

Abstract
This paper reports that the acetylation of lysine ε-NH3 + groups of α-amylase—one of the most important hydrolytic enzymes used in industry—produces highly negatively charged variants that are enzymatically active, thermostable, and more resistant than the wild-type enzyme to irreversible inactivation on exposure to denaturing conditions (e.g., 1 h at 90°C in solutions containing 100-mM sodium dodecyl sulfate). Acetylation also protected the enzyme against irreversible inactivation by the neutral surfactant TRITON X-100 (polyethylene glycol p-(1,1,3,3-tetramethylbutyl)phenyl ether), but not by the cationic surfactant, dodecyltrimethylammonium bromide (DTAB). The increased resistance of acetylated α-amylase toward inactivation is attributed to the increased net negative charge of α-amylase that resulted from the acetylation of lysine ammonium groups (lysine ε-NH3 + → ε-NHCOCH3). Increases in the net negative charge of proteins can decrease the rate of unfolding by anionic surfactants, and can also decrease the rate of protein aggregation. The acetylation of lysine represents a simple, inexpensive method for stabilizing bacterial α-amylase against irreversible inactivation in the presence of the anionic and neutral surfactants that are commonly used in industrial applications.
Keywords: amylase, charge ladders, industrial biotechnology, protein aggregation, protein engineering, sodium dodecyl sulfate, thermostability
This paper describes a strategy for generating chemically modified derivatives of industrially important enzymes with unusually high net negative charge in order to increase their resistance toward irreversible inactivation by negatively charged and neutral surfactants (Vendruscolo and Dobson 2007). The acetylation of multiple lysine residues (∼17) in a thermostable α-amylase (from Bacillus licheniformis; E.C. 3.2.2.1, denoted BLA) protected this enzyme from irreversible inactivation by heat and surfactant, and did not perturb the native structure or decrease the thermostability of the enzyme. Using inexpensive chemistry to produce highly charged enzymes is a practical method for improving the resistance of industrially important hydrolases (e.g., α-amylase) and other enzymes (e.g., carbonic anhydrase, E.C. 4.2.1.1) against irreversible, surfactant- and heat-mediated inactivation. Achieving similarly large increases in net charge with mutagenesis is time consuming, and the effects of multiple amino acid substitutions on the structure, thermostability, and activity of the protein can be difficult to predict.
Enzymes are used as catalysts in various industrial, commercial, and pharmaceutical applications (Kirk et al. 2002; Soetaert and Vandamme 2006); they are more selective, more efficient, and less toxic than many synthetic catalysts. Among their disadvantages (in commercial or industrial applications) are their low thermostability, their propensity to aggregate, their high cost, and their potential to act as allergens. Efforts to develop variants of industrially important enzymes that have increased resistance toward inactivation have involved: (1) the discovery of new enzymes from thermophilic, halophilic, or alkaliphilic bacteria such as Bacillus clausii (Salameh and Wiegel 2007), (2) the engineering of existing enzymes by site-directed mutagenesis (Korkegian et al. 2005; Reetz et al. 2006), and directed evolution (Johannes and Zhao 2006).
The largest application of enzymes in industrial biotechnology, both in terms of quantity and cost, is the use of α-amylases, cellulases, lipases, and proteases in commercial cleaning solutions, e.g., fabric cleaners (Kirk et al. 2002). Among the surfactants commonly used in commercial cleaners are the anionic surfactants sodium dodecyl sulfate (SDS, also known as sodium lauryl sulfate, or SLS), sodium lauryl ether sulfate (SLES), and the nonionic surfactant Triton X-100 (i.e., 4-[1,1,3,3-tetramethylbutyl]phenyl-polyethylene glycol). Cationic surfactants are not commonly used because of their toxicity. The concentration of surfactant in liquid formulations of detergent is typically 5%–20% by weight (Saeki et al. 2007). Enzymes used in these applications must therefore retain (or regain) activity after storage in high concentrations of surfactants.
The inactivation of enzymes (in vivo and in vitro) is a result of: (1) protein unfolding and/or loss of a cofactor, (2) the misfolding and aggregation of proteins, (3) the covalent modification of amino acids needed for catalysis, (4) the binding of inhibitors at the active site, and (5) the hydrolysis of peptide bonds.
The inactivation of industrial enzymes in applications in vitro, and particularly in surfactant solutions, involves principally the first two factors: unfolding and aggregation (or misfolding). Four factors determine the rate of aggregation of proteins: thermostability of the native state, net charge, hydrophobicity, and the propensity to form β-sheet secondary structure (Chiti et al. 2002, 2003). In general, the rate of aggregation correlates directly with both hydrophobicity and propensity to form β-sheets, and inversely with both net charge (positive or negative) and thermostability (Chiti and Dobson 2006). The potent effect of net charge, in particular, on the rate of protein aggregation is illustrated by single amino acid substitutions that reduce net charge and promote protein aggregation without significantly changing the hydrophobicity, thermostability, solution structure, or activity of the protein (e.g., the E100K, D101N, S139K, and D90A variants of superoxide dismutase that cause the protein aggregation disease, amyotrophic lateral sclerosis) (Shaw and Valentine 2007). Liu and coworkers have shown recently that “supercharged” variants of green fluorescent protein and glutathione-S-transferase, with increases in formal charge between ∼20 and 50 units (generated by multiple rounds of mutagenesis), are especially resilient to irreversible inactivation and aggregation (Lawrence et al. 2007).
Net charge, hydrophobicity, and thermostability also govern the rate of surfactant-mediated unfolding. Recent work from our laboratory using protein charge ladders has shown that increases in net charge, and decreases in hydrophobicity, can increase the kinetic stability (e.g., the half-life) of native bovine carbonic anhydrase II (BCA II) against denaturation in SDS by factors of up to 1000 (Gitlin et al. 2006b,c; Gudiksen et al. 2006). This kinetic stabilization against denaturation by SDS was most pronounced at intermediate degrees of acetylation (i.e., with the intermediate rungs of the charge ladder), and occurred in spite of large decreases in the thermostability of BCA II (e.g., peracetylation of BCA II decreased the temperature of unfolding by ∼20°C) (Gitlin et al. 2006b).
Results and Discussion
This study focused on α-amylase, a calcium-containing metallo-enzyme that catalyzes the hydrolysis of α-1,4 glycosidic bonds in starch. α-Amylase has at least six different industrial applications (including a part of the process for the production of ethanol from corn starch) (Kirk et al. 2002). The heat-stable α-amylase protein that we used (from Bacillus licheniformis, and denoted BLA), is a single subunit protein with a molecular weight (MW) of 55 kDa; it has no disulfide bonds and has 28 lysine residues. The isoelectric point (pI) is ∼6.0.
Chemical modification of α-amylase
By adding 20 μL of 800-mM acetic anhydride to 10.0 mL of a solution of BLA (1.5 mg/mL BLA, 100 mM HEPES, pH 9.0, 23°C), we obtained a mixture of derivatives of BLA that had 16–19 R-NHCOCH3 groups (ΔMW = +42 Da per acetyl modification). The predominant species contained 17 acetyl modifications according to electrospray ionization (ESI) mass spectrometry (MS) (Fig. 1A,B). We studied this mixture of acetylated species, which we refer to as “BLA-Ac(∼17),” without further separation. Since BLA does not naturally undergo acetylation at the N terminus, it is possible that some regioisomers in the BLA-Ac(∼17) mixture were acetylated at the N terminus by acetic anhydride (discussed below; see also Supplemental Fig. S1).
Figure 1.

Mass spectrometry of acetylated BLA and capillary electrophoresis of a charge ladder of BLA. (A,B) Electrospray ionization mass spectra of unmodified BLA (denoted “BLA-Ac[0]”) and acetylated BLA (denoted “BLA-Ac=[∼17]”). (C) Capillary electropherogram of a charge ladder of BLA prepared by acetylating lysine-ε-NH3 + (and possibly the α-amino terminus). The ruler above the charge ladder correlates the number of acetyl modifications, n, with each rung of the ladder; dashed lines in the ruler denote the shoulders of some rungs that constitute a faint second and third charge ladder (the chemical and biophysical nature of these ladders are unknown). The peak marked “DMF” represents the electrically neutral marker, dimethyl formamide. (D) Plot of the electrophoretic mobility (μ) of each rung of the charge ladder of BLA as a function of n (i.e., the number of acetyl modifications). A line was fit by linear least squares to the linear region of the plot; the x-intercept is equal to the ratio Z0BLA-Ac(0)/ΔZAc; i.e., the ratio between the net charge of the rung of lowest mobility (Z0BLA-Ac[0]) and the change in charge resulting from a single acetylation (ΔZAc). The net charge (Zo) of BLA-Ac(0) (μ = 1.3 cm2 kV−1 min−1) is equal to the x-intercept. The inset table shows Z0BLA-Ac(0) as a function of ΔZAc.
In order to determine if the acetylation of lysine residues in BLA-Ac(∼17) were nonspecific in the lysine residues that were modified, and to detect N-terminal actetylation, we used tandem ESI-MS (ESI-MS/MS) to identify modified residues in BLA-Ac(∼17). Nonspecific acetylation of lysine residues in BLA-Ac(∼17) would result in the detection of acetyl modifications on all 28 lysine residues in BLA.
Because the acetylation of lysine ε-NH3 + groups in BLA might prevent trypsin from proteolyzing BLA at lysine residues, we performed two separate proteolyses of BLA-Ac(∼17) using trypsin and elastase (elastase hydrolyzes peptide bonds at hydrophobic residues). Peptides representing ∼89% of the BLA sequence were sequenced by ESI-MS/MS (Fig. S1; see Supplemental material). Eighteen different lysine residues had a +42 Da modification: lys70, 80, 88, 154, 176, 213, 234, 237, 251, 306, 315, 319, 370, 381, 383, 389, 392, and 436. Five lysine residues were not modified: lys47, 76, 106, 180, and 344. Three lysine residues, lys23, 136, and 170 and the ala1 N-terminal alanine residue were not contained in the sequence coverage; we therefore do not know if these residues were acetylated. Nevertheless, the results of ESI-MS/MS suggest that the acetylation of lysine residues in BLA were not entirely random under our reaction conditions.
Measuring the net charge of α-amylase with protein charge ladders and capillary electrophoresis
From studies of carbonic anhydrase at pH 8.4, we have estimated that each acetyl modification increases the net negative charge by 0.9, instead of 1.0 (Gitlin et al. 2006a). This difference in charge is attributed to charge regulation. Charge regulation can be thought of as a consequence of differences between the local pH (pHlocal) at a particular residue and the pH of the bulk solvent (pHsolv); such differences in pHlocal and pHsolv will affect the ionization of acidic or basic residues that have pKa values within ±3 units of pHsolv. (Menon and Zydney first discussed charge regulation in the context of protein charge ladders) (Menon and Zydney 2000). Alternatively and equivalently, charge regulation can be considered to reflect changes in the values of pKa of ionizable groups due to changes in local electrostatic fields. In the case of the acetylation of lysine-ε-NH3 +, charge regulation arises from a local decrease in pH, and/or an increase in pKa (largely in histidine residues).
After taking into account the effects of charge regulation (as measured from charge ladders of BCA II) (Gitlin et al. 2006b), we estimated that the proteins in the mixture BLA-Ac(∼17) had an average net charge (at pH 8.5) that was ∼15 units more negative (i.e., 17 × 0.9) than that of unmodified BLA (referred to as “BLA-Ac[0]”). In order to estimate the net charge of BLA-Ac(0) and the net charge of the BLA derivatives in the BLA-Ac(∼17) mixture, we prepared a charge ladder of BLA by acetylating lysine-ε-NH3 + functional groups, and analyzed this charge ladder using capillary electrophoresis (CE) (Fig. 1C). CE separates molecular species according to their electrophoretic mobility (μ), which is proportional to the net charge (Z) of the protein and inversely proportional to its hydrodynamic drag (Equation 1) (Grossman 1992). The analysis of protein charge ladders by CE is a convenient tool for the estimation of the net charge of a protein (Gao and Whitesides 1997). We have previously reviewed methods for measuring the net charge of proteins with protein charge ladders and capillary electrophoresis (Gitlin et al. 2006a), and we also include a brief discussion here.
Equation 1 is the generally used approximation that expresses the electrophoretic mobility (μ) of a molecular species in terms of its net charge (Z) and molecular weight (M). The terms Cp and α in Equation 1 are constants. The theoretical value of α varies from 0.3 to 1.0, and depends on the model used to derive the hydrodynamic drag from the molecular weight; typically for globular proteins α ≈ 2/3 (Rickard et al. 1991; Basak and Ladisch 1995). Equation 1 expresses the mobility of rungs of a charge ladder by expressing the net charge, Z, as a function of the net charge of the unmodified protein (Z°BLA-[Ac0]) and the change in charge (n · ΔZ) that results from n acetylations.
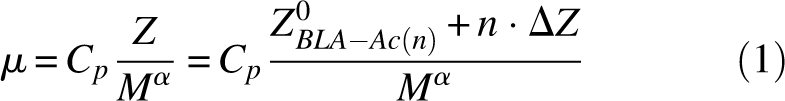 |
The charge ladder for BLA (Fig. 1C) consisted of 23 observable rungs (including the “zeroth” rung, which represents the unmodified protein, BLA-Ac[0]). Each rung consisted of a mixture of proteins derived from BLA with the same number (n) of acetylated R-NH3 + functionalities, and thus approximately the same net charge. For an R-NH3 +-based charge ladder of BLA, there might theoretically be 30 rungs (BLA has 28 lysine residues and one nonacetylated α-amino terminus; the unmodified protein counts as another rung). The observed charge ladder (Fig. 1C) has, however, only 23 observable rungs. The rungs at n ≥ 22 were not well resolved; the 23rd rung (i.e., n = 22 in Fig. 1C) appeared as a shoulder and not a well-resolved peak. We expected these higher rungs to have lower resolution because of the nonlinear relationship between n and μ at very high values of n (Carbeck et al. 1999).
If the change in net charge (ΔZ) that results from acetylation is known, then the net charge of the lowest rung (e.g., ZoBLA-[Ac0]) can be estimated by fitting a line to the linear region of the plot and extrapolating the line to the X-axis. Figure 1D shows this type of plot: Here the electrophoretic mobility of each rung of the charge ladder of BLA is plotted as a function of the number of acetyl modifications (n). Extrapolation of the best fit line to the X-axis determined that ZoBLA-Ac(0) = –1.8 (assuming ΔZ = 0.9 per acetyl modification).
In contrast to the net charge of BLA-Ac(0) determined by protein charge ladders and capillary electrophoresis (i.e., ZoBLA-Ac[0] = –1.8), the net charge of BLA-Ac(0) at pH 8.5 is predicted from its sequence to be –13 (i.e., BLA contains 21 Arg, 28 Lys, 23 His, 25 Glu, and 37 Asp residues; one nonacetylated α-amino terminus and one α-carboxy terminus). A difference of this magnitude between values is surprising (although the net charge of proteins as measured by CE (ZCE) and their net charge as calculated from the amino acid sequence (Zseq) are often different). For example, a survey of 23 proteins found that the values of Zseq and ZCE differed in magnitude by an average of 26% (Colton et al. 1997).
We have two explanations (which are not mutually exclusive) for why the measured net negative charge of BLA-Ac(0) is ∼11 units lower than the value predicted from its amino acid sequence: (1) the coordination of metal cations, and (2) nonstandard values of pKa of some ionizable functionalities in BLA. The binding of Ca2+ to BLA is essential for its activity (Juncosa et al. 1994) and X-ray crystallography has shown that the active site of BLA exhibits a unique Ca2+–Na+–Ca2+ triad that is coordinated to side-chain carboxylate groups and the oxygen of main-chain carbonyl groups. A third Ca2+ cation is bound distal to the active site (Lee et al. 1991; Machius et al. 1998). The binding of these metal cations might reduce the net negative charge of BLA by as much as 7 units of formal charge. The most plausible candidates for anomalous values of pKa are histidine residues. A survey of pKa values of 39 different histidine residues from 13 different proteins (Edgcomb and Murphy 2002) showed that the pKa of imidazole groups (as measured by NMR spectroscopy) differed greatly from the regularly cited (“standard”) value of 6.3 (Thurlkill et al. 2006) and ranged from 4.6 (His40 of bovine chymotrypsinogen) to 9.2 (His72 in tyrosine phosphatase). The physical-chemical basis for this variation is not completely understood (Edgcomb and Murphy 2002), but may be due in part to local variations in electrostatic potential. The value of pKa of aspartate-β-CO2H and glutamate-γ-CO2H also vary widely in proteins (García-Moreno et al. 1997).
Measuring the effects of acetylation on the structure and thermostability of α-amylase
In order to determine if the acetyl modifications changed the secondary structure of native BLA, we analyzed BLA-Ac(0) and BLA-Ac(∼17) with circular dichroism (CD) spectroscopy. The CD spectra of BLA-Ac(0) and BLA-Ac(∼17) (Fig. 2A) were nearly superimposable; this similarity suggests that the extensive acetylation did not perturb the overall secondary structure of BLA (under reaction conditions that favored the native state of BLA; 25°C, pH 8.4).
Figure 2.

Lysine acetylation did not alter the secondary structure or decrease the thermostability of BLA. (A) Circular dichroism of unmodified BLA (denoted “BLA-Ac[0]”) and acetylated BLA (denoted “BLA-Ac[∼17]”). CD spectra of acetylated and nonacetylated proteins were nearly superimposable (0 mM surfactant, pH 8.5, 25 mM Tris). (B) Thermal unfolding of acetylated and unmodified BLA monitored by differential scanning calorimetry (pH 7.4, 10 mM phosphate, 0 mM surfactant). BLA-Ac(0) had a single transition at Tm = 83°C; the thermal unfolding of BLA-Ac(∼17) yielded two transitions at 84°C and 93°C.
Differential scanning calorimetry (DSC) demonstrated that lysine acetylation did not decrease the thermostability of BLA (Fig. 2B; 0 mM surfactant, 10 mM phosphate, pH 7.4). The thermal unfolding of BLA-Ac(0) produced a single endothermic transition with a melting temperature (Tm) at 83°C. This value is consistent with previously published reports (Declerck et al. 2003). The thermal unfolding of BLA-Ac(∼17) produced two unresolved, overlapping endothermic transitions at ∼84°C and 93°C. We cannot fully explain the overlapping transitions, but it is reasonable to attribute them to (1) a uniform decrease in the cooperativity of unfolding of all proteins in the mixture, (2) distinct transitions for different derivatives in the mixture (e.g., BLA-Ac[16] vs. BLA-Ac[19]), or (3) distinct transitions for regioisomers of derivatives in the mixture. Nevertheless, the increase in thermostability upon acetylation is not completely surprising, and is not necessarily due to increases in net charge. For example, lysine acetylation also increases the hydrophobicity of the lysine side chain. The Hansch π-parameter (logP) for NH3 + groups is log P = –2.12; and for NHCOCH3 groups log P = –1.21 (Hansch and Steward 1964; Hansch and Coats 1970). Although it is a general rule of protein engineering that increasing the surface hydrophobicity (e.g., by introducing hydrophobic residues via site-directed mutagenesis) decreases the thermostability of the native state (Matthews 1993; Lee and Vasmatzis 1997), BLA represents a well-known exception to this rule (Declerck et al. 2003; Machius et al. 2003). Over nine different hydrophobic variants of BLA have been shown to be more thermostable than the wild-type (WT) protein. For example, the hepta-variant of BLA (H133I/H156Y/A181T/N190F/A209V/Q264S/N265Y) exhibited a Tm = 106°C, whereas the WT protein exhibited a Tm = 83°C (Declerck et al. 2003). X-ray crystallographic analysis of hydrophobic variants of BLA suggested that the stabilizing effects of these bulky substitutions could be due to an increase in the molecular packing at surface indentations in BLA (Declerck et al. 2003). The stabilizing effects of lysine acetylation in BLA (against thermal unfolding) could be due to a similar effect.
We do not expect the increase in thermostability that we observed for acetylated α-amylase to be a general outcome of acetylating proteins. For example, analysis of derivatives of BCA II using DSC showed that peracetylation lowered the Tm from 70°C to 50°C (although peracetylation did increase the kinetic stability of BCA II toward unfolding by SDS) (Gitlin et al. 2006b). Similarly, the acetylation of ∼60% of the lysine residues in cytochrome c reduced the Tm from 85°C to 65°C (Jain and Hamilton 2002).
Lysine acetylation protects α-amylase from irreversible inactivation by heat and surfactant.
We measured the enzymatic activity of BLA and its derivatives by measuring an increase in absorbance at 400 nm due to the formation of p-nitrophenol (λmax = 400 nm) by hydrolysis of 4,6-o-benzilidine-p-nitrophenyl maltoheptaoside in the presence of α-glucosidase (McCleary et al. 2002). The activity of BLA was measured after cooling an aliquot of BLA solution (25°C or 90°C, 0–100 mM SDS, Triton X-100 or DTAB) on ice and diluting 1:181 into an assay solution of maltoheptaoside and α-glucosidase that contained no surfactant and was at room temperature (see Materials and Methods for additional details).
After 1 h in 100 mM SDS, 25°C (and after a 1:181 dilution into the surfactant-free assay solution), both proteins had equal activity (Fig. 3A); that is, before heating, the activities of BLA-Ac(0) and BLA-Ac(∼17) were indistinguishable. On incubating at 90°C, however, the activity of BLA-Ac(0) decreased by ∼60% after 20 min and by 95% after 60 min (Fig. 3B; see Supplemental material for raw kinetic data). The BLA-Ac(∼17) protein was more resistant to inactivation: It lost only ∼30% of its initial activity after 20 min, and ∼60% after 60 min (Fig. 3B). We fit the inactivation plot in Figure 3B with a single exponential function (y = Ae−x/k). It is difficult to measure values of initial activity that are <5%; we therefore only fit points in Figure 3B with values >5% initial activity. This fit yielded time constants (k) of 94 ± 4 min for BLA-Ac(∼17) (R 2 = 0.969) and 17 ± 1 min for BLA-Ac(0) (R 2 = 0.946). The ratio of time constants, k BLA-Ac(∼17):k BLA-Ac(0), is 5.5 ± 0.1; this ratio indicates that BLA-Ac(∼17) had a 5.5-fold greater half-life than BLA-Ac(0) under the experimental conditions. Numerical integration of the inactivation plots for BLA-Ac(0) and BLA-Ac(∼17) in Figure 3B yielded a total product ratio (BLA-Ac[∼17]: BLA-Ac[0]) of 5.2; that is, BLA-Ac(∼17) produced 5.2-fold more product than did BLA-Ac(0) during the experimental time course.
Figure 3.

Lysine acetylation inhibited the irreversible inactivation of BLA under denaturing conditions (90°C, 100 mM SDS, 25 mM Tris, 192 mM glycine, pH 8.5). Activity assays were initiated by diluting an aliquot of heated BLA solutions 1:181 into the α-glucosidase/4,6-o-benzilidine-p-nitrophenyl maltoheptaoside assay solution. (A) Activities of BLA-Ac(∼17) and BLA-Ac(0) were approximately equivalent after exposure to 100 mM SDS at room temperature (pH 8.5, 25 mM Tris) with turnover numbers equaling 847 ± 16 min−1 for BLA-Ac(∼17) and 885 ± 20 min−1 for BLA-Ac(0). Error bars represent the standard deviation of absorbance values from the mean (N = 7). (B) Inactivation of BLA-Ac(∼17) and BLA-Ac(0) after incubation in 100 mM SDS (90°C, pH 8.5), expressed as a percent of the initial activity (i.e., activity in 100 mM SDS at room temperature, before heating at 90°C). Values <5% of the initial activity were ignored due to unknown reliability. Both data sets were fit to an exponential (y = Ae−x/k) yielding time constants (k) of 94 ± 4 min for BLA-Ac(∼17) (R 2 = 0.969) and 17 ± 1 min for BLA-Ac(0) (R 2 = 0.946); BLA-Ac(∼17) therefore had a 5.5-fold greater half-life than BLA-Ac(0). Numerical integration yielded a total product ratio (BLA-Ac[∼17]: BLA-Ac[0]) of 5.2. Error bars in B represent the standard deviation of percent of activity from the mean (N = 7).
Surfactant aids the refolding of acetylated α-amylase.
Lysine acetylation did not protect BLA against thermal inactivation in the absence of surfactant (Fig. 4). Both acetylated and unmodified BLA were inactive (i.e., retained <5% of the initial activity) after 60 min at 90°C in 0 mM surfactant. The requirement for surfactant in order for denatured acetylated BLA to refold into its active form is consistent with previous reports that surfactants aid in the refolding of chemically or thermally unfolded proteins (including α-amylase) (Cleland et al. 1992; Khodagholi and Yazdanparast 2005; Swietnicki 2006).
Figure 4.

The protective effects of lysine acetylation against BLA inactivation in anionic (SDS), cationic (DTAB), and nonionic (TRITON X-100) surfactants. In the absence of detergent (denoted 0 mM detergent), both BLA-Ac(0) and BLA-Ac(∼17) were almost completely inactivated after 1 h at 90°C. In the presence of 1 mM SDS, the BLA-Ac(∼17) retained ∼20% of the initial activity, whereas BLA-Ac(0) retained <5% of the initial activity after 1 h at 90°C. At 10 or 100 mM SDS, the percent of initial activity retained by BLA-Ac(∼17) was nearly an order of magnitude higher than that of BLA-Ac(0). Both BLA-Ac(0) and BLA-Ac(∼17) retained ≤5% initial activity after exposure to 1, 10, or 100 mM DTAB for 1 h, 90°C. In 1–100 mM Triton X-100, BLA-Ac(∼17) retained 45%–55% activity and the unmodified protein retained ∼5% activity. Error bars represent the standard deviation of activity values from the mean (N = 7).
Surfactant-mediated refolding is not completely understood, but one hypothesis is that the hydrophobic tail of the surfactant shields hydrophobic regions of the protein from aggregating with other hydrophobic polypeptides, and thus prevents irreversible aggregation and precipitation of the protein (Cleland and Randolph 1992; Lu et al. 2005). Surfactants are sometimes used in tandem with α/β-cyclodextrins in order to increase the yield of refolding of thermally or chemically denatured proteins (Rozema and Gellman 1996; Daugherty et al. 1998). The mode of action in these tandem systems is thought to involve the binding of surfactant to the protein, followed by the stripping of the surfactant by the cyclodextrin (these tandem systems are even considered to be simple alternatives to conventional denaturation/renaturation methods for studying protein folding) (Otzen and Oliveberg 2001). The typical yield of recovered activity for thermally or chemically denatured proteins, when surfactant and cyclodextrins are used, is 50%–100%; excluding the cyclodextrin step, however, can result in recovery of only ∼1% (Rozema and Gellman 1996; Daugherty et al. 1998; Khodarahmi and Yazdanparast 2004; Khodagholi and Yazdanparast 2005; Yazdanparast et al. 2005a,b). After 1 h under harsh conditions (90°C, 100 mM surfactant), up to 50% of the activity of BLA-Ac(∼17) could be recovered (without cyclodextrins), whereas <5% of the activity of BLA-Ac(0) was recovered.
Effects of surfactant charge on inactivation of acetylated α-amylase
In order to investigate how the charge of the surfactant affects the inactivation of BLA, and in order to test further the hypothesis that electrostatic interactions between the surfactant and the polypeptide govern (at least in part) the rate of inactivation, we measured the activity of BLA after exposure to positively charged and neutral surfactants at three concentrations (1, 10, and 100 mM surfactant; Fig. 4). BLA-Ac(0) was completely inactivated after 60 min at 90°C in cationic, neutral or anionic surfactants at these concentrations (Fig. 4). The inability of BLA-Ac(0) to regain enzymatic activity when cooled and diluted from denaturing to nondenaturing concentrations of neutral, cationic, or anionic surfactant is consistent with previous studies involving chemically denatured α-amylase (Khodarahmi and Yazdanparast 2004; Yazdanparast et al. 2005a).
Acetylation did not protect BLA from inactivation in the presence of the cationic surfactant DTAB. Both BLA-Ac(0) and BLA-Ac(∼17) retained <5% of the initial activity after heating in DTAB and dilution into the assay solution (which was at room temperature; Fig. 4). The low activity of BLA-Ac(∼17) in DTAB might be due to favorable electrostatic interactions between the positively charged DTAB and the negatively charged BLA-Ac(∼17) polypeptide. Several groups have shown that charged macromolecules, as well as charged surfactants, bind more favorably (kinetically or thermodynamically) to polymers (e.g., DNA or polypeptides) that have the opposite sign of net charge than to polymers with the same sign of net charge (La Mesa 2005; Liu and Guo 2007).
Lysine acetylation did protect BLA from inactivation in the presence of the neutral surfactant, Triton X-100 (Fig. 4), and the protection was slightly more pronounced in Triton than in the anionic surfactant SDS. For example, after 60 min at 90°C, BLA-Ac(∼17) retained ∼55% initial activity in 10 mM Triton, but only ∼37% initial activity in 10 mM SDS. We hypothesize that the activity retained by BLA-Ac(∼17) in neutral surfactant was greater than that in anionic or cationic surfactant because the neutral surfactant bound more weakly than the anionic or cationic surfactant. We note that in the presence of Triton, the difference in retained activity between BLA-Ac(∼17) and BLA-Ac(0) cannot be explained by differing electrostatic interactions between the two proteins and surfactant because TRITON is uncharged.
Lysine acetylation does not affect the rate of inactivation by sodium perborate
We also explored the possibility that lysine acetylation might protect BLA from inactivation by sodium perborate (NaBO3), an oxidant used along with α-amylase in many commercial and industrial processes. We found, however, that the kinetic profiles of inactivation for both BLA-Ac(0) and BLA-Ac(∼17) were indistinguishable (Supplemental Fig. S2).
Lysine acetylation inhibits the precipitation of α-amylase
The acetylation of BLA also resulted in a decrease in its propensity to form insoluble aggregates. Supplemental Figure S3 compares the results of assays of the thermal aggregation of BLA-Ac(0) and BLA-Ac(∼17) (see Supplemental material for experimental details). Aggregation was initiated by heating solutions of BLA at 90°C (9 μM BLA, pH 8.5, 25 mM Tris, 192 mM glycine, 10% trifluoroethanol); aggregation was assayed by centrifuging protein solutions and measuring the absorbance of the supernatant. Supplemental Figure S3 shows that ∼90% of BLA-Ac(0) aggregated after 15 min at 90°C. In contrast, only ∼15% of BLA-Ac(∼17) aggregated after 15 min; after 200 min the amount of aggregated BLA-Ac(∼17) had not increased and ∼80% of the protein remained soluble. We note that our result regarding the increased solubility of BLA-Ac(∼17) is similar to the claims of Eriksen and Pedersen (N. Eriksen and G. Pedersen, U.S. Patent 5,286,404, Feb. 15, 1994); they claimed that the solubility of an engineered alkaline protease (Alcalase) could be increased by the acylation of its lys-ε-NH3 +.
Conclusions
Acetylation of α-amylase with acetic anhydride generated derivatives with high net negative charge, apparently without perturbing the structure of the enzyme, and without decreasing its enzymatic activity or thermostability. These derivatives of BLA have a net charge ranging from approximately –16 to –19 (at pH 8.5). The acetylation reaction is easily performed with stoichiometric amounts of acetic anhydride. The by-product of the acetylation reaction is acetic acid, and the acetylated lysine functionality is a common post-translational modification found in biological systems (Kim et al. 2002; O'Shea et al. 2005).
Lysine acetylation represents a useful method for increasing the net charge of α-amylase and improving its resistance against irreversible inactivation; these changes may make it more useful in some applications in biotechnology. The methods that we described for α-amylase should be applicable to other proteins used in industrial biotechnology, especially those used in other enzyme–surfactant systems; for example, surfactants are being considered for use in the production of ethanol from lignocellulose sources such as soft wood or corn stover (Kaar and Holtzapple 1998), as well as in the degradation of paper-based waste (Kim and Chun 2004). In both of these processes, the presence of a nonionic surfactant (e.g., Triton X-100) can increase the activity of cellulase and inhibit the thermal inactivation of cellulase (Kaar and Holtzapple 1998; Kim and Chun 2004). Up to 60% of the production cost of fermentation of corn stover can come from the cost of the cellulase enzyme (Galbe and Zacchi 2002). Therefore, an inexpensive method for prolonging the activity of enzyme catalysts, such as the chemical modification method described here, might improve cost-sensitive industrial processes that involve enzymes (Kaar and Holtzapple 1998).
Materials and Methods
Chemical modification of α-amylase
Heat-stable α-amylase from Bacillus licheniformis (BLA) was purchased from Sigma as a lyophilized powder (∼95% pure, Sigma Cat. No. A4551). Acetylated variants of BLA were prepared using protocols for the acetylation of other proteins (Gitlin et al. 2006c). Briefly, solutions of BLA (1.5 mg/mL or 27 μM BLA, 100 mM HEPBS buffer, pH 9.0, 10.0 mL total volume) were allowed to react with various volumes of acetic anhydride (800 mM acetic anhydride in dioxane) ranging from 0 to 300 μL; the pH of the reaction mixture was monitored with a pH electrode and kept at 9.0 by the addition of NaOH. We then dialyzed BLA proteins (both BLA-Ac(∼17) and the nonacetylated BLA-Ac(0) control) against Tris buffer (25 mM Tris, 192 mM glycine pH 8.5, 2 mM CaCl2) using Slide-A-Lyzer dialysis cartridges (Pierce; 10,000 MWCO). For storage, protein solutions were adjusted to 10.8 μM of BLA and stored at 4°C. The concentration of BLA was determined using UV-vis absorption spectroscopy with the extinction coefficient, ε = 137,000 M · cm−1 (280 nm).
Electrospray ionization mass spectrometry (ESI-MS)
Acetylated BLA proteins were analyzed with HPLC directly coupled to an electrospray ionization triple quadrupole mass spectrometer (API III Perkin-Elmer Sciex). During LC-ESI-MS the scanning range of the ESI-MS was set at 300–2300 m/z. The ESI-MS was calibrated by flow injection of a mixture of polypropylene glycol (PPG) 425, 1000, and 2000 Da (3.3 × 10−5, 1 × 10−4, and 2 × 10−4 M, respectively) in water/methanol (1:1, v/v) containing 2 mM ammonium formate and 0.1% (v/v) acetonitrile. Normal spectra were obtained by scanning at instrument conditions sufficient to resolve the isotopes of the PPG/NH4 + singly charged ion at m/z 906 with 40% valley and with a 0.3-Da step size during data acquisition. Calculation of molecular masses from the series of multiply charged ions found in the spectra, and deconvolution of the ion series into a molecular mass spectrum, was achieved with the Bio MultiView computer programs (version 1.3.1 PE Sciex).
Charge ladders and capillary electrophoresis
Charge ladders of BLA were made by reacting lys-ε-NH3 + groups with increasing amounts of acetic anhydride as described above and also as previously described (Gitlin et al. 2006b). Solutions of BLA charge ladders were dialyzed against the running buffer before analysis. The neutral marker dimethyl formamide (DMF) was added to each sample (1 μM) in order to calibrate the electrophoretic flow during each CE experiment.
Capillary electrophoresis (CE) was performed on a Beckman Coulter P/ACE Electrophoresis apparatus that was fitted with a fused-silica capillary (length = 110 cm; inner diameter = 50 μm; Polymicro Technologies). Protein species were detected by absorbance at 214 nm. CE experiments were conducted for 1 h with an applied voltage of 30 kV. A Tris-Glycine buffer was used as the running buffer (10 mM Tris, 192 mM glycine, pH 8.5). This buffer is ideal for experiments involving capillary electrophoresis because it has a low absorbance at 214 nm, and has a low conductivity (which allows it to be subjected to high voltages without overheating the apparatus) (Hjerten et al. 1995; Righetti et al. 2000).
Circular dichroism spectroscopy
Circular dichroism (CD) spectra were obtained on a JASCO J-7 spectropolarimeter. Buffer conditions were 0 and 3 mM SDS, 25 mM Tris, 192 mM glycine, pH 8.5. Mean residue ellipticities, θ, (deg cm2 dmol−1) were calculated by using the relation: [θ] = θobs × (MRW / 10 × l × c), where θobs is the measured signal (ellipticity) in millidegrees, l is the optical path length of the cell in cm, c is concentration of the protein in mg/mL, and MRW is the mean residue molecular weight (molecular weight of the peptide divided by the number of amino acid residues).
Differential scanning calorimetry
Differential scanning calorimetry (DSC) was carried out on a VP-DSC instrument (MicroCal) with a scan rate of 1°C/min. Acetylated and unmodified BLA samples (∼10 μM; pH 7.4, 10 mM phosphate) were degassed prior to analysis. Raw DSC data were smoothed and deconvoluted using Origin 5.0 (MicroCal). Phosphate buffer was used for analysis of BLA with DSC (instead of Tris buffer, which was used for all other experiments). Tris is often avoided in experiments involving DSC because the buffering capacity of Tris is more dependent upon the temperature than other buffers (e.g., phosphate or acetate).
Thermal inactivation of α-amylase in the presence of detergent
Stock solutions of BLA-Ac(0) and BLA-Ac(∼17) (20 μL, 10.8 μM BLA, 25 mM Tris, 192 mM glycine, pH 8.5HHHHhhh) were diluted 1:25 into 480 μL of surfactant solution (1, 10 or 100 mM surfactant; 25 mM Tris, 192 mM glycine, pH 8.5). The three surfactants used in this study were the anionic surfactant sodium dodecyl sulfate (SDS), the nonionic surfactant Triton X-100, and cationic surfactant dodecyltrimethyl ammonium bromide (DTAB). Solutions of BLA were then heated at 90°C in closed microcentrifuge tubes using a Peltier-effect heating device. At various times, up to 520 min, microliter-scale aliquots were removed, cooled on ice for 2 min, diluted 1:181 into the activity assay buffer (50 mM maleic acid, 1 mM CaCl2, 50 mM NaCl, NaN3 0.005% w/v, pH 6.67, room temperature; Megazyme, Inc.), and the activity of BLA was measured as described below. To determine whether any loss of activity was reversible, the solutions of BLA were cooled to 25°C for 1 h at the end of the 520-min, 90°C incubation, and the activity was measured. We chose to use Tris-glycine buffer because of its strong buffer capacity at alkaline pH (many enzyme-surfactant systems operate at alkaline pH) and also because Tris is an ideal buffer for electrophoretic experiments (Hjerten et al. 1995; Righetti et al. 2000).
Measurement of α-amylase activity
All reagents used for measuring the activity of BLA were purchased as a kit from Megazyme, Inc. (product number: KCERA). The activity of BLA was determined by measuring the increase in absorbance at 400 nm, resulting from the hydrolysis of blocked 4,6-o-benzilidine-p-nitrophenyl maltoheptaoside (by BLA) and the subsequent hydrolysis of the resulting oligosaccharide (by α-glucosidase) to yield glucose and p-nitrophenol (λmax of p-nitrophenol = 400 nm; ε = 18,290 M · cm−1). Enzymatic assays were initiated by combining 90 μL of solution “A” (thermostable α-glucosidase, blocked p-nitrophenyl maltoheptaoside, pH 6.73) with 90 μL of solution “B” (100 mM maleic acid, 2 mM CaCl2, 100 mM NaCl, NaN3 0.01% w/v, pH 6.58) followed by the addition of 1 μL of BLA-surfactant or BLA-perborate solution (final assay conditions: 50 mM maleic acid, 1 mM CaCl2, 50 mM NaCl, NaN3 0.005% w/v, pH 6.67, room temperature). This mixture was immediately loaded into a cuvette for measurement by UV-vis spectroscopy. Spectra were collected at 10-sec intervals for 1000 sec.
Electronic supplemental material
Supplemental data are available online (at http://www.proteinscience.org/supplemental) including: the results of proteolytic digestion of BLA-Ac(∼17) with trypsin and elastase and the sequencing of BLA-Ac(∼17) with ESI-MS/MS; raw kinetic plots of the inactivation of BLA-Ac(0) and BLA-Ac(17) by heat and surfactant; kinetic plots of the inactivation of BLA-Ac(0) and BLA-Ac(17) by sodium perborate; aggregation assays for BLA-Ac(0) and BLA-Ac(17). A Materials and Methods section that describes these experiments is also included online.
Acknowledgments
The authors acknowledge a National Institutes of Health Grant for Financial Support (GM051559). The authors gratefully acknowledge Debby Pheasant of the Biophysical Instrumentation Facility (Massachusetts Institute of Technology) for technical assistance operating the DSC instrument. B.F.S. thanks an NIH Ruth Kirchstein National Research Service Award (GM081055) for post-doctoral support; G.F.S. acknowledges support from the Bourse Lavoisier Générale du Ministères des Affaires Etrangères Français.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: George M. Whitesides, Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA; e-mail: gwhitesides@gmwgroup.harvard.edu; fax: (617) 495-9857.
Abbreviations: BLA, α-amylase from bacillus licheniformis; CD, circular dichroism; CE, capillary electrophoresis; DSC, differential scanning calorimetry; DTAB, dodecyltrimethylammonium bromide; ESI-MS, electrospray ionization mass spectrometry; HEPBS, N-(2-Hydroxyethyl)piperazine-N′-(4-butanesulfonic acid); SDS, sodium dodecyl sulfate; Tris, tris-hydroxymethylaminomethane.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.035154.108.
References
- Basak, S.K., Ladisch, M.R. Correlation of electrophoretic mobilities of proteins and peptides with their physicochemical properties. Anal. Biochem. 1995;226:51–58. doi: 10.1006/abio.1995.1190. [DOI] [PubMed] [Google Scholar]
- Carbeck, J.D., Colton, I.J., Anderson, J.R., Deutch, J.M., Whitesides, G.M. Correlations between the charge of proteins and the number of ionizable groups they incorporate: Studies using protein charge ladders, capillary electrophoresis, and Debye-Huckel theory. J. Am. Chem. Soc. 1999;121:10671–10679. [Google Scholar]
- Chiti, F., Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Chiti, F., Calamai, M., Taddei, N., Stefani, M., Ramponi, G., Dobson, C.M. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc. Natl. Acad. Sci. 2002;99(Suppl. 4):16419–16426. doi: 10.1073/pnas.212527999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti, F., Stefani, M., Taddei, N., Ramponi, G., Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- Cleland, J.L., Randolph, T.W. Mechanism of polyethylene glycol interaction with the molten globule folding intermediate of bovine carbonic anhydrase B. J. Biol. Chem. 1992;267:3147–3153. [PubMed] [Google Scholar]
- Cleland, J.L., Hedgepeth, C., Wang, D.I. Polyethylene glycol enhanced refolding of bovine carbonic anhydrase B. Reaction stoichiometry and refolding model. J. Biol. Chem. 1992;267:13327–13334. [PubMed] [Google Scholar]
- Colton, I.J., Anderson, J.R., Gao, J.M., Chapman, R.G., Isaacs, L., Whitesides, G.M. Formation of protein charge ladders by acylation of amino groups on proteins. J. Am. Chem. Soc. 1997;119:12701–12709. [Google Scholar]
- Daugherty, D.L., Rozema, D., Hanson, P.E., Gellman, S.H. Artificial chaperone-assisted refolding of citrate synthase. J. Biol. Chem. 1998;273:33961–33971. doi: 10.1074/jbc.273.51.33961. [DOI] [PubMed] [Google Scholar]
- Declerck, N., Machius, M., Joyet, P., Wiegand, G., Huber, R., Gaillardin, C. Hyperthermostabilization of Bacillus licheniformis α-amylase and modulation of its stability over a 50°C temperature range. Protein Eng. 2003;16:287–293. doi: 10.1093/proeng/gzg032. [DOI] [PubMed] [Google Scholar]
- Edgcomb, S.P., Murphy, K.P. Variability in the pKa of histidine side chains correlates with burial within proteins. Proteins. 2002;49:1–6. doi: 10.1002/prot.10177. [DOI] [PubMed] [Google Scholar]
- Galbe, M., Zacchi, G. A review of the production of ethanol from softwood. Appl. Microbiol. Biotechnol. 2002;59:618–628. doi: 10.1007/s00253-002-1058-9. [DOI] [PubMed] [Google Scholar]
- Gao, J.M., Whitesides, G.M. Using protein charge ladders to estimate the effective charges and molecular weights of proteins in solution. Anal. Chem. 1997;69:575–580. doi: 10.1021/ac9608073. [DOI] [PubMed] [Google Scholar]
- García-Moreno, B., Dwyer, J.J., Gittis, A.G., Lattman, E.E., Spencer, D.S., Stites, W.E. Experimental measurement of the effective dielectric in the hydrophobic core of a protein. Biophys. Chem. 1997;64:211–224. doi: 10.1016/s0301-4622(96)02238-7. [DOI] [PubMed] [Google Scholar]
- Gitlin, I., Carbeck, J.D., Whitesides, G.M. Why are proteins charged? Networks of charge–charge interactions in proteins measured by charge ladders and capillary electrophoresis. Angew. Chem. Int. Ed. Engl. 2006a;45:3022–3060. doi: 10.1002/anie.200502530. [DOI] [PubMed] [Google Scholar]
- Gitlin, I., Gudiksen, K.L., Whitesides, G.M. Effects of surface charge on denaturation of bovine carbonic anhydrase. ChemBioChem. 2006b;7:1241–1250. doi: 10.1002/cbic.200600191. [DOI] [PubMed] [Google Scholar]
- Gitlin, I., Gudiksen, K.L., Whitesides, G.M. Peracetylated bovine carbonic anhydrase (BCA-Ac18) is kinetically more stable than native BCA to sodium dodecyl sulfate. J. Phys. Chem. B. 2006c;110:2372–2377. doi: 10.1021/jp055699f. [DOI] [PubMed] [Google Scholar]
- Grossman, P.D. Capillary electrophoresis: Theory and practice. Academic Press; San Diego, CA: 1992. pp. 111–132. [Google Scholar]
- Gudiksen, K.L., Gitlin, I., Moustakas, D.T., Whitesides, G.M. Increasing the net charge and decreasing the hydrophobicity of bovine carbonic anhydrase decreases the rate of denaturation with sodium dodecyl sulfate. Biophys. J. 2006;91:298–310. doi: 10.1529/biophysj.106.081547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch, C., Coats, E. α-Chymotrypsin: A case study of substituent constants and regression analysis in enzymic structure–activity relationships. J. Pharm. Sci. 1970;59:731–743. doi: 10.1002/jps.2600590602. [DOI] [PubMed] [Google Scholar]
- Hansch, C., Steward, A.R. The use of substituent constants in the analysis of the structure–activity relationship in penicillin derivatives. J. Med. Chem. 1964;7:691–694. doi: 10.1021/jm00336a001. [DOI] [PubMed] [Google Scholar]
- Hjerten, S., Valtcheva, L., Elenbring, K., Liao, J.L. Fast, high-resolution (capillary) electrophoresis in buffers designed for high-field strengths. Electrophoresis. 1995;16:584–594. doi: 10.1002/elps.1150160195. [DOI] [PubMed] [Google Scholar]
- Jain, R., Hamilton, A.D. Designing protein denaturants: Synthetic agents induce cytochrome c unfolding at low concentrations and stoichiometries. Angew. Chem. Int. Ed. Engl. 2002;41:641–643. [Google Scholar]
- Johannes, T.W., Zhao, H. Directed evolution of enzymes and biosynthetic pathways. Curr. Opin. Microbiol. 2006;9:261–267. doi: 10.1016/j.mib.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Juncosa, M., Pons, J., Dot, T., Querol, E., Planas, A. Identification of active site carboxylic residues in Bacillus licheniformis 1,3-1,4-β-D-glucan 4-glucanohydrolase by site-directed mutagenesis. J. Biol. Chem. 1994;269:14530–14535. [PubMed] [Google Scholar]
- Kaar, W.E., Holtzapple, M.T. Benefits from tween during enzymic hydrolysis of corn stover. Biotechnol. Bioeng. 1998;59:419–427. doi: 10.1002/(sici)1097-0290(19980820)59:4<419::aid-bit4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Khodagholi, F., Yazdanparast, R. Artificial chaperone-assisted refolding of GuHCl-denatured α-amylase at low temperature: Refolding versus aggregation. Protein J. 2005;24:303–313. doi: 10.1007/s10930-005-6751-y. [DOI] [PubMed] [Google Scholar]
- Khodarahmi, R., Yazdanparast, R. Refolding of chemically denatured α-amylase in dilution additive mode. Biochim. Biophys. Acta. 2004;1674:175–181. doi: 10.1016/j.bbagen.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Kim, J.Y., Kim, K.W., Kwon, H.J., Lee, D.W., Yoo, J.S. Probing lysine acetylation with a modification-specific marker ion using high-performance liquid chromatography/electrospray-mass spectrometry with collision-induced dissociation. Anal. Chem. 2002;74:5443–5449. doi: 10.1021/ac0256080. [DOI] [PubMed] [Google Scholar]
- Kim, S.B., Chun, J.W. Enhancement of enzymatic digestibility of recycled newspaper by addition of surfactant in ammonia-hydrogen peroxide pretreatment. Appl. Biochem. Biotechnol. 2004;113-116:1023–1031. doi: 10.1385/abab:115:1-3:1023. [DOI] [PubMed] [Google Scholar]
- Kirk, O., Borchert, T.V., Fuglsang, C.C. Industrial enzyme applications. Curr. Opin. Biotechnol. 2002;13:345–351. doi: 10.1016/s0958-1669(02)00328-2. [DOI] [PubMed] [Google Scholar]
- Korkegian, A., Black, M.E., Baker, D., Stoddard, B.L. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Mesa, C. Polymer–surfactant and protein–surfactant interactions. J. Colloid Interface Sci. 2005;286:148–157. doi: 10.1016/j.jcis.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Lawrence, M.S., Phillips, K.J., Liu, D.R. Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, B., Vasmatzis, G. Stabilization of protein structures. Curr. Opin. Biotechnol. 1997;8:423–428. doi: 10.1016/s0958-1669(97)80063-8. [DOI] [PubMed] [Google Scholar]
- Lee, S.Y., Kim, S., Sweet, R.M., Suh, S.W. Crystallization and a preliminary X-ray crystallographic study of α-amylase from Bacillus licheniformis . Arch. Biochem. Biophys. 1991;291:255–257. doi: 10.1016/0003-9861(91)90131-2. [DOI] [PubMed] [Google Scholar]
- Liu, Y., Guo, R. Interaction between casein and the oppositely charged surfactant. Biomacromolecules. 2007;8:2902–2908. doi: 10.1021/bm7006136. [DOI] [PubMed] [Google Scholar]
- Lu, D., Liu, Z., Liu, Z., Zhang, M., Ouyang, P. Molecular simulation of surfactant-assisted protein refolding. J. Chem. Phys. 2005;122:134902. doi: 10.1063/1.1866052. [DOI] [PubMed] [Google Scholar]
- Machius, M., Declerck, N., Huber, R., Wiegand, G. Activation of Bacillus licheniformis α-amylase through a disorder → order transition of the substrate-binding site mediated by a calcium–sodium–calcium metal triad. Structure. 1998;6:281–292. doi: 10.1016/s0969-2126(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Machius, M., Declerck, N., Huber, R., Wiegand, G. Kinetic stabilization of Bacillus licheniformis α-amylase through introduction of hydrophobic residues at the surface. J. Biol. Chem. 2003;278:11546–11553. doi: 10.1074/jbc.M212618200. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. Structural and genetic analysis of protein stability. Annu. Rev. Biochem. 1993;62:139–160. doi: 10.1146/annurev.bi.62.070193.001035. [DOI] [PubMed] [Google Scholar]
- McCleary, B.V., McNally, M., Monaghan, D., Mugford, D.C. Measurement of α-amylase activity in white wheat flour, milled malt, and microbial enzyme preparations, using the Ceralpha assay: Collaborative study. J. AOAC Int. 2002;85:1096–1102. [PubMed] [Google Scholar]
- Menon, M.K., Zydney, A.L. Determination of effective protein charge by capillary electrophoresis: Effects of charge regulation in the analysis of charge ladders. Anal. Chem. 2000;72:5714–5717. doi: 10.1021/ac000752b. [DOI] [PubMed] [Google Scholar]
- O'Shea, J.J., Kanno, Y., Chen, X., Levy, D.E. Cell signaling. Stat acetylation—a key facet of cytokine signaling? Science. 2005;307:217–218. doi: 10.1126/science.1108164. [DOI] [PubMed] [Google Scholar]
- Otzen, D.E., Oliveberg, M. A simple way to measure protein refolding rates in water. J. Mol. Biol. 2001;313:479–483. doi: 10.1006/jmbi.2001.5039. [DOI] [PubMed] [Google Scholar]
- Reetz, M.T., Carballeira, J.D., Vogel, A. Iterative saturation mutagenesis on the basis of B factors as a strategy for increasing protein thermostability. Angew. Chem. Int. Ed. Engl. 2006;45:7745–7751. doi: 10.1002/anie.200602795. [DOI] [PubMed] [Google Scholar]
- Rickard, E.C., Strohl, M.M., Nielsen, R.G. Correlation of electrophoretic mobilities from capillary electrophoresis with physicochemical properties of proteins and peptides. Anal. Biochem. 1991;197:197–207. doi: 10.1016/0003-2697(91)90379-8. [DOI] [PubMed] [Google Scholar]
- Righetti, P.G., Gelfi, C., Bossi, A., Olivieri, E., Castelletti, L., Verzola, B., Stoyanov, A.V. Capillary electrophoresis of peptides and proteins in isoelectric buffers: An update. Electrophoresis. 2000;21:4046–4053. doi: 10.1002/1522-2683(200012)21:18<4046::AID-ELPS4046>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Rozema, D., Gellman, S.H. Artificial chaperone-assisted refolding of carbonic anhydrase B. J. Biol. Chem. 1996;271:3478–3487. doi: 10.1074/jbc.271.7.3478. [DOI] [PubMed] [Google Scholar]
- Saeki, K., Ozaki, K., Kobayashi, T., Ito, S. Detergent alkaline proteases: Enzymatic properties, genes, and crystal structures. J. Biosci. Bioeng. 2007;103:501–508. doi: 10.1263/jbb.103.501. [DOI] [PubMed] [Google Scholar]
- Salameh, M., Wiegel, J. Lipases from extremophiles and potential for industrial applications. Adv. Appl. Microbiol. 2007;61:253–283. doi: 10.1016/S0065-2164(06)61007-1. [DOI] [PubMed] [Google Scholar]
- Shaw, B.F., Valentine, J.S. How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem. Sci. 2007;32:78–85. doi: 10.1016/j.tibs.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Soetaert, W., Vandamme, E. The impact of industrial biotechnology. Biotechnol. J. 2006;1:756–769. doi: 10.1002/biot.200600066. [DOI] [PubMed] [Google Scholar]
- Swietnicki, W. Folding aggregated proteins into functionally active forms. Curr. Opin. Biotechnol. 2006;17:367–372. doi: 10.1016/j.copbio.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Thurlkill, R.L., Grimsley, G.R., Scholtz, J.M., Pace, C.N. pK values of the ionizable groups of proteins. Protein Sci. 2006;15:1214–1218. doi: 10.1110/ps.051840806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendruscolo, M., Dobson, C.M. Chemical biology: More charges against aggregation. Nature. 2007;449:555. doi: 10.1038/449555a. [DOI] [PubMed] [Google Scholar]
- Yazdanparast, R., Khodagholi, F., Khodarahmi, R. Artificial chaperone-assisted refolding of chemically denatured α-amylase. Int. J. Biol. Macromol. 2005a;35:257–263. doi: 10.1016/j.ijbiomac.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Yazdanparast, R., Khodarahmi, R., Soori, E. Comparative studies of the artificial chaperone-assisted refolding of thermally denatured bovine carbonic anhydrase using different capturing ionic detergents and β-cyclodextrin. Arch. Biochem. Biophys. 2005b;437:178–185. doi: 10.1016/j.abb.2005.03.003. [DOI] [PubMed] [Google Scholar]