at 4 kHz by a 12-bit resolution analog-to-digital converter and stored on the hard disk of a computer. All experiments were done at room temperature (22–24 °C).
Outward K+ currents were evoked in each cell by 500 ms depolarizing pulses (0.2 Hz) to test potentials from −40 to +60 mV. The holding potential was −80 mV and a 100 ms pre-pulse was applied to −60 mV to inactivate the fast Na+ current. For each test pulse, the peak outward current (Ipeak) and steady-state current (Iss) at the end of the clamp pulse were measured. Data were normalized as current densities by dividing measured current amplitude by whole-cell capacitance. In addition to measuring I-V relations, voltage-dependent steady-state parameters of Ipeak were also determined. First, steady-state activation parameters were derived from recorded I-V relations by calculating the conductance (G) at each test potential (Vm), normalized to maximum conductance at +60 mV (G/Gmax), and plotting these values as a function of Vm. Data were fitted by a Boltzmann distribution to determine the steady-state activation parameters V1/2 and k according to the relationship:
where V1/2 is the voltage at half-maximal activation, and k the slope factor at Vm = V1/2. Steady-state inactivation was examined by applying 500 ms pre-pulses from −100 to 0 mV prior to pulsing to +60 mV and inactivation curves constructed by plotting normalized current (I/Imax) against pre-pulse voltage. These data were also fitted by a Boltzmann distribution to derive the steady-state inactivation parameters V1/2 and k.
The present study utilized the diazine dicarbonyl compound diamide to elicit acute oxidative conditions in isolated myocytes superfused with standard extracellular solution. This thiol-specific molecule rapidly enters cells and primarily oxidizes GSH to its disulfide form (GSSG), thereby perturbing cellular redox balance independent of ROS [20]. In some experiments, the electrophysiological effects of diamide were compared with H2O2. The redox regulation of K+ currents was studied using an experimental protocol consisting of 20 min exposure to diamide followed by washout with standard external solution. Ionic currents were recorded during the washout period. This pre-treatment protocol was used because diamide typically destabilized the tight seal, which precluded longer recording periods needed to characterize the time course of current changes in a given cell.
Enzyme activities
TrxR activity was measured by a modification of the insulin disulfide reduction assay [9]. Briefly, aliquots of cell lysate from isolated myocytes were added to a reaction mixture containing 80 mmol/l HEPES (pH 7.5), 6 mmol/l EDTA, 0.9 mg/ml NADPH and 2 mg/ml insulin. The reaction was started by adding 5 µmol/l E. coli Trx and the samples heated to 37 °C for 20 min. The reaction was stopped by adding 500 µl of 0.4 mg/ml DTNB/6 mol/l guanidine-HCl in 0.2 mol/l Tris-HCl, pH 8.0, and the absorbance read at 412 nm. Measured absorbance from samples were compared to standard curves generated with known amounts of rat liver TrxR.
GR activity was measured by the technique of Carlberg and Mannervik [21]. For this assay, cell suspensions were homogenized in ice-cold Tris buffer (0.1 M, pH 8.0 with 2 mmol/l EDTA) and centrifuged at 4 °C (6000 g) for 30 min. A 200 µl aliquot of the supernatant was added to a cuvette containing KH2PO4 buffer (0.2 mol/l, pH 7.0) plus 2 mmol/l EDTA, 20 mmol/l GSSG and 2 mmol/l NADPH. The change in absorbance at 340 nm was monitored for 5 min at 30 °C. GR activity was expressed in mU defined as the amount of enzyme catalyzing the reduction of 1 nmol NADPH per minute. Unless stated otherwise, all other compounds used in these experiments were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO).
Statistical analysis
All results are expressed as mean ± SEM. Statistical comparisons of two groups were made using an unpaired Student’s t-test, whereas more than two groups were compared by analysis of variance. When a significant difference among groups was indicated by the initial analysis, individual comparisons were made using a Student-Newman-Keuls modified t-test. Differences were considered significant at p<0.05.
RESULTS
Inhibition of K+ currents by diamide
Previous fluorescence microscopy studies from our laboratory have shown that 100 µmol/l diamide significantly decreases intracellular GSH concentration by 41% compared with untreated myocytes [19], which is consistent with cell oxidation. The electrophysiological effects of diamide are illustrated in Figure 1A which compares superimposed currents elicited at test potentials from −40 to +60 mV in an untreated, control myocyte (upper traces) with another cell pre-treated with 100 µmol/l diamide. The bottom traces show that in the diamide-treated myocyte, Ipeak and Iss were markedly decreased compared with control. Mean data from several cells are summarized in Figures 1B and 1C which also show the concentration-dependence of diamide. In our experiments, 10 µmol/l diamide elicited a consistent but relatively small decrease in Ipeak and Iss (p>0.05) whereas 100 µmol/l diamide significantly decreased both currents over the voltage range of −10 to +60 mV. At +60 mV, 100 µmol/l diamide decreased Ipeak and Iss density from control by 41% and 45%, respectively. Given the concentration-response of K+ currents shown in Figures 1B and 1C, we used a test concentration of 100 µmol/l diamide for all subsequent experiments.
Figure 1.
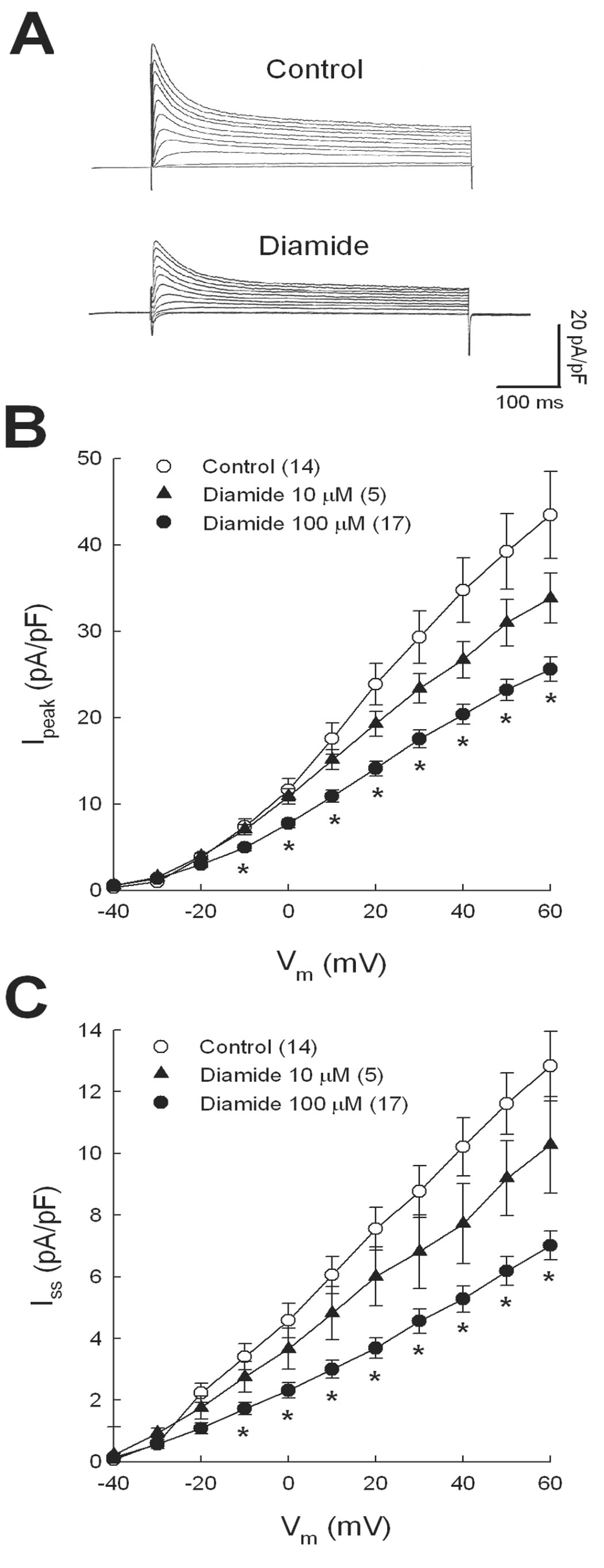
Concentration-dependent inhibition of K+ currents by diamide. Ventricular myocytes were superfused with 10 or 100 µmol/l diamide and K+ currents recorded after washout. A: superimposed current traces recorded at test potentials from −40 to +60 mV. Mean I-V relations for Ipeak (B) and Iss (C) were measured from standard clamp protocols described in Methods. * = p<0.05 compared with control. Numbers in parentheses represent number of myocytes studied.
Since diamide perturbs cellular redox balance independent of ROS, we compared its electrophysiological effects with H2O2, which is produced endogenously during the dismutation of superoxide anion [22, 23]. As summarized in Figure 2A, pre-treating myocytes with H2O2 for 20 min decreased Ipeak to a similar extent as diamide. Specifically, 10 µmol/l H2O2 decreased Ipeak at +60 mV by 48% from control (p<0.05), whereas 1 µmol/l H2O2 decrease Ipeak by 24% (p<0.05). In contrast to diamide however, neither concentration of H2O2 significantly altered Iss except at −30 mV where 10 µmol/l H2O2 increased Iss compared with control.
Figure 2.
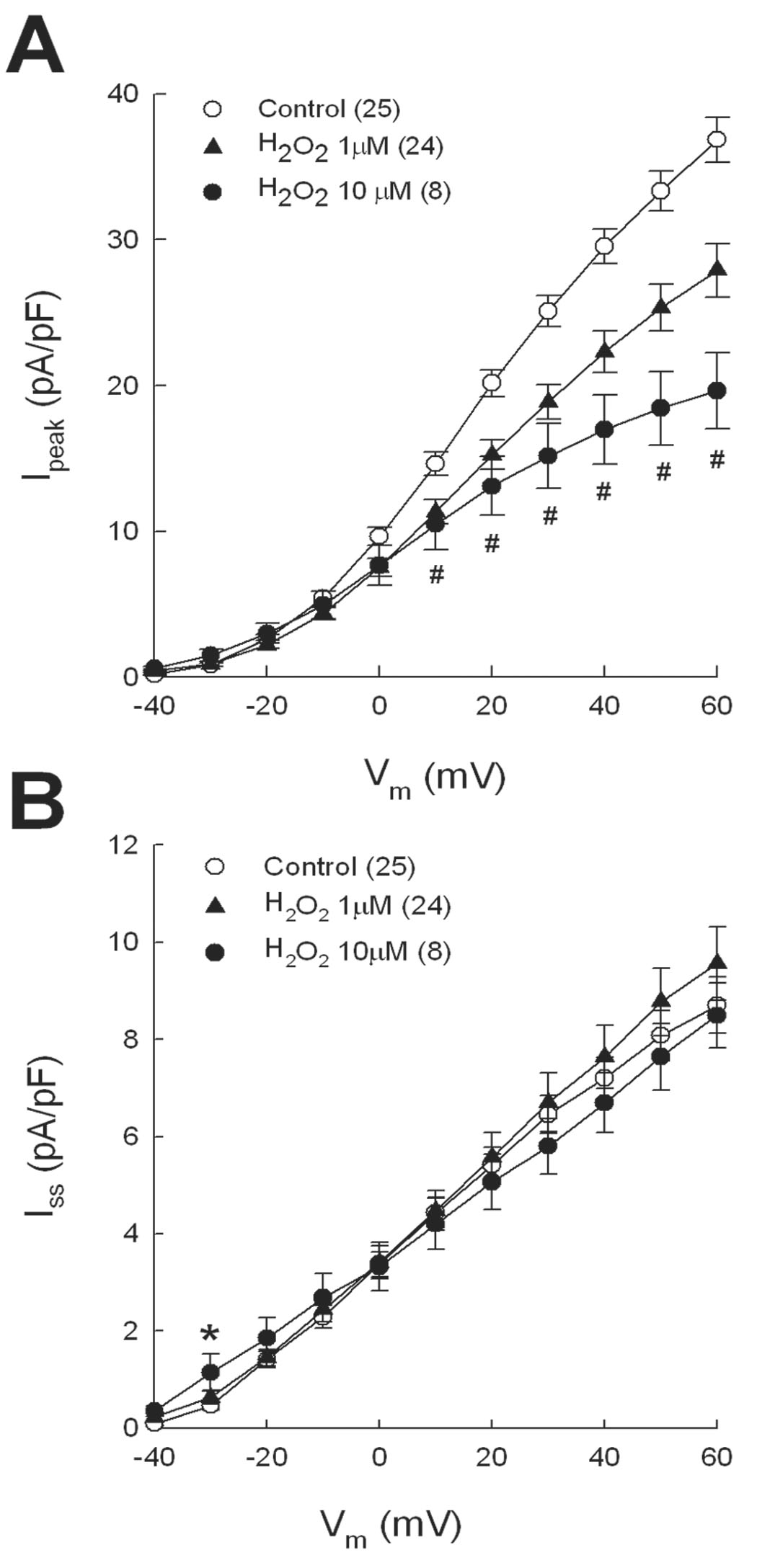
Inhibition of Ipeak by H2O2. The same experimental protocol used for diamide was repeated with 1 or 10 µmol/l H2O2. Mean I-V relations are shown for Ipeak (A) and Iss (B). # = p<0.05 for both 1 and 10 µM H2O2 compared with control (panel A). * = p<0.05 compared with control (panel B).
The covalent modification of free sulfhydryl groups by thiol-reactive agents generally results in stable intermediates, and thus it was expected that inhibition of K+ currents by diamide or H2O2 would persist after washout of oxidant. To assess this, currents were recorded from several myocytes in nine different test protocols at various time points during washout of diamide and mean data were compared between an arbitrarily chosen early and late time period. In 20 myocytes studied after 15–45 min of washout, mean Ipeak and Iss densities measured at +60 mV were 23.4±1.8 and 7.3±0.7 pA/pF, respectively. With a longer washout period (46–100 min), the respective mean current densities were 25.9±2.3 and 8.0±0.9 pA/pF (n=16). There was no significant difference in maximum Ipeak or Iss between the two washout periods (p>0.05), but both sets of data were significantly less than control (43.4±5.0 and 12.8±1.1 pA/pF, respectively; p<0.05), indicating there was no spontaneous recovery of K+ currents on washout alone. A similar stable inhibition of Ipeak was also observed in myocytes pre-treated with 10 µmol/l H2O2 (data not shown).
To further examine the impact of diamide on Ipeak properties, time- and voltage-dependent parameters were measured and are summarized in Table 1. Compared with control, diamide caused a significant shift in the steady-state activation curve to more negative potentials by approximately 8 mV (p<0.05). The steady state inactivation curve for diamide treated cells was also significantly shifted leftward compared with control by ~12 mV (p<0.05). The slope factor k for the activation and inactivation curves was not altered by diamide.
Table 1.
Diamide-induced changes in time- and voltage-dependent properties of Ipeak
| Control | Diamide | |
|---|---|---|
| Inactivation kinetics | ||
| n | 11 | 13 |
| T, ms | 38.5 ± 2.2 | 33.7 ± 1.2 |
| Steady-state activation | ||
| n | 12 | 12 |
| V½, mV | 4.0 ± 2.0 | −3.9 ± 2.2* |
| k,mV | 12.6 ± 0.5 | 13.6 ± 1.0 |
| Steady-state inactivation | ||
| n | 7 | 16 |
| V½, mV | −46.4 ± 3.1 | −58.8 ± 2.2* |
| k,mV | −11.6 ± 5.2 | −14.9 ± 1.6 |
Data were collected from untreated, control myocytes and a separate group pre-treated with 100 µM diamide. Values are means ± SEM. Inactivation kinetics (T) were measured at +60 mV. n, number of myocytes. V½, voltage at 50% activation or inactivation. k, slope factor.
p<0.05 compared with control.
Mediators of K+ current inhibition
It has previously been shown that activation of protein kinase C (PKC) decreases Ipeak and Iss in rat ventricular myocytes [24, 25] and that oxidants reacting with the N-terminal regulatory domain of PKC stimulate its activity [26]. Although not studied as extensively as PKC, it is also known that activation of other kinase pathways, such as tyrosine kinase, can modulate K+ channels in heart [27]. Thus, to determine if K+ current inhibition by diamide was mediated by a phosphorylase mechanism, a first series of experiments was done where cells were pre-treated with the pan-specific PKC inhibitors calphostin C (100 nmol/l) or GF109203x (50 nmol/l) for 30 min before adding diamide for 20 min. As in our other experiments, Ipeak and Iss were recorded after washout of diamide. Figure 3A shows that pre-treating cells with PKC inhibitors did not significantly alter the effect of diamide to inhibit Ipeak: mean Ipeak densities at +60 mV for diamide, calphostin C+diamide, and GF109203x+diamide groups were decreased from control by 41, 35, and 28%, respectively (p<0.05 compared with control). Similarly, PKC inhibitors did not affect suppression of Iss by diamide (Fig. 3B): mean Iss densities at +60 mV for diamide, calphostin C+diamide and GF109203x+diamide groups were decreased from control by 45, 35, and 48%, respectively (p<0.05 compared with control). However, the electrophysiological effects of diamide were blocked by pan-specific inhibitors of protein tyrosine kinases. Thus in myocytes treated with genistein (10 µmol/l) or lavendustin A (1 µmol/l) prior to diamide, neither Ipeak (Fig. 3A) nor Iss (Fig. 3B) was different from control, suggesting that protein tyrosine kinases or phosphatases mediated the inhibitory actions of diamide on K+ currents. None of the kinase inhibitors alone had direct effects on Ipeak or Iss when examined in control myocytes pre-treated for 45–60 min: Ipeak densities at +60 mV in untreated myocytes and those treated with 100 nmol/l calphostin C, 50 nmol/l GF109203x, 10 µmol/l genistein or 1 µmol/l lavendustin A were: 43.4±5.0 (n=14), 42.6±2.9 (n=7), 43.1±4.6 (n=6), 46.7±3.3 (n=6), and 42.1±4.8 (n=8) pA/pF, respectively (p>0.05); mean Iss densities in the same groups were 12.8±1.1, 11.0±2.2, 10.8±2.3, 13.2±1.5, and 12.5±2.3 pA/pF, respectively (p>0.05).
Figure 3.
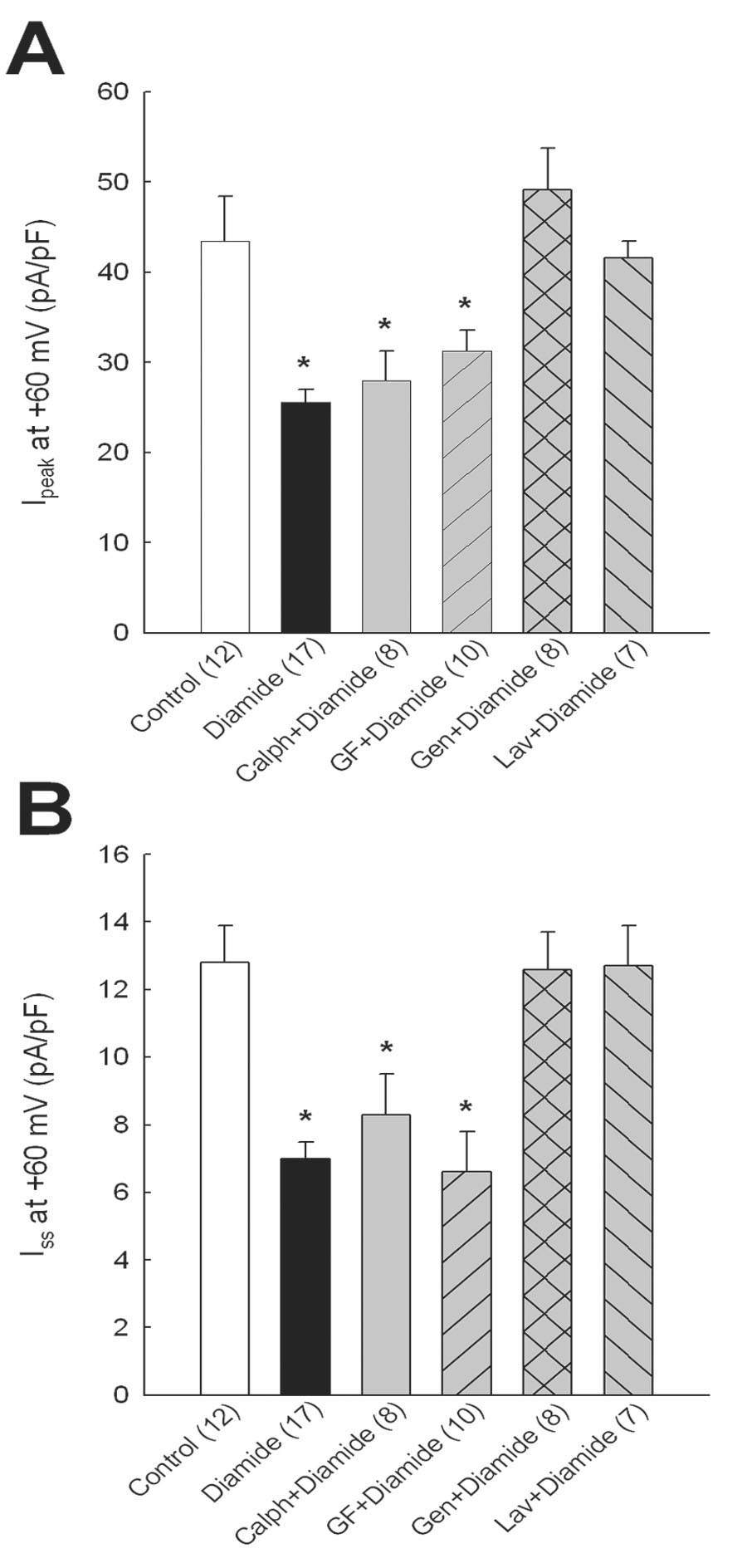
Effects of kinase inhibitors on diamide-induced K+ current inhibition. Myocytes were treated with calphostin C (100 nmol/l), GF109203x (50 nmol/l), genistein (10 µmol/l) or lavendustin A (1 µmol/l) for 30 min before adding 100 µmol/l diamide for 20 min. Ipeak (A) and Iss (B) were recorded after washout of diamide. * = p<0.05 compared with control.
Regulation of K+ currents by oxidoreductase systems
In previous studies of myocytes from infarcted or diabetic rat hearts, we showed that activators of glucose metabolism up-regulate K+ currents through redox-mediated mechanisms [18, 28, 29]. In the present study we tested the effects of dichloroacetate (DCA) and the insulin mimetic bis-peroxovanadium-1, 10-phenanthroline (bpV(phen)) to reverse K+ current inhibition by diamide. DCA stimulates pyruvate dehydrogenase activity as well as glucose-6-phosphate dehydrogenase (G6PD), both of which are key regulators of NADPH production via glucose metabolism [30, 31]. By comparison bpV(phen) inhibits receptor tyrosine phosphatases, thereby activating the insulin receptor kinase and downstream pathways that include G6PD [32]. After incubating myocytes with diamide, the oxidant was washed out with external solution containing 1.5 mmol/l DCA or 10 μmol/l bpV (phen). As shown by I-V curves in Figures 4A and 4B, Ipeak and Iss densities were significantly increased by the addition of DCA (open triangles) or bpV (phen) compared with myocytes treated with diamide alone (filled triangles). For purposes of comparison, data from untreated control myocytes (open circles) are also shown. Of the compounds tested, DCA more completely normalized both currents after diamide treatment, and was thus used in subsequent experiments. It should also be noted that DCA alone had no effect on K+ currents in control myocytes: Ipeak at +60 mV in DCA-treated and untreated control cells was 43.9±3.7 (n=8) and 43.4±5.0 (n=12) pA/pF, respectively (p>0.05); mean Iss at the same test voltage was 12.1±2.0 and 12.8±1.1 pA/pF, respectively (p>0.05).
Figure 4.
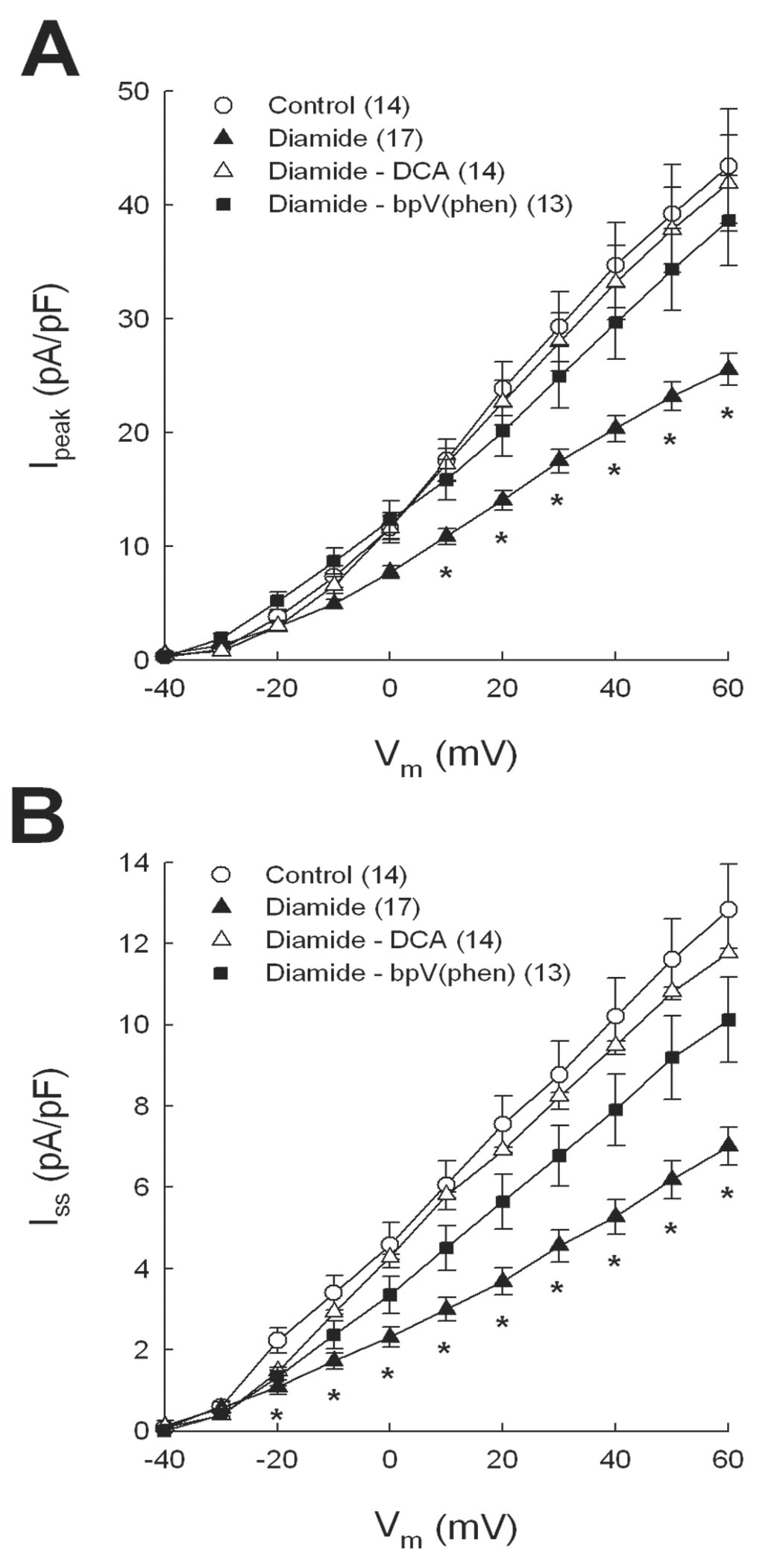
Recovery of Ipeak (A) and Iss (B) after diamide treatment by metabolic activators. Myocytes were exposed to 100 µmol/l diamide and then superfused with 1.5 mmol/l dichloroacetate (DCA) or 10 µmol/l bis-peroxovanadium- 1,10-phenanthroline (bpV(phen)). K+ currents were recorded after 15 min of exposure to metabolic activators. * = p<0.05 compared with control.
Regulation of K+ channels by oxidoreductase systems was examined by using the response of diamide-treated myocytes to DCA as an experimental end point to test the efficacy of inhibitors of the thioredoxin and glutaredoxin systems. Since specific pharmacological blockers of Trx and Grx have not been developed, we used known inhibitors of their upstream regulators, TrxR and GR, respectively. For these studies, myocytes were incubated with inhibitor for 30 min before adding diamide for 20 min. Diamide was then washed out with external solution containing DCA plus reductase inhibitor. Figure 5A compares the response of Ipeak to DCA in diamide-treated myocytes without and with the TrxR inhibitor auranofin (AF) [33]. In the absence of TrxR inhibition DCA significantly increased Ipeak density to control levels (open triangles) after diamide treatment, but in the presence of the 10 nmol/l AF, Ipeak density was not increased by DCA. In contrast, AF did not block the effect of DCA to increase Iss after diamide (Fig. 5B), such that I-V relations for the DCA (open triangles) and AF+DCA (filled squares) groups of myocytes were nearly identical. The effects of DCA and TrxR inhibition were also examined in myocytes treated with H2O2. Figure 5C shows that DCA up-regulated Ipeak after 10 µmol/l H2O2 treatment and this response was blocked by AF.
Figure 5.
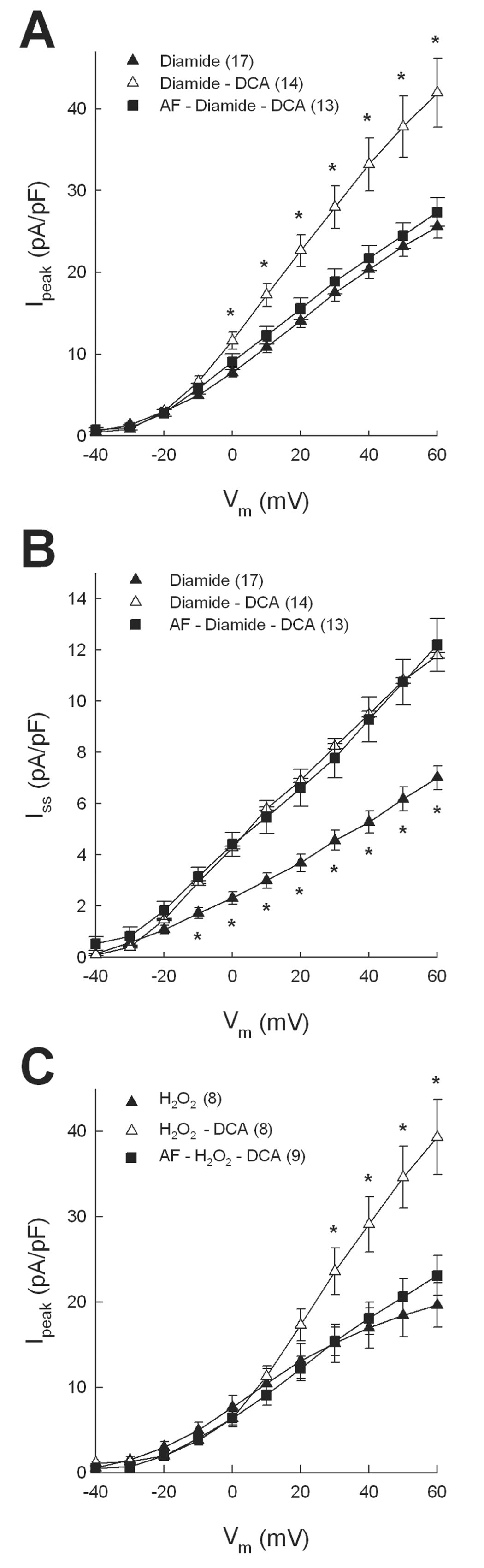
Block of DCA-mediated recovery of Ipeak by the TrxR inhibitor auranofin (AF). Myocytes were pretreated with 10 nmol/l AF before superfusion with diamide followed by washout with DCA. AF blocked the effect of DCA to increase Ipeak (A) but not Iss (B). AF also blocked the effect of DCA to up-regulate Ipeak after 10 µmol/l H2O2 (C). In panels A and C * = p<0.05 compared with diamide alone. In panel B * = p<0.05 compared with diamide-DCA.
Experiments similar to those summarized in Figure 5 also tested the effects of another TrxR inhibitor, 13-cis-retinoic acid (RA) [34], and a pan-specific reductase inhibitor 1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU) [35]. Figures 6A and 6B show that both RA (1µmol/l) and BCNU (100 µmol/l) completely blocked the response of Ipeak to DCA in both diamide- and H2O2-treated myocytes. Figure 6C further shows that RA did not block the effect of DCA on Iss in diamide-treated cells, similar to AF studies (Fig. 5B), but that the pan-specific inhibitor BCNU partially blocked the DCA effect. Thus, in the presence of BCNU plus DCA, maximum Iss density was significantly less than control but significantly greater than diamide alone. As with kinase blockers, reductase inhibitors alone had no direct effect on K+ currents in control myocytes: Ipeak densities at +60 mV in untreated myocytes and those treated with 10 nmol/l AF, 1 µmol/l RA or 100 µmol/l BCNU were: 43.4±5.0 (n=14), 41.9±5.2 (n=7), 47.3±2.3 (n=11), and 40.9±4.5 (n=8) pA/pF, respectively (p>0.05); mean Iss densities in the same groups were 12.8±1.1, 13.8±1.5, 14.0±1.2, and 10.9±0.8 pA/pF, respectively (p>0.05). Finally, to verify the specificity of oxidoreductase inhibitors separate groups of control myocytes were treated with 10 nmol/l AF, 1µmol/l RA or 100 µmol/l BCNU for 30 min and assayed for TrxR and GR activities. Figure 6D shows that AF, RA and BCNU all significantly inhibited TrxR activity by 50–70% (p<0.05), whereas only BCNU significantly inhibited GR. These analyses therefore indicate that AF and RA are specific inhibitors of TrxR and support the conclusion that the thioredoxin system is involved in regulating Kv channels that underlie Ipeak but not those channels carrying Iss.
Figure 6.
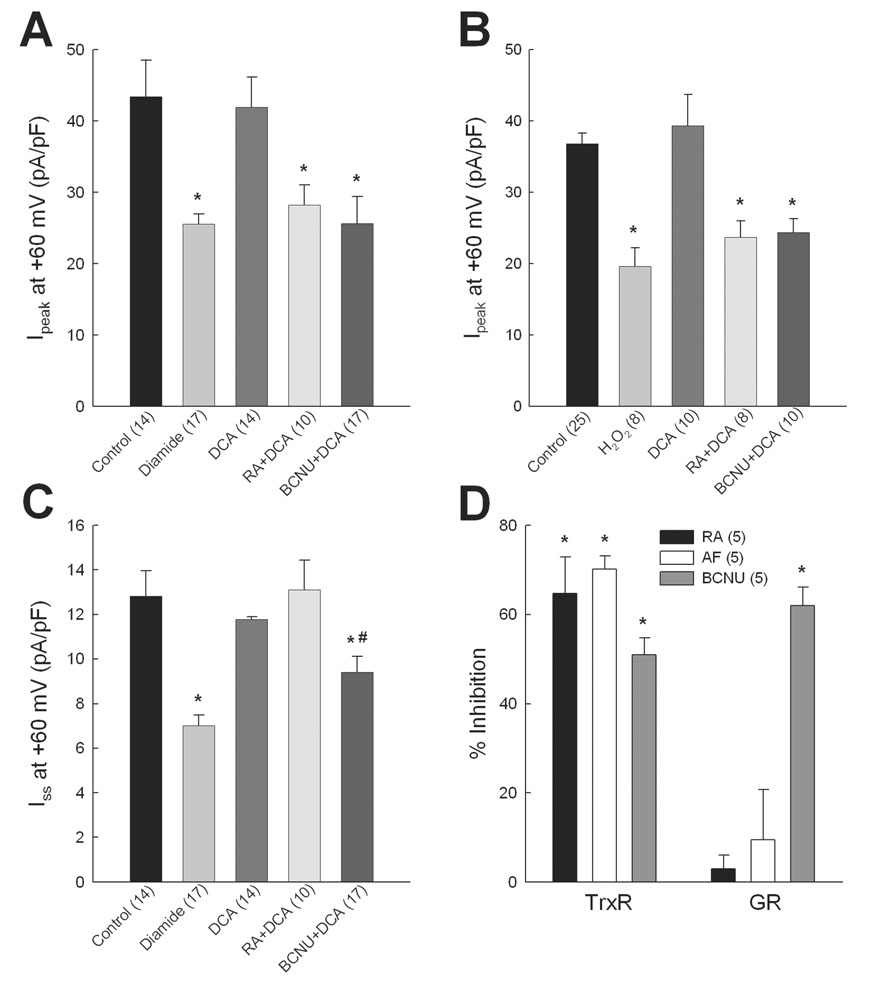
Regulation of K+ currents by oxidoreductase systems. DCA-mediated up-regulation of Ipeak (A) and Iss (C) was assessed in myocytes pre-treated with 13-cis-retinoic acid (RA; 1 µmol/l) or 1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU; 100µmol/l) for 30 min before diamide exposure. Up-regulation of Ipeak by DCA was also assessed in myocytes exposed to 10 µmol/l H2O2 in place of diamide (B). * = p<0.05 compared with control. # = p<0.05 compared with diamide alone (panel C). D: TrxR and GR activities were assayed in control myocyte suspensions after treatment with inhibitors for 30 min. * = p<0.05 compared with no inhibition.
DISCUSSION
Redox modulation of protein thiols
An oxidative shift in cellular redox balance was elicited in the present study by the diazine dicarbonyl compound diamide, which rapidly oxidizes intracellular GSH to GSSG [20]. Increased GSSG levels can modify protein function via reaction with free cysteine -SH groups to form intramolecular or intermolecular disulfides, or protein-mixed disulfides [1, 2, 3]. Although GSH is considered the main target of diamide, protein thiols may also be directly modified [20, 36]. Indeed, comparison of the present data with previous studies from our laboratory [16] suggests that some electrophysiological effects of diamide may be independent of cellular GSSG accumulation. In particular, dialyzing ventricular myocytes with GSSG decreases Ipeak density without changing Iss and without significant changes in voltage-dependent parameters [16]. By comparison, diamide markedly inhibited Ipeak and Iss (Fig. 1) and shifted the activation and inactivation curves for Ipeak to more negative potentials (Table 1). Differences were also observed between the effects of diamide and H2O2 on Iss (Fig. 1C and Fig. 2B). Although H2O2 increases GSSG formation via glutathione peroxidase [1, 2, 11, 22, 23], it can also directly attack protein thiols to produce oxidized intermediates [37]. Our data thus suggest that the different effects of diamide and H2O2 on Iss may be due to different thiol intermediates produced by these oxidants. For example, diamide can directly cause the formation of thiyl radical (-S.) or thiolate anion (-S−) intermediates [37], whereas H2O2 forms sulfenic acid (-SOH) intermediates in target proteins [37]. Whether such mechanisms explain the electrophysiological differences between diamide and H2O2 in our study warrants further experimentation on a molecular level.
The return of oxidized thiol intermediates to their reduced state by cytosolic oxidoreductases requires reducing equivalents supplied by NADPH, which is generated mainly by the pentose pathway where G6PDis the rate-limiting step [38, 39]. The redox function of this enzyme in heart is supported by studies in rat ventricular myocytes treated with G6PD inhibitors, showing that intracellular GSH levels are significantly decreased and the contractile phenotype is depressed [38]. Moreover, G6PD-deficient mice show more pronounced GSH depletion and contractile dysfunction after ischemia-reperfusion compared with wild-type mice [40]. Although most studies have examined the relationship of G6PD with the redox status of GSH, the NADPH produced by the pentose pathway similarly affects the function of the thioredoxin system [39].
Given the requirement of NADPH by oxidoreductase systems, we reasoned that increased flux through the pentose pathway would promote protein reduction and reverse inhibition of K+ currents elicited by diamide (Fig. 4). We tested DCA and bpV(phen) based on previous studies from our laboratory showing that these agents normalize GSH levels in depleted myocytes [19, 28, 41], further supporting the functional link between glucose metabolism and cellular redox mechanisms. DCA stimulates mitochondrial pyruvate dehydrogenase while also promoting the cellular uptake of glucose and pyruvate [30, 31]. Increased pyruvate dehydrogenase activity is proposed to lead to citrate accumulation in the cytosol which increases NADPH production by inhibiting phosphofructokinase and diverting glycolytic flux to G6PD [42]. By comparison, bpV(phen) activates insulin receptor kinase and its downstream effectors by inhibiting receptor tyrosine phosphatase [32]. In relation to redox-mediated mechanisms, it is likely that bpV(phen) increases glucose uptake [32] and the availability of glucose-6-phosphate for the pentose pathway, while it may also stimulate pyruvate dehydrogenase [31]. Although DCA and bpV(phen) would seem to have similar metabolic effects, the former compound elicited a more complete normalization of K+ currents after diamide treatment (Fig. 4). This apparent difference in efficacy may be due to a dose effect or to a quantitative difference in the metabolic response of myocytes to these agents [31].
Differential control of K+ currents by thioredoxin and glutaredoxin systems
Using the electrophysiological response of diamide-treated cells to DCA as an experimental end point, our studies of TrxR and GR inhibitors indicate that Kv channels are differentially regulated by thioredoxin and glutaredoxin systems. Since AF (Fig. 5A) and RA (Fig. 6A) blocked the effect of DCA to up-regulate Ipeak after diamide it is likely that the thioredoxin system controls Kv channels underlying this current, most likely Kv4.2, Kv4.3, and possibly Kv1.4 [43]. In contrast, the response of Iss to DCA after diamide was not altered by TrxR inhibitors (Fig. 5B and Fig. 6B), suggesting that the thioredoxin system was not involved. Instead, a pan-specific reductase inhibitor, BCNU, partially blocked the DCA effect on Iss (Fig. 6B). Since Kv1.5 and Kv2.1 channels largely contribute to Iss in rat myocytes [43], it may be postulated that one or both are regulated by the glutaredoxin system, although accessory subunits may also be involved. Given that BCNU inhibits TrxR and GR to a similar degree (Fig. 6C) [35], it follows that this compound also blocked the effect of DCA to up-regulate Ipeak (Fig. 6A). However, the reason that BCNU only partially blocked the response of Iss to DCA (Fig. 6B) is not clear from these studies. It is possible that residual GR activity not blocked by BCNU allowed sufficient GSH to be generated to reduce a portion of the proteins oxidized by diamide, or that a particular Kv isoform contributing to Iss is not controlled by the glutaredoxin system.
An important mechanism to define is whether oxidoreductase regulation of K+ channels in our experimental model was by direct thiol modification of Kv α-/accessory subunits, or by indirect effects on redox-sensitive signaling molecules. Hence, we addressed the possibility that diamide-induced electrophysiological changes were indirectly mediated by activation of kinase pathways, some of which acutely regulate K+ currents in ventricular myocytes [24, 25, 27]. In our studies, neither calphostin C nor GF109203x affected the ability of diamide to inhibit Ipeak or Iss, indicating that PKC activation was not involved. However, the protein tyrosine kinase inhibitors genistein and lavendustin A each blocked electrophysiological changes elicited by diamide (Fig. 3), suggesting a key role for tyrosine kinase activation and/or tyrosine phosphatase inhibition. Studies in various cell types suggest that protein tyrosine kinase can modulate Kv channels, although the net effect of kinase activation is unclear. For example, genistein has been shown to inhibit Ito in rat ventricular [27] and human atrial [44] myocytes. In expressed Shaker channels (Kv1.5, Kv1.4), Src tyrosine kinase binds to the α-subunit and inhibits macroscopic currents [45, 46], although rat Kv1.5 does not co-immunoprecipitate with Src as does the human isoform [46]. Finally, inhibition of specific tyrosine phosphatases has been shown to increase currents carried by Kv2.1 channels [47]. Given the diverse isoforms expressed in mammalian cells, it is clear more experimentation is needed to identify the specific tyrosine kinase pathways regulating cardiac Kv channels. Moreover, kinase regulation may involve accessory proteins such as KChIP, as has been shown for neuronal Kv4.2 channels phosphorylated by ERK [48].
While the role of tyrosine kinase/phosphatase pathways in controlling Kv channels is still unclear, there is more compelling evidence for their redox sensitivity. In general, transient oxidation of thiols in protein tyrosine phosphatases leads to their inactivation, whereas oxidative conditions activate protein tyrosine kinases [49]. In the latter case, oxidants may act by direct thiol modification or indirectly by concomitant inhibition of protein tyrosine phosphatase, which can lead to phosphorylation and sustained activation of tyrosine kinase [49]. Importantly, oxidative modifications of thiols in tyrosine phosphatases are reversed by intracellular reducing molecules such as Trx, Grx and GSH [50]. In our experimental model therefore, it may be postulated that diamide inhibited Ipeak and Iss through activation of tyrosine kinases and/or inhibition of tyrosine phosphatases, and that these effects were reversed by oxidoreductase systems stimulated by DCA. Since data in Figure 5 and Figure 6 support the notion that Ipeak and Iss are differentially regulated by the thioredoxin and glutartedoxin systems, it is reasonable to propose that the kinase/phosphatase pathways controlling their respective channels are also different.
Limitations
Although our study implicates oxidoreductase systems as important regulators of cardiac K+ channels, certain limitations must be kept in mind. First, due to the effect of diamide on seal stability, we compared independent groups of myocytes and did not characterize the time course of current inhibition in a given cell. Ideally, the inhibitory effects of diamide and the recovery of currents by DCA or bpV(phen) would be studied in the same cell, so that paired comparisons could be made with each cell serving as its own control. Second, we did not completely characterize the impact of diamide on the intracellular redox status of myocytes. Although we had previously shown that diamide depletes GSH in myocytes [19], we did not measure cellular levels of GSSG or other redox-related molecules such as NADPH. Third, our experiments were conducted on a mixed population of myocytes from the left ventricle and septum, which likely influenced the variability of measured currents. It is well known that regional and species differences in expression of certain Kv channels exist in the normal heart [43], and it is possible that oxidoreductase systems and/or kinase pathways also show regional or species differences. Finally, our studies were carried out at room temperature to maximize the stability of voltage-clamp recordings and we did not examine regulation of K+ currents at 37 °C. Thus, we cannot rule out the possibility that oxidoreductase systems operating at physiological temperature regulate Kv channels differently than what was observed in the present study.
In summary, our data suggest that Kv channels in ventricular myocytes are differentially regulated by oxidoreductase systems that are metabolically linked to glucose utilization and that act via tyrosine kinase/phosphatase pathways. Endogenous cardiac oxidoreductase networks may therefore be important factors controlling ion channels and associated regulatory proteins affected by pathogenic oxidative stress.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by a grant from the National Heart, Lung and Blood Institute (HL 66446).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
REFERENCES
- 1.Powis G, Briehl M, Oblong L. Redox signaling and the control of cell growth and death. Pharm Ther. 1995;68(1):149–173. doi: 10.1016/0163-7258(95)02004-7. [DOI] [PubMed] [Google Scholar]
- 2.Sen CK. Redox signaling and the emerging therapeutic potential of thiol antioxidants. Biochem Pharmacol. 1998;55:1747–1758. doi: 10.1016/s0006-2952(97)00672-2. [DOI] [PubMed] [Google Scholar]
- 3.Paget MSB, Buttner MJ. Thiol-based regulatory switches. Ann Rev Genet. 2003;37:91–121. doi: 10.1146/annurev.genet.37.110801.142538. [DOI] [PubMed] [Google Scholar]
- 4.Bigelow DJ, Squier TC. Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim Biophys Acta. 2005;1703:121–134. doi: 10.1016/j.bbapap.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka T, Nakamura H, Nishiyama A, Hosoi F, Masutani H, Wada H, Yodoi J. Redox regulation by thioredoxin superfamily; Protection against oxidative stress and aging. Free Radical Res. 2000;33:851–855. doi: 10.1080/10715760000301361. [DOI] [PubMed] [Google Scholar]
- 6.Shioji K, Kishimoto C, Nakamura H, Masutani H, Yuan Z, Oka S, Yodoi J. Overexpression of thioredoxin-1 in transgenic mice attenuates adriamycin-induced cardiotoxicity. Circulation. 2002;106:1403–1409. doi: 10.1161/01.cir.0000027817.55925.b4. [DOI] [PubMed] [Google Scholar]
- 7.Yamawaki H, Jaendeler J, Berk BC. Thioredoxin. A key regulator of cardiovascular homeostasis. Circ Res. 2003;93:1029–1033. doi: 10.1161/01.RES.0000102869.39150.23. [DOI] [PubMed] [Google Scholar]
- 8.Arnér ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 9.Holmgren A, Bjornstedt M. Thioredoxin and thioredoxin reductase. Methods Enzymol. 1995;252:199–208. doi: 10.1016/0076-6879(95)52023-6. [DOI] [PubMed] [Google Scholar]
- 10.Powis G, Montfort WR. Properties and biological activities of thioredoxins. Ann Rev Pharmacol Toxicol. 2001;41:261–295. doi: 10.1146/annurev.pharmtox.41.1.261. [DOI] [PubMed] [Google Scholar]
- 11.Filomeni G, Rotilio G, Ciriolo MR. Cell signaling and the glutathione redox system. Biochem Pharmacol. 2002;64:1057–1064. doi: 10.1016/s0006-2952(02)01176-0. [DOI] [PubMed] [Google Scholar]
- 12.Hirota K, Nakamura H, Masutani H, Yodoi J. Thioredoxin superfamily and thioredoxin-inducing agents. Ann NY Acad Sci. 2002;957:189–199. doi: 10.1111/j.1749-6632.2002.tb02916.x. [DOI] [PubMed] [Google Scholar]
- 13.Yoshitake S, Nanri H, Fernando MR, Minakami S. Possible differences in the regenerative roles played by thioltransferase and thioredoxin for oxidatively damaged proteins. J Biochem. 1994;116:42–46. doi: 10.1093/oxfordjournals.jbchem.a124500. [DOI] [PubMed] [Google Scholar]
- 14.Chiamvimonvat V, O'Rourke B, Kamp TJ, Kallen RG, Hofmann F, Flockerzi V, Marban E. Functional consequences of sulfhydryl modification in the pore forming subunits of cardiovascular Ca2+ and Na+ channels. Circ Res. 1995;76:325–334. doi: 10.1161/01.res.76.3.325. [DOI] [PubMed] [Google Scholar]
- 15.Hool LC. Differential regulation of the slow and rapid components of guinea pig cardiac delayed rectifier channels by hypoxia. J Physiol. 2003;554.3:743–754. doi: 10.1113/jphysiol.2003.055442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rozanski GJ, Xu Z. Sulfhydryl Modulation of K+ channels in rat ventricular myocytes. J Mol Cell Cardiol. 2002;34:1623–1632. doi: 10.1006/jmcc.2002.2112. [DOI] [PubMed] [Google Scholar]
- 17.Sims C, Harvey RD. Redox modulation of basal and β-adrenergically stimulated cardiac L-type Ca2+ channel activity by phenylarsine oxide. Brit J Pharmacol. 2004;142:797–807. doi: 10.1038/sj.bjp.0705845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Li S, Rozanski GJ. Redox regulation of K+ channel remodeling in diabetic rat heart. Am J Physiol, Heart Circ Physiol. 2005;288:H1417–H1424. doi: 10.1152/ajpheart.00559.2004. [DOI] [PubMed] [Google Scholar]
- 19.Li S, Li X, Rozanski GJ. Regulation of glutathione in cardiac myocytes. J Mol Cell Cardiol. 2003;35:1145–1152. doi: 10.1016/s0022-2828(03)00230-x. [DOI] [PubMed] [Google Scholar]
- 20.Kosower NS, Kosower EM. Diamide: an oxidant probe for thiols. Methods Enzymol. 1995;251:123–133. doi: 10.1016/0076-6879(95)51116-4. [DOI] [PubMed] [Google Scholar]
- 21.Carlberg I, Mannervik B. Glutathione reductase. Methods Enzymol. 1985;113:484–490. doi: 10.1016/s0076-6879(85)13062-4. [DOI] [PubMed] [Google Scholar]
- 22.Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Rad Biol Med. 2001;31(11):1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 23.Singal PK, Khaper N, Palance V, Kumar D. The role of oxidative stress in the genesis of heart disease. Cardiovasc Res. 1998;40:426–432. doi: 10.1016/s0008-6363(98)00244-2. [DOI] [PubMed] [Google Scholar]
- 24.Apkon M, Nerbonne JM. α1-Adrenergic agonists selectively suppress voltage-dependent K+ currents in rat ventricular myocytes. Proc Natl Acad Sci. 1988;85:8756–8760. doi: 10.1073/pnas.85.22.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thorneloe KS, Liu XF, Walsh MP, Shimoni Y. Transmural differences in rat ventricular protein kinase C epsilon correlate with its functional regulation of a transient cardiac K+ current. J Physiol. 2001;533.1:145–154. doi: 10.1111/j.1469-7793.2001.0145b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radical Biol Med. 2000;28:1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 27.Gao Z, Lau CP, Wong TM, Li GR. Protein tyrosine kinase-dependent modulation of voltage-dependent potassium channels by genistein in rat cardiac ventricular myocytes. Cell Signal. 2004;16:333–341. doi: 10.1016/j.cellsig.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Li S, Xu Z, Lou MF, Anding P, Liu D, Roy SK, Rozanski GJ. Redox control of K+ channel remodeling in rat ventricle. J Mol Cell Cardiol. 2006;40:339–349. doi: 10.1016/j.yjmcc.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 29.Rozanski GJ, Xu Z. A metabolic mechanism for cardiac K+ channel remodeling. Clin Exp Pharmacol Physiol. 2002;29:132–137. doi: 10.1046/j.1440-1681.2002.03618.x. [DOI] [PubMed] [Google Scholar]
- 30.Bersin RM, Stacpoole PW. Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am Heart J. 1997;134(5):841–855. doi: 10.1016/s0002-8703(97)80007-5. [DOI] [PubMed] [Google Scholar]
- 31.Lloyd S, Brocks C, Chatham JC. Differential modulation of glucose, lactate, and pyruvate oxidation by insulin and dichloroacetate in the rat heart. Am J Physiol Heart Circ Physiol. 2003;285:H163–H172. doi: 10.1152/ajpheart.01117.2002. [DOI] [PubMed] [Google Scholar]
- 32.Posner BJ, Faure R, Burgess JW, Bevan AP, Lachance D, Zhang-Sun G, Fantus IG, Ng JB, Hall DA, Lum BS, Shaver A. Peroxovanadium compounds. A new class of potent phosphotyrosine phosphatase inhibitors which are insulin mimetics. J Biol Chem. 1994;296(6):4596–4604. [PubMed] [Google Scholar]
- 33.Venardos K, Harrison G, Headrick J, Perkins A. Auranofin increases apoptosis and ischemia-reperfusion injury in the rat isolated heart. Clin Exp Pharmacol Physiol. 2004;31:289–294. doi: 10.1111/j.1440-1681.2004.03993.x. [DOI] [PubMed] [Google Scholar]
- 34.Schallreuter KU, Wood JM. The stereospecific suicide inhibition of human melanoma thioredoxin reductase by 13-cis-retinoic acid. Biochem Biophys Res Comm. 1989;160:573–579. doi: 10.1016/0006-291x(89)92471-6. [DOI] [PubMed] [Google Scholar]
- 35.Schallreuter KU, Gleason FK, Wood JM. The mechanism of action of the nitrosourea anti-tumor drugs on thioredoxin reductase, glutathione reductase and ribonucleotide reductase. Biochim Biophys Acta. 1990;1054:14–20. doi: 10.1016/0167-4889(90)90199-n. [DOI] [PubMed] [Google Scholar]
- 36.Dalle-Donne I, Giustarini, Colombo R, Milzani A, Rossi R. S-glutathionylation in human platelets by a thiol-disulfide exchange-independent mechanism. Free Radical Biol Med. 2005;38:1501–1510. doi: 10.1016/j.freeradbiomed.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 37.Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Nat Acad Sci. 2004;101:17982–17989. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain M, Brenner DA, Cui L, Lim CC, Wang B, Pimentel DR, Koh S, Sawyer DB, Leopold JA, Handy DE, Loscalzo J, Apstein CS, Liao R. Glucose-6-phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ Res. 2003;93:e9–e16. doi: 10.1161/01.RES.0000083489.83704.76. [DOI] [PubMed] [Google Scholar]
- 39.Biaglow JE, Ayene IS, Koch CJ, Donahue J, Stamato TD, Mieyal JJ, Tuttle SW. Radiation response of cells during altered protein thiol redox. Radiation Res. 2003;159:484–494. doi: 10.1667/0033-7587(2003)159[0484:rrocda]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 40.Jain M, Cui L, Brenner DA, Wang B, Handy DE, Leopold JA, Loscalzo J, Apstein CS, Liao R. Increased myocardial dysfunction after ischemia-reperfusion in mice lacking glucose-6-phosphate dehydrogenase. Circulation. 2004;109:898–903. doi: 10.1161/01.CIR.0000112605.43318.CA. [DOI] [PubMed] [Google Scholar]
- 41.Li S, Li X, Li Y-L, Shao C-H, Bidasee KR, Rozanski GJ. Insulin regulation of glutathione and contractile phenotype in diabetic ventricular myocytes. Am J Physiol Heart Circ Physiol. 2007;292:H1619–H1629. doi: 10.1152/ajpheart.00140.2006. [DOI] [PubMed] [Google Scholar]
- 42.Mallet RT, Sun J. Antioxidant properties of myocardial fuels. Mol Cell Biochem. 2003;253:103–111. doi: 10.1023/a:1026009519783. [DOI] [PubMed] [Google Scholar]
- 43.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Kumar R, Wagner MB, Cheng J, Mishra M, Joyner RW. Regulation of transient outward current in human atrial myocytes by protein tyrosine kinase pathway. J Cardiovasc Electrophysiol. 2002;13:927–935. doi: 10.1046/j.1540-8167.2002.00927.x. [DOI] [PubMed] [Google Scholar]
- 45.Nitabach MN, Llamas DA, Thompson IJ, Collins KA, Holmes TC. Phosphorylation-dependent and phosphorylation-independent modes of modulation of shaker family voltage-gated potassium channels by SRC family protein tyrosine kinases. J Neurosci. 2002;22:7913–7922. doi: 10.1523/JNEUROSCI.22-18-07913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holmes TC, Fadool DA, Ren R, Levitan IB. Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science. 1996;274:2089–2091. doi: 10.1126/science.274.5295.2089. [DOI] [PubMed] [Google Scholar]
- 47.Tiran Z, Peretz A, Sines T, Shinder V, Sap J, Attali B, Elson A. Tyrosine phosphatases epsilon and alpha perform specific and overlapping functions in regulation of voltage-gated potassium channels in Schwann cells. Mol Biol Cell. 2006;17:4330–4342. doi: 10.1091/mbc.E06-02-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schrader LA, Birnbaum SG, Nadin BM, Ren Y, Bui D, Anerson AE, Sweatt JD. ERK/MAPK regulates Kv4.2 potassium current by direct phosphorylation of the pore-forming subunit. Am J Physiol Cell Physiol. 2006;290:C852–C861. doi: 10.1152/ajpcell.00358.2005. [DOI] [PubMed] [Google Scholar]
- 49.Chiarugi P. PTPs versus PTKs: The redox side of the coin. Free Radical Res. 2005;39:353–364. doi: 10.1080/10715760400027987. [DOI] [PubMed] [Google Scholar]
- 50.Cho SH, Lee CH, Ahn Y, Kim H, Kim H, Ahn CY, Yang KS, Lee SR. Redox regulation of PTEN and protein tyrosine phosphatases in H2O2-mediated cell signaling. FEBS Lett. 2004;560:7–13. doi: 10.1016/s0014-5793(04)00112-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.