

Abstract
A novel short-chain (S)-1-phenyl-1,2-ethanediol dehydrogenase (SCR) from Candida parapsilosis exhibits coenzyme specificity for NADPH over NADH. It catalyzes an anti-Prelog type reaction to reduce 2-hydroxyacetophenone into (S)-1-phenyl-1,2-ethanediol. The coding gene was overexpressed in Escherichia coli and the purified protein was crystallized. The crystal structure of the apo-form was solved to 2.7 Å resolution. This protein forms a homo-tetramer with a broken 2-2-2 symmetry. The overall fold of each SCR subunit is similar to that of the known structures of other homologous alcohol dehydrogenases, although the latter usually form tetramers with perfect 2-2-2 symmetries. Additionally, in the apo-SCR structure, the entrance of the NADPH pocket is blocked by a surface loop. In order to understand the structure–function relationship of SCR, we carried out a number of mutagenesis–enzymatic analyses based on the new structural information. First, mutations of the putative catalytic Ser-Tyr-Lys triad confirmed their functional role. Second, truncation of an N-terminal 31-residue peptide indicated its role in oligomerization, but not in catalytic activity. Similarly, a V270D point mutation rendered the SCR as a dimer, rather than a tetramer, without affecting the enzymatic activity. Moreover, the S67D/H68D double-point mutation inside the coenzyme-binding pocket resulted in a nearly 10-fold increase and a 20-fold decrease in the kcat/KM value when NADH and NADPH were used as cofactors, respectively, with kcat remaining essentially the same. This latter result provides a new example of a protein engineering approach to modify the coenzyme specificity in SCR and short-chain dehydrogenases/reductases in general.
Keywords: alcohol dehydrogenase, Candida parapsilosis, short-chain dehydrogenases/reductases (SDR), X-ray crystallography, site-directed mutagenesis
The (S)-specific carbonyl reductase (SCR, also known as S-specific alcohol dehydrogenase) from Candida parapsilosis is an NADPH-dependent member of the classical enzyme family of short-chain dehydrogenases/reductases (SDRs) and has strong potential for biotechnological applications (Wei et al. 2000). It is the only known SDR that catalyzes the enantioselective reduction of 2-hydroxyacetophenone to produce the valuable, optically active 1-phenyl-1, 2-ethanediol with high yield and chiral purity (Wei et al. 2000; Cao et al. 2006; Supplemental Fig. 1). In turn, 1-phenyl-1, 2-ethanediol is a versatile chiral building block for the synthesis of numerous pharmaceuticals, agrochemicals, pheromones, and liquid crystal products. Using microbial expression systems to prepare the pure and active enzyme in large quantities should enable the commercial production of pure chiral alcohol compounds for industrial usage. Given the importance of SCR in organic synthesis, the scr gene of C. parapsilosis has been cloned and overexpressed in Escherichia coli. A review on the properties of this enzyme has recently become available (Nie et al. 2007).
A number of structural studies have been performed on SDR enzymes from different sources, including mannitol-2-dehydrogenase from Agaricus bisporus (MtDH) (Hörer et al. 2001), R-specific alcohol dehydrogenase from Lactobacillus brevis (RADH) (Schlieben et al. 2005), β-ketoacyl reductase from E. coli (FabG) (Price et al. 2001), and Drosophila alcohol dehydrogenase (ADH), the latter of which is the most widely characterized member of the SDR family in terms of substrate specificities (Benach et al. 1999; Smilda et al. 2001). Most SDR enzymes have a core structure of 250–350 residues in length and are often found in either dimeric or tetrameric forms (Jornvall et al. 1995). In the tetrameric form, the subunits are always observed to assume 2-2-2 symmetry in which subunit interfaces are described by three orthogonal dyad axes, conventionally termed P, Q, and R (Tanaka et al. 2001). On the other hand, dimeric SDR proteins are found to mimic the dimer interface of either the P- or the Q-symmetry-related interface in the tetramer (Grimm et al. 2000; Kristan et al. 2005). All available three-dimensional (3D) structures of SDR subunits display a characteristic Rossmann-fold structure consisting of a central seven-strand parallel β-sheet flanked by α-helices (Liese et al. 1996; Oppermann et al. 2003). The active site, located at the C-terminal end of the major parallel β-sheet, includes the Ser-Tyr-Lys catalytic triad, as well as both a coenzyme- and a substrate-binding site (Lesk 1995). This region of the 3D structure is highly conserved at the primary sequence level, including an N-terminal TGxxxGxG motif (Hörer et al. 2001) as part of the nucleotide-binding region, and the Ser-Tyr-Lys catalytic triad that forms a proton relay system (Oppermann et al. 2003).
In this work, we report the crystal structure of SCR from C. parapsilosis. We identified three novel features from this SCR structure: (1) an extended N-terminal peptide that stabilizes the Q-axis-related dimerization; (2) an unusual tetramerization that is devoid of 2-2-2 point-group symmetry, particularly in the R-axis-related dimer interface; and (3) two of the four potential NADPH-binding sites in the tetramer are occupied by surrounding peptides in the apo-enzyme form. Using site-directed mutagenesis, we constructed several mutants involved either in intersubunit interactions, the conserved catalytic triad, or the phosphate-binding loop of the coenzyme-binding site. The results of these mutations provide greater understanding of the catalytic mechanism underlying the stereo-selective reduction of S-specific alcohol dehydrogenase.
Results and Discussion
Crystallography study and overall structure of an SCR molecule
SCR is a member of the classical SDR family of enzymes (Liese et al.1996; Oppermann et al. 2003). A single subunit of SCR comprises 279 residues. The crystal structure of apo-SCR was determined using molecular replacement methods and refined at a 2.69 Å resolution to a final Rwork of 20.5% (Rfree of 26.4%), with good stereochemical properties (Table 1). The crystal form belongs to the space group P212121 with cell dimensions of a = 104.7 Å, b = 142.8 Å, and c = 151.8 Å. Eight SCR molecules (i.e., chains A–H) were identified per asymmetric unit (asu) and were organized into two tetramers, namely ABCD and EFGH. The cores of these subunits superimpose well with each other; the root-mean-square deviation (rmsd) between 204 pairs of Cα atoms ranges from 0.3–0.5 Å (including residues 14–116, 146–169, and 196–272). However, their surface loop regions surrounding the active site assumed drastically different conformations. Accordingly, they can be divided into two groups: A, C, E, and G belong to one group, and B, D, F, and H belong to the other. Each SCR subunit has approximate dimensions of 48 × 55 × 65 Å.
Table 1.
Data collection and refinement statistics

The final refined model is comprised of most of the crystallized polypeptide, with the exception of some terminal or loop regions (Fig. 1). One asymmetric unit contains 113 ordered solvent molecules (modeled as water) with refined B-factors <49 Å2. Each subunit is composed of a coenzyme-binding domain, which folds into a classical α/β Rossmann-fold, i.e., a seven-strand parallel β-sheet (βC-βB-βA-βD-βE-βF-βG) flanked on both sides by α-helices (Ghosh et al. 1991) as shown in Figure 2 and in the sequence alignment of SCR with selected members of the SDR family shown in Figure 3. Based on a comparison with homologous structures, the putative catalytic triad (Oppermann et al. 2003) of SCR is believed to comprise of Ser172, Tyr187, and Lys191, which are located at the C-terminal end of the parallel β-sheet. Although they are conserved between MtDH and SCR, their side chain conformations in the NADP-free crystal form differ dramatically from those in the MtDH–NADP+ complex structure (PDB code: 1H5Q). The loop between βF and αG contains one small helix (αFG, residues 230–238), which is responsible for substrate specificity among the different enzymes within the SDR family (Ghosh et al. 1994; Tanaka et al. 1996; Benach et al. 1998; Mazza et al. 1998; Yamashita et al. 1999). Furthermore, SCR contains an extended N-terminal peptide (i.e., residues 1–25) and a short helix (residues 26–30) projecting out from the core domain that might stabilize the oligomer (see below). In the refined structure, the A, C, E, and G subunits contained two mobile regions within loop regions around residues 125 and 180, respectively. In contrast, the B, D, F, and H subunits contained one mobile loop in the region of residues 129–138. There are five Cys residues in each SCR molecule, but none of them are involved in disulfide bond formation. There are 12 Pro residues in each SCR subunit. In each subunit of the final refined model, only Pro16 of the N-terminal peptide has a cis conformation and is directly involved in the SCR dimerization (Nordling et al. 2001).
Figure 1.

Quaternary structure of the tetramer of SCR. The four subunits (A–D) are labeled and colored differently. (Arrows) Local dyad symmetry axes; the tetramer is viewed along the peudo R-axis. Disordered regions are labeled with boundary residue numbers and implicated with dots. Structural figures were prepared with the program PyMOL (DeLano Scientific).
Figure 2.

Structural overview of the SCR subunit. (Red) The novel N-terminal peptide, (cyan) α-helices, (magenta) β-strands. Secondary structures and both N and C termini are labeled.
Figure 3.

Sequence alignment of SCR with selected members of the SDR family. (Red highlighting) Identical residues, (blue boxes) conserved residues. Selected residue numbers of SCR are labeled above its sequence. Secondary structure elements of apo-SCR (molecule A) are marked on the top of alignment; (blue dots) regions showing different conformations between molecules A and B. The following highly conserved regions are indicated: the glycine-rich consensus sequence that is in contact with the pyrophospho bridge of NAD(P) in SDRs, and the substrate-binding loop. (Hollow arrows) The three residues of the catalytic triad, Ser172, Tyr187, and Lys191. (Green highlighting) Residues invisible in molecule A of the refined structure; (solid triangles) residues located in the A–B dimer interface (<3.0 Å). The abbreviations of protein names used in the alignment are as follows: SCR, (S)-1-phenyl-1, 2-ethanediol dehydrogenase from Candida parapsilosis (GenBank ID: DQ675534; PDB ID: 3CTM); MtDH, mannitol-2-dehydrogenase from Agaricus bisporus (AFO53764; 1H5Q); RADH, R-specific alcohol dehydrogenase from Lactobacillus brevis (AJ544275; 1NXQ); ACP, β-ketoacyl reductase from E. coli (19714462; 1I01); and PED, (S)-specific 1-phenylenthanol dehydrogenase of the denitrifying bacterium strain Ebn1 (56312783; 2EW8). This figure was prepared with the program ESPript (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi/), and the alignment was manually adjusted based on 3D structural superposition.
Sequence alignments reveal that SCR shares the same level of structural similarity with MtDH, FabG, RADH, and PED (Fig. 3), although SCR possesses a longer N-terminal peptide. In general, we observe more conserved residues at the beginning and ends of secondary structural elements than in their interior, indicating that one role of these conserved residues might be to define boundaries of the core of the Rossmann-fold where the active site and other functional apparatus can be formed.
As discussed earlier, the highly conserved sequence regions include an N-terminal Gly-rich motif with sequence TGxxxGxG (around position 45 in SCR), the N(A)NAG motif (about position 120), and the catalytic triad of Ser172, Tyr178, and Lys191 residues (Price et al. 2001; Schlieben et al. 2005; Hoffken et al. 2006). These three primary regions cluster together in the 3D structure around the active site, and their functional importance has been probed by several mutational and structural studies (Oppermann et al. 1997; Filling et al. 2002). The other side of the active site is lined by the insertion loop between βE and αF. In addition, structural comparison with MtDH suggests that the NADPH specificity (relative to NADH) in SCR is likely to be imposed by the backbone amide groups of Asn66, Ser67, and His68 and the side chains of Ser43, Ser67, and His68.
Oligomerization
The eight SCR molecules in an asu form four dimers: A–B, C–D, E–F, and G–H. Each is related by the so-called Q-axis dyad symmetry (Fig. 4) and has approximate dimensions of 50 × 70 × 70 Å. These A–B type dimers can be superimposed well with each other; the rmsd for 408 Cα pairs of their core domains also ranged from 0.3 to 0.5 Å. Three major regions in the primary sequence are involved in this dimer interface: the N-terminal peptide, the helix αG, and the strand βG (∼Val 270). The major β-sheets from the two subunits appeared to join together at the edge of βG, forming an extended one; however, no hydrogen bond was formed. Each subunit in such a dimer buries a solvent-accessible surface area (SAS) of 2700–2850 Å2. This interface is structurally conserved among most SDR proteins. For example, the A–D dimer in MtDH (1H5Q) may superimpose to the A–B dimer of SCR with an rmsd of 1.3 Å for 401 Cα atoms (using a 3.0 Å cutoff). Nevertheless, the N-terminal peptide in SCR is 27 residues longer than that of MtDH. In the case of SCR, 1850 Å2 (i.e., about one-third) of the buried SAS was contributed by the N-terminal peptide (residues 1–30) and its counterpart. This extra N-terminal peptide seals the dimer interface, presumably providing additional stability to the Q-axis-related A–B type dimer.
Figure 4.

Ribbon diagram of the Q-dimer of SCR. The dimer is viewed along the Q-axis. (Salmon) Subunit A, (orange) subunit B, (green highlighting) the αE and αF regions and other structural differences between A and B molecules. α-Helices, N and C termini are labeled. The disordered regions are implicated with dots.
The A–B type dimers further dimerize to form two tetramers in the asu, namely (A–B)–(C–D) and (E–F)–(G–H). The dimer–dimer interface is formed via the helices αE and αF at the A–C end and via the loop around Leu183 at the B–D end (Fig. 3). A total SAS of 1500 Å2 from each dimer is buried in the interface. The tetramer has approximate dimensions of 70 × 70 × 90 Å. The two tetramers superimpose well with each other (rmsd of 816 Cα pairs was only 0.4 Å), suggesting that the tetramer is a preexisting species in solution under the crystallization condition. However, the interface between subunits A and C, which is more similar to the canonical tetramer interface of SDR proteins, visibly differs from the interface between subunits B and D. The two dyad axes of the A–B and C–D dimers are off by ∼15° and 4 Å (Fig. 1). The total surface area buried during tetramerization by the A type subunit is ∼3950 Å2 (28%), and that by the B type subunit is 3200 Å2 (22%). Therefore, the tetramer has a broken 2-2-2 symmetry, and there were three types of dimer interfaces in the tetramer, namely A–B, A–C, and B–D. We also observed a slightly different crystal form (data not shown) having a similar crystal packing as the above-discussed crystal form, but with cell parameters a = 101.1 Å, b = 146.0 Å, and c = 159.8 Å. The corresponding cell parameter change in the c direction was 5% between the two forms. The ABCD and EFGH tetramers from the two crystal forms had rmsds of 0.49 and 0.45 Å (for all Cα) with their counterparts, respectively, further supporting the preexistence of the tetramer in solution.
This structurally observed tetramer formation is consistent with results from our molecular sizing exclusion chromatography and analytical ultracentrifugation studies, which showed that SCR forms a homo-tetramer in solution (Fig. 5).
Figure 5.

Analytical ultracentrifugation analysis of SCR variants. (Top two panels) Sedimentation velocity data and the systematic noise decomposition. (Bottom three panels) Transformation to molar mass distributions from the sedimentation velocity profile: 112 kDa, 100 kDa, and 60 kDa for wild type (WT), Δ31, and V270D, respectively. The former two correspond to tetramers; the latter one corresponds to a dimer in a buffer containing 20 mM Tris (pH 8.0) and 150 mM NaCl.
Nonfunctional catalytic site in apo-SCR
The present crystal structure represents inactive forms of the SCR enzyme. There are four structurally independent active sites per tetramer, one in each subunit. The active site distance (i.e., Cα distance between the two Ser127 residues) is 28 Å for the A–B dimer and 25 Å for both A–C and B–D. The entrance of each subunit faces toward the solvent. The two types of subunits exemplified by A and B subunits assume two different conformations, both of which are nonfunctional. In the A molecule, the catalytic triad is fairly close to the active form, while the NADPH-binding pocket is severely collapsed. In contrast, in the B molecule, the catalytic triad is completely rearranged, while the NADPH-binding pocket remains fairly intact.
In the A molecule, three primary sequence regions block NADPH binding: The loop around residues in the 120s-loop C-terminal to βD occupies the adenine-binding pocket; the 170s-loop C-terminal to βE is situated in the nicotinamide-binding site; and a short 310 helix in the region of 220s, C-terminal to βF, is rotated toward the nicotinamide-binding site. In particular, Val121 occupies the nicotinamide ribose position, and Thr122 and Ala123 would overlap with both the ribose of adenine and its phosphate group. Near the active site, the A–C type dimer interface remained the same as that in the canonical SDR tetramer as exemplified by MtDH (1H5Q). Although the backbones of two of the catalytic triad residues, Tyr187 and Lys191, remain in the “normal” conformation, their side chains assume nonfunctional rotamers. On the other hand, a backbone trace of the 170s-loop, where the catalytic Ser172 resides, showed a major difference from that of the canonical SDR, and the loop blocks the entrance of the NADPH coenzyme. The side chain of Ser172 moves by ∼5 Å relative to the canonical SDR and potentially forms hydrogen bonds with the backbone carbonyl oxygen of Gly120 and the side chain of Asp144. The conformational change around Ser172 forces the side chains of the other two catalytic residues, Tyr187 and Lys191, to adapt their nonfunctional rotamers.
In molecule B, the NADP+-binding site is less scrutinized. The only major backbone conformational change around the pocket is that of the short 310 helix C-terminal to βF rotating toward the nicotinamide-binding pocket, as in molecule A. In the new conformation, Cα of Ile223 is shifted by 5.5 Å relative to the canonical position (1H5Q), and the side chain overlaps with nicotinamide and its proximal phosphate group. The helix αF N terminus, where two of the three catalytic triad residues are located, is shortened by one turn compared with molecule A. Consequently, the catalytic triad in molecule B is arranged in a nonfunctional conformation. In particular, the Tyr187 side chain points toward the B–D dimer interface within the tetramer instead of the potential nicotinamide-binding site. Similarly, the catalytic Lys191 moves toward the protein surface and forms a hydrogen bond with the side chain of Asp269. While αDE (residues 132–145) remains the canonical length in molecule A, as in MtDH (1H5Q), the helix is completely dissolved in molecule B. Instead, the corresponding peptide forms a small β-sheet with strand Gly120–Trp123 on one side and strand Ile177–Ile180 on the other side. The hydrogen bond network is further extended by one backbone hydrogen bond between the carbonyl oxygen of Val178 and the amide group of Thr189, thus forming a mini, four-strand, mixed β-sheet.
While neither the inactive form of A nor B affected the A–B type dimer interface, the canonical P-axis-related dimer interface was interrupted by the B form (Fig. 1). The P-dyad symmetry-related A–C interface is likely to be functional for the following reasons: First, it uses the conserved four-helix bundle of αE and αF from the two subunits. The interface is fairly extensive, with each subunit burying ∼1500 Å2 SAS in the dimer interface. Second, compared with MtDH, SCR has a shorter 180s-loop. This truncation allows a structural complementation of the unique, longer N-terminal peptide that extends to the A–C type dimer interface. Third, the buried interface is composed of many hydrophobic residues (including Tyr134, Trp137, Ile141, Leu145, Tyr149, Tyr150, Ala185, Ala192, Ala193, Leu197, and Leu201) that would otherwise be exposed to the solvent. In contrast, the B–D interface is minor; each subunit buries only ∼360 Å2 SAS (Fig. 1). Formation of a functional catalytic triad in molecule B would require reorganization of the B–D type interface.
Comparing our SCR structure with other known SDR structures, we conclude that it contains two different snapshots of the SCR inactive state in the absence of a coenzyme. While the recombinant SCR protein clearly showed full enzymatic activity (see below), it is not clear why each Q-axis-related dimer is formed by a pair of distinct A and B SCR molecules. Their interface has near-perfect dyad symmetry, and thus it is unlikely that the two molecules correlate their conformational changes through communication across the interface. It is more probable that the two molecules in a given A–B dimer may change their two active site conformations independently in solution, and the observed tetramer in the crystal is a storage form for the apo-SCR. The substrate-binding loop in SCR consists of residues 220–240. It is flexible in the SCR structure, which is typical among SDR enzymes.
To test the probable effects of the cofactor on tetramerization, we compared the size exclusion chromatography profiles of SCR samples in the presence and the absence of NADPH. The results showed that, in both cases, SCR formed homo-tetramers (Supplemental Fig. 2). It further suggests that a conformational change in the SCR subunits induced by NADPH binding converts the tetramer into an active form, probably similar to the canonical 2-2-2 symmetrical one.
The conserved Ser-Tyr-Lys triad is essential for catalysis
In our apo-SCR crystal structure, the conserved catalytic triad, Ser172-Tyr178-Lys191, has varied, noncanonical, and, presumably, nonfunctional conformations. To verify their functional role, we constructed four mutants of single-residue substitutions, S172A, S172T, Y187A, and Y187F. The CD temperature scan showed that their melting temperatures were 46°C, 48°C, 45°C, and 46°C, respectively, all lower than the wild-type melting temperature (52°C). While the activity of the conserved S172T mutant was essentially the same as the wild type, the enzymatic activities of S172A, Y187A, and Y187F were undetectable (Table 2). These results indicate that the crystallized recombinant protein of SCR is enzymatically active in the presence of coenzymes and that it uses the same conserved catalytic triad as other SDR proteins (Winberg et al. 1999; Tanaka et al. 2001).
Table 2.
Kinetic parameters for 2-hydroxyacetophenone reduction by wild-type and mutant enzymes

The N-terminal peptide stabilizes the SCR oligomer
Among the SDR proteins compared in Figure 3, SCR is unique in possessing an extra long N-terminal peptide. This region contains two short helical structures and packs against neighboring subunits through extensive interactions. We constructed a truncation mutant, Δ31, by deleting the N-terminal 31 residues to verify the functional role of this peptide region. From an enzymatic assay of 2-hydroxyacetophenone reduction (Table 2), the kinetic parameters KM and k cat of this mutant were essentially the same as those of the wild type, suggesting that this additional N-terminal peptide has little to do with active site formation and substrate binding. Furthermore, the molecular weight distribution of the Δ31 recombinant protein in analytic ultracentrifugation (AUC) analysis showed a single tetramer peak (∼100 kDa), which was broader than the equivalent peak for the wild-type protein (Fig. 5). This result indicates that Δ31 also forms a tetramer in solution, which is similar to the wild type (∼112-kDa position) but is less stable. A CD temperature scan indicated that the melting temperature of the Δ31 variant was 48°C, slightly lower than that of the wild-type enzyme (52°C). Combined with the structural observations, these results indicate that the N-terminal peptide of SCR contributes marginally to the stability of the SCR tetramer but is not essential for its enzymatic activity.
Tetramerization is not required for SCR activity
Among the two major types of subunit interfaces, A–B and A–C, the Q-symmetry-related interface between subunits A and B is significantly more extensive (∼5700 Å2 total buried SAS); there is more than twice the SAS buried in this interface as in the P-symmetry-related A–C interface (∼2250 Å2 total buried SAS). Previously, effects of mutants that break the dimer at the A–C type interface have been investigated in 17β-HSDcl (Kristan et al. 2005). Like most dimeric members of the SDR family, the functional form of 17β-HSDcl is an A–C type dimer; breaking this dimer by point mutations results in inactive monomers. To investigate a possible functional role of the A–B interface, we introduced a dimer-breaking mutation in the dyad symmetrical hydrophobic interface. In particular, a tightly packed Val residue at position 270 in the βG strand was replaced with a negatively charged residue, Asp, which was expected to disrupt the A–B type dimerization without affecting the A–C interface. Consistent with our prediction, AUC measurements confirmed that the V270D mutant of SCR exists as a homodimer in solution (Fig. 5). Gel-filtration also showed a single peak at the 58 kDa position (data not shown). Thermal stability of the V270D variant was analyzed using a CD temperature scan, resulting in a 45°C melting temperature, 7°C lower than that of the wild-type SCR. Furthermore, kinetic analysis showed that the KM and k cat of V270D had slightly lower enzymatic activity than the wild type (Table 2). Our results indicate that SCR may maintain its enzymatic activity in a dimer form. This dimer likely uses the A–C type interface, because the A–B type interface is disrupted in the V270D variant and the B–D type interaction would be too weak to maintain a dimer independently.
S67D/H68D mutation switches cofactor specificity from NADPH to NADH
For industrial applications, converting the cofactor specificity of an enzyme from NADPH to NADH would be of great significance, as NADH is substantially cheaper than NADPH. To explore the possibility of converting SCR from a NADPH-dependent enzyme into an NADH-dependent one, we constructed a double-point mutant, S67D/H68D, making changes inside the so-called phosphate-binding loop between βB and αC. As suggested by previous studies on other SDRs (Mittl et al. 1994), residues located in this loop play a critical role in determining the coenzyme specificity for NADH or NADPH. In particular, carefully introduced aspartic acid residues would be energetically unfavorable for the negatively charged 2′-phosphate group of NADPH to bind, thus favoring NADH over NADPH.
Compared with the wild-type SCR, this S67D/H68D variant showed an increase in KM for NADPH and a decrease in KM for NADH; meanwhile, it maintained kcat essentially the same for both reactions (Table 2). The net results of the S67D/H68D double mutation are a nearly 10-fold increase in the kcat/KM value for NADH but a 20-fold decrease in that for NADPH. The kcat/KM ratio between NADH and NADPH changes from 0.068 for wild type to 13.8 for the mutant (∼200-fold). Meanwhile, in the CD thermal denaturation assay, the recombinant protein of the S67D/H68D mutant showed similar thermal stability to wild-type SCR. Therefore, we have successfully changed the coenzyme specificity of this enzyme without disturbing its stability.
Our apo-SCR crystal structure does not provide a direct structural explanation for the substrate specificity change of the S67D/H68D mutant due to being trapped in a nonfunctional conformation, and we were unable to determine a crystal structure of SCR in complex with the NADPH cofactor. Therefore, spectroscopic techniques were employed to monitor structural changes during cofactor binding. In a CD assay, the molar residue ellipticity at 209 nm was used as a measurement of structural transition from random coils to ordered secondary structures (such as helices) (Nakamura et al. 2006). As shown in Figure 6, the absolute value of the CD signal increases as the coenzyme concentration increases, suggesting that coenzyme binding induces ordered structural components in the enzyme (Nakamura et al. 2006). The saturated molar residue ellipticities of the S67D/H68D variant for NADH and NADPH were 3.22 × 10−4 and 2.92 × 10−4 degree.cm2.dmol−1, respectively; and those of wild-type SCR were 3.07 × 10−4 and 3.47 × 10−4 degree.cm2.dmol−1, respectively. These data indicate that both coenzymes have the ability to induce the random-to-ordered transition in SCR variants. More importantly, NADPH has higher inducing ability than NADH for the wild-type SCR, but NADH has stronger inducing ability than NADPH for the S67D/H68D variant. As a complementary approach, we measured the formation of the enzyme–cofactor binary complexes for the wild-type and S67D/H68D variants using the fluorescence titration method. The fluorescence in both cases was quenched by the addition of the coenzyme. The data were analyzed by a modified model from a simple Hill equation (Sakoda and Hiromi 1976; Nakanishi et al. 1997) with consideration of cooperativity. The ratio of Kd values between NADH and NADPH for the S67D/H68D mutant was 0.28, indicating a stronger preference for NADH; in contrast, the ratio became 1.9 for wild type, indicating a weaker binding for NADH (Table 2). Moreover, the parameters obtained from the cooperative Hill equation are <1 (Table 2), suggesting a negative cooperativity in the NAD(P)H binding to both SCR variants. These spectroscopic results indicate that the adenine ribose moiety of the coenzymes (particularly of NADH) favorably interacts with the substituting aspartate residues (Nakamura et al. 2006). For example, Asp67 may form a hydrogen bond with the hydroxyl groups of the adenine ribose group of the coenzyme, as illustrated by previous crystal structures of NADH-dependent SDRs (Ghosh et al. 1994; Varughese et al. 1994; Jornvall et al. 1995; Tanaka et al. 1996).
Figure 6.

CD analysis of cofactor binding in wild type and the S67D/H68D variant. (A) CD spectra of SCR and S67D/H68D with NAD(P)H. The spectra are colored by concentration of NADH/NADPH from pink (lowest) to dark red (highest). (Black line) CD spectrum of SCR only. (B) The molar ellipticities at 209 nm against coenzyme concentrations. The error bars represent the standard deviations estimated from data of three independent experiments.
Our results show that the double-point mutation S67D/H68D inside the coenzyme-binding pocket converts the coenzyme specificity of SCR from NADP(H) to NAD(H). Our work thus provides a new example of a protein engineering approach to modify coenzyme specificity in short-chain dehydrogenases/reductases and will likely have valuable industrial applications.
Materials and Methods
Microorganisms and chemicals
C. parapsilosis strain CCTCC M203011 was obtained from the American Type Culture Collection (ATCC). The organisms were cultivated as described previously (Liese et al.1996; Barzegar et al. 2007). The enzyme and cofactors were purchased from the Sigma-Aldrich Chemical Co., Inc. The substrate β-hydroxyacetophenone was prepared using the method described by Liese et al. (1996).
Protein expression and site-directed mutagenesis
The scr gene (GenBank ID: DQ675534) expression system in E. coli was previously described by Nie et al. (2007). The pETSCR plasmid was used as a template to generate recombinant plasmids coding for SCR mutants. Site-directed mutagenesis was performed using standard protocols (Baik et al. 2005). The mutations were verified by DNA sequencing. The oligonucleotides used for mutations were summarized in Table 1, with the mutated bases underlined.
Purification of recombinant enzymes
All recombinant proteins used were expressed as His6-tagged proteins in E. coli strain BL21 (DE3) and purified first by affinity chromatography on a Ni2+-Sepharose column (His-Trap Kit, Pharmacia). The pooled fractions were further loaded on a Resource Q column (1 × 1 cm) equilibrated with the buffer (20 mM Tris-HCl, pH 8.5) using an ÄKTA Protein Purifier system (Pharmacia). It was followed by Superdex 200 (10/300 GL) chromatography in a buffer containing 20 mM Tris-HCl (pH 8.5) and 150 mM NaCl in the presence or absence of 0.5 mM NADPH. All mutant enzymes were purified to homogeneity as judged by Coomassie Brilliant Blue staining of SDS-polyacrylamide gel electrophoresis (PAGE) gels. The extinction coefficient of 1.9 AU/M was used for all SCR variants.
Protein crystallization and data collection
The apo-SCR crystals were grown by the hanging-drop vapor diffusion method in a buffer containing 18% (w/v) Polyethylene glycol 2000 monomethyl ether (PEG2K MME) and 8% (v/v) isopropanol, pH 8.5. A droplet was prepared by mixing an equal volume (1.0 + 1.0 μL) of a protein solution containing 20 mg/mL SCR in a buffer of 20 mM Tris-HCl (pH 8.5) and 150 mM NaCl and a reservoir solution. Crystals obtained after 5 d of growth were soaked into the cryo-protectant solution of 24% (w/v) PEG2K MME and 6% (w/v) sucrose prior to data collection. Diffraction data were collected at beamline BL5A of the Photon Factory synchrotron facility (KEK, Tsukuba, Japan) and processed with the HKL2000 program package (Otwinowski and Minor1997). The data collection and processing statistics are summarized in Table 1.
Structure determination and refinement
The crystal structure of mannitol 2-dehydrogenase (MtDH, PDB file 1H5Q) from Agaricus bisporus (Hörer et al. 2001) was used as a search model for molecular replacement. MtDH forms a homotetramer with 2-2-2 symmetry, and one of its P-axis-related dimers was successfully employed as the search model. Nonconserved residues were changed to alanine with the SEAMAN program (Kleywegt 1996). The structure was initially refined with CNS (Brunger et al. 1998), and the program REFMAC (Murshudov et al. 1997) from the CCP4 suite (Collaborative Computational Project, Number 4 1994) was used for final refinement. Manual model building and adjustments were carried out with the programs Coot (Emsley and Cowtan 2004) and O (Jones et al. 1991). Refinement statistics are summarized in Table 1. Amino acid sequences of short-chain dehydrogenases/reductases were aligned using the program CLUSTALW (Thompson et al. 1994). SAS were calculated with the program EdPDB (Zhang and Matthews 1995). The stereochemical quality of the final model was examined with the program PROCHECK (Laskowski et al.1993).
Enzyme assay and kinetic experiments
Enzymatic activity of the SCR variants for reduction of 2-hydroxyacetophenone was measured at 35°C according to the method described by Nie et al. (2007). One unit of enzyme activity is defined as the amount of enzyme catalyzing the reduction/oxidation of 1 μmol of NAD(P)H/ NAD(P)+ per minute under the assay conditions. Kinetic parameters were measured and calculated using a Beckman DU-7500 spectrophotometer, equipped with a Multicomponent/SCA/Kinetics Plus software package and a thermostated circulating water bath. Varied concentrations of substrate 2-hydroxyacetophenone (0.5–20 mM), enzyme (10–200 μM), and cofactors NAD(P)H (0.5–5.0 mM) in 100 mM phosphate buffer (pH 7.5) were used. The reported values represent the average and standard deviation calculated from at least three independent measurements.
Circular dichroism (CD)
CD measurements were carried out using a Jasco J720 spectropolarimeter (JASCO, Inc.). Wavelength scan data were collected from 190 to 260 nm in a phosphate buffer (pH 7.5) with the following instrument settings: response, 1 s; sensitivity, 100 mdeg; speed, 50 nm/min; average of 30 scans. Scans were repeated every 2°C or 5°C between 20°C and 70°C. The protein concentration was ∼3 μM in 50 mM Na/K phosphate buffer (pH 6.8). The decrease in CD signal with the increase of temperature was recorded at the local minima of 209 nm.
Analytical ultracentrifugation (AUC)
SCR variants at a concentration of 0.5–1.0 mg/mL were used for the AUC analysis with a buffer of 20 mM Tris-HCl (pH 8.0) and 150 mM NaCl. Sedimentation experiments were performed using an XL-A analytical ultracentrifuge (Beckman Coulter) equipped with a four-cell An-60 Ti rotor at 20°C. Sedimentation velocity analysis was performed at 58,000 rpm. Data were analyzed with the SEDFIT program (Pekar and Sukumar 2007).
Fluorescence experiment
Nucleotide binding to SCR was measured by monitoring the quenching of intrinsic enzyme fluorescence upon incremental addition of nucleotides. Emission spectra (300–500 nm) were recorded on an F-4500 fluorescence spectrophotometer (Hitachi) at 280 nm excitation. The samples for measurements were prepared at the final concentration of 3 μM enzyme and 30–400 μM NAD(P)H in 50 mM Na/K phosphate buffer (pH 7.5). The Kd value was determined by the nonlinear least squares method (Sakoda and Hiromi 1976; Nakamura et al. 2006). The model (Equation 1) is expressed by a simple Hill equation with consideration of Hill coefficient h and dissociation constant Kd.
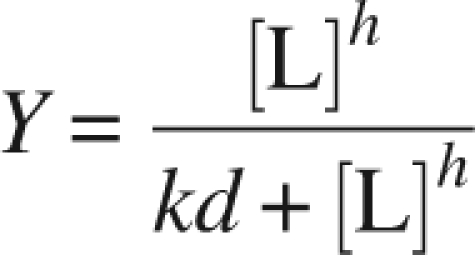 |
where Y is fractional saturation of the enzyme (i.e., [occupied sites]/[total sites]) (Bell and Dalziel 1975; Nakamura et al. 2006), and [L] (Lesk 1995) is the ligand concentration. The Hill coefficient reflects the degree of cooperativity: The value h = 21 indicates the absence of cooperativity, h > 1 and h < 1 mean positive and negative cooperativity, respectively (Nakamura et al. 2006). The nonlinear fitting using this model was performed with the software Origin version 7.5.
Protein Data Bank accession code
The SCR structure has been deposited in the RSCB Protein Data Bank with the accession number 3CTM.
Acknowledgments
We thank Z. Lou for synchrotron data collection; X. Wang and H. Zhang for help in CD and fluorescence experiments; and X. Yu, Z. Zhou, and M. Zhang for technical assistance. This project was supported in part by the National Key Basic Research and Development Program of China (973 Program Nos. 2003CB716008, 2006DFB32420), the Hi-Tech Research and Development Program of China (863 Program Nos. 2006AA02A322 and 2007AA02Z226), Chinese Academy of Science grant (Nos. KSCX2-YW-05 and 2006CB911002), and grants from the National Science Foundation of China (NSFC) (Nos. 30221003 and 20776060). This work was also carried out as part of the Project for the Ministry of Education, People's Republic of China under the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT, IRT0532) and Jiangsu Development Strategies of Science and Technology (BG2007008).
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Yan Xu, Jiangnan University, Wuxi 214122, China; e-mail: biosean@yahoo.com.cn; fax: 86-10-8586411; or Xuejun C. Zhang, Oklahoma Medical Research Foundation, Oklahoma City, Oklahoma 73104, USA; e-mail: Cai-Zhang@omrf.org; fax: (405) 271-7953.
Abbreviations: ADH, alcohol dehydrogenase; FabG, β-ketoacyl reductase (from Escherichia coli); MtDH, mannitol-2-dehydrogenase (from Agaricus bisporus); PDB, Protein Data Bank; PED, (S)-specific 1-phenylenthanol dehydrogenase (of the denitrifying bacterium strain Ebn1); RADH, R-specific alcohol dehydrogenase (from Lactobacillus brevis); rmsd, root-mean-square deviation; SCR, (S)-specific carbonyl reductase (from Candida parapsilosis); SDR, short-chain dehydrogenases/reductases.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.035089.108.
References
- Baik, S.H., Michel, F., Aghajari, N., Haser, R., Harayama, S. Cooperative effect of two surface amino acid mutations (Q252L and E170K) in glucose dehydrogenase from Bacillus megaterium IWG3 on stabilization of its oligomeric state. Appl. Environ. Microbiol. 2005;71:3285–3293. doi: 10.1128/AEM.71.6.3285-3293.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzegar, A., Moosavi-Movahedi, A.A., Rezaei-Zarchi, S., Saboury, A.A., Ganjali, M.R., Norouzi, P., Hakimelahi, G.H., Tsai, F.Y. The aggregation and stability mechanistic studies on alcohol dehydrogenase by α-CyD. Biotechnol. Appl. Biochem. 2007 doi: 10.1042/BA20070031. http://lib.bioinfo.pl/pmid:17685894. [DOI] [PubMed] [Google Scholar]
- Bell, J.E., Dalziel, K. Conformational changes of glyceraldehyde-3-phosphate dehydrogenase induced by the binding of NAD. A unified model for positive and negative cooperativity. Biochim. Biophys. Acta. 1975;410:243–251. doi: 10.1016/0005-2744(75)90226-0. [DOI] [PubMed] [Google Scholar]
- Benach, J., Atrian, S., Gonzalez-Duarte, R., Ladenstein, R. The refined crystal structure of Drosophila lebanonensis alcohol dehydrogenase at 1.9 Å resolution. J. Mol. Biol. 1998;282:383–399. doi: 10.1006/jmbi.1998.2015. [DOI] [PubMed] [Google Scholar]
- Benach, J., Atrian, S., Gonzalez-Duarte, R., Ladenstein, R. The catalytic reaction and inhibition mechanism of Drosophila alcohol dehydrogenase: Observation of an enzyme-bound NAD-ketone adduct at 1.4 Å resolution by X-ray crystallography. J. Mol. Biol. 1999;289:335–355. doi: 10.1006/jmbi.1999.2765. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cao, L., Lee, J., Chen, W., Wood, T.K. Enantioconvergent production of (R)-1-phenyl-1,2-ethanediol from styrene oxide by combining the Solanum tuberosum and an evolved Agrobacterium radiobacter AD1 epoxide hydrolases. Biotechnol. Bioeng. 2006;94:522–529. doi: 10.1002/bit.20860. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Emsley, P., Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Filling, C., Berndt, K.D., Benach, J., Knapp, S., Prozorovski, T., Nordling, E., Ladenstein, R., Jornvall, H., Oppermann, U. Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J. Biol. Chem. 2002;277:25677–25684. doi: 10.1074/jbc.M202160200. [DOI] [PubMed] [Google Scholar]
- Ghosh, D., Weeks, C.M., Grochulski, P., Duax, W.L., Erman, M., Rimsay, R.L., Orr, J.C. Three-dimensional structure of holo 3 α,20 β-hydroxysteroid dehydrogenase: A member of a short-chain dehydrogenase family. Proc. Natl. Acad. Sci. 1991;88:10064–10068. doi: 10.1073/pnas.88.22.10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, D., Wawrzak, Z., Weeks, C.M., Duax, W.L., Erman, M. The refined three-dimensional structure of 3 α,20 β-hydroxysteroid dehydrogenase and possible roles of the residues conserved in short-chain dehydrogenases. Structure. 1994;2:629–640. doi: 10.1016/s0969-2126(00)00064-2. [DOI] [PubMed] [Google Scholar]
- Grimm, C., Maser, E., Mobus, E., Klebe, G., Reuter, K., Ficner, R. The crystal structure of α-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni shows a novel oligomerization pattern within the short chain dehydrogenase/reductase family. J. Biol. Chem. 2000;275:41333–41339. doi: 10.1074/jbc.M007559200. [DOI] [PubMed] [Google Scholar]
- Hoffken, H.W., Duong, M., Friedrich, T., Breuer, M., Hauer, B., Reinhardt, R., Rabus, R., Heider, J. Crystal structure and enzyme kinetics of the (S)-specific 1-phenylethanol dehydrogenase of the denitrifying bacterium strain EbN1. Biochemistry. 2006;45:82–93. doi: 10.1021/bi051596b. [DOI] [PubMed] [Google Scholar]
- Hörer, S., Stoop, J., Mooibroek, H., Baumann, U., Sassoon, J. The crystallographic structure of the mannitol 2-dehydrogenase NADP+ binary complex from Agaricus bisporus . J. Biol. Chem. 2001;276:27555–27561. doi: 10.1074/jbc.M102850200. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., Kjeldgaard, M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Jornvall, H., Persson, B., Krook, M., Atrian, S., Gonzalez-Duarte, R., Jeffery, J., Ghosh, D. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J. SERC Daresbury Laboratory; Warrington, UK: 1996. Joint CCP4 and ESF–EACBM newsletter on protein crystallography, no. 32; pp. 32–36. [Google Scholar]
- Kristan, K., Deluca, D., Adamski, J., Stojan, J., Lanisnik Rizner, T. Dimerization and enzymatic activity of fungal 17β-hydroxysteroid dehydrogenase from the short-chain dehydrogenase/reductase superfamily. BMC Biochem. 2005;6:28. doi: 10.1186/1471-2091-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, M.D., Thorton, J.M. PROCHECK—a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- Lesk, A.M. NAD-binding domains of dehydrogenases. Curr. Opin. Struct. Biol. 1995;5:775–783. doi: 10.1016/0959-440x(95)80010-7. [DOI] [PubMed] [Google Scholar]
- Liese, A., Karutz, M., Kamphuis, J., Wandrey, C., Kragl, U. Enzymatic resolution of 1-phenyl-1,2-ethanediol by enantioselective oxidation: Overcoming product inhibition by continuous extraction. Biotechnol. Bioeng. 1996;51:544–550. doi: 10.1002/(SICI)1097-0290(19960905)51:5<544::AID-BIT6>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Mazza, C., Breton, R., Housset, D., Fontecilla-Camps, J.C. Unusual charge stabilization of NADP+ in 17β-hydroxysteroid dehydrogenase. J. Biol. Chem. 1998;273:8145–8152. doi: 10.1074/jbc.273.14.8145. [DOI] [PubMed] [Google Scholar]
- Mittl, P.R., Berry, A., Scrutton, N.S., Perham, R.N., Schulz, G.E. Anatomy of an engineered NAD-binding site. Protein Sci. 1994;3:1504–1514. doi: 10.1002/pro.5560030916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nakamura, S., Oda, M., Kataoka, S., Ueda, S., Uchiyama, S., Yoshida, T., Kobayashi, Y., Ohkubo, T. Apo- and holo-structures of 3α-hydroxysteroid dehydrogenase from Pseudomonas sp. B-0831. Loop–helix transition induced by coenzyme binding. J. Biol. Chem. 2006;281:31876–31884. doi: 10.1074/jbc.M604226200. [DOI] [PubMed] [Google Scholar]
- Nakanishi, M., Matsuura, K., Kaibe, H., Tanaka, N., Nonaka, T., Mitsui, Y., Hara, A. Switch of coenzyme specificity of mouse lung carbonyl reductase by substitution of threonine 38 with aspartic acid. J. Biol. Chem. 1997;272:2218–2222. doi: 10.1074/jbc.272.4.2218. [DOI] [PubMed] [Google Scholar]
- Nie, Y., Xu, Y., Mu, X.Q., Wang, H.Y., Yang, M., Xiao, R. Purification, characterization, gene cloning, and expression of a novel alcohol dehydrogenase with anti-prelog stereospecificity from Candida parapsilosis . Appl. Environ. Microbiol. 2007;73:3759–3764. doi: 10.1128/AEM.02185-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordling, E., Oppermann, U.C., Jörnvall, H., Persson, B. Human type 10 17 β-hydroxysteroid dehydrogenase: Molecular modelling and substrate docking. J. Mol. Graph. Model. 2001;19:514–520. doi: 10.1016/s1093-3263(00)00098-x. 591–513. [DOI] [PubMed] [Google Scholar]
- Oppermann, U.C., Filling, C., Berndt, K.D., Persson, B., Benach, J., Ladenstein, R., Jornvall, H. Active site directed mutagenesis of 3 β/17 β-hydroxysteroid dehydrogenase establishes differential effects on short-chain dehydrogenase/reductase reactions. Biochemistry. 1997;36:34–40. doi: 10.1021/bi961803v. [DOI] [PubMed] [Google Scholar]
- Oppermann, U., Filling, C., Hult, M., Shafqat, N., Wu, X., Lindh, M., Shafqat, J., Nordling, E., Kallberg, Y., Persson, B., et al. Short-chain dehydrogenases/reductases (SDR): The 2002 update. Chem. Biol. Interact. 2003;143–144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z., Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pekar, A., Sukumar, M. Quantitation of aggregates in therapeutic proteins using sedimentation velocity analytical ultracentrifugation: Practical considerations that affect precision and accuracy. Anal. Biochem. 2007;367:225–237. doi: 10.1016/j.ab.2007.04.035. [DOI] [PubMed] [Google Scholar]
- Price, A.C., Zhang, Y.M., Rock, C.O., White, S.W. Structure of β-ketoacyl-[acyl carrier protein] reductase from Escherichia coli: Negative cooperativity and its structural basis. Biochemistry. 2001;40:12772–12781. doi: 10.1021/bi010737g. [DOI] [PubMed] [Google Scholar]
- Sakoda, M., Hiromi, K. Determination of the best-fit values of kinetic parameters of the Michaelis-Menten equation by the method of least squares with the Taylor expansion. J. Biochem. 1976;80:547–555. doi: 10.1093/oxfordjournals.jbchem.a131310. [DOI] [PubMed] [Google Scholar]
- Schlieben, N.H., Niefind, K., Muller, J., Riebel, B., Hummel, W., Schomburg, D. Atomic resolution structures of R-specific alcohol dehydrogenase from Lactobacillus brevis provide the structural bases of its substrate and cosubstrate specificity. J. Mol. Biol. 2005;349:801–813. doi: 10.1016/j.jmb.2005.04.029. [DOI] [PubMed] [Google Scholar]
- Smilda, T., Kamminga, A.H., Reinders, P., Baron, W., van Hylckama Vlieg, J.E., Beintema, J.J. Enzymic and structural studies on Drosophila alcohol dehydrogenase and other short-chain dehydrogenases/reductases. J. Mol. Evol. 2001;52:457–466. doi: 10.1007/s002390010175. [DOI] [PubMed] [Google Scholar]
- Tanaka, N., Nonaka, T., Nakanishi, M., Deyashiki, Y., Hara, A., Mitsui, Y. Crystal structure of the ternary complex of mouse lung carbonyl reductase at 1.8 Å resolution: The structural origin of coenzyme specificity in the short-chain dehydrogenase/reductase family. Structure. 1996;4:33–45. doi: 10.1016/s0969-2126(96)00007-x. [DOI] [PubMed] [Google Scholar]
- Tanaka, N., Nonaka, T., Nakamura, K.T., Hara, A. SDR structure, mechanism of action, and substrate recognition. Curr. Org. Chem. 2001;5:89–111. [Google Scholar]
- Thompson, J.D., Higgins, D.G., Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varughese, K.I., Xuong, N.H., Kiefer, P.M., Matthews, D.A., Whiteley, J.M. Structural and mechanistic characteristics of dihydropteridine reductase: A member of the Tyr-(Xaa)3-Lys-containing family of reductases and dehydrogenases. Proc. Natl. Acad. Sci. 1994;91:5582–5586. doi: 10.1073/pnas.91.12.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Z.L., Lin, G.Q., Li, Z.Y. Microbial transformation of 2-hydroxy and 2-acetoxy ketones with Geotrichum sp . Bioorg. Med. Chem. 2000;8:1129–1137. doi: 10.1016/s0968-0896(00)00023-7. [DOI] [PubMed] [Google Scholar]
- Winberg, J.O., Brendskag, M.K., Sylte, I., Lindstad, R.I., McKinley-McKee, J.S. The catalytic triad in Drosophila alcohol dehydrogenase: pH, temperature and molecular modelling studies. J. Mol. Biol. 1999;294:601–616. doi: 10.1006/jmbi.1999.3235. [DOI] [PubMed] [Google Scholar]
- Yamashita, A., Kato, H., Wakatsuki, S., Tomizaki, T., Nakatsu, T., Nakajima, K., Hashimoto, T., Yamada, Y., Oda, J. Structure of tropinone reductase-II complexed with NADP+ and pseudotropine at 1.9 Å resolution: Implication for stereospecific substrate binding and catalysis. Biochemistry. 1999;38:7630–7637. doi: 10.1021/bi9825044. [DOI] [PubMed] [Google Scholar]
- Zhang, X.J., Matthews, B.W. EDPDB: A multifunctional tool for protein structure ananlysis. J. Appl. Crystallogr. 1995;28:624–630. [Google Scholar]