

Abstract
The mitochondrial RNA-binding proteins (MRP) 1 and 2 play a regulatory role in RNA editing and putative role(s) in RNA processing in Trypanosoma brucei. Here, we report the purification of a high molecular weight protein complex consisting solely of the MRP1 and MRP2 proteins from the mitochondrion of T. brucei. The MRP1/MRP2 complex natively purified from T. brucei and the one reconstituted in Escherichia coli in vivo bind guide (g) RNAs and pre-mRNAs with dissociation constants in the nanomolar range, and efficiently promote annealing of pre-mRNAs with their cognate gRNAs. In addition, the MRP1/MRP2 complex stimulates annealing between two non-cognate RNA molecules suggesting that along with the cognate_duplexes, spuriously mismatched RNA hybrids may be formed at some rate in vivo._A mechanism of catalyzed annealing of gRNA/pre-mRNA by the MRP1/MRP2 complex is proposed.
Keywords: Trypanosoma brucei, Mitochondrion, Kinetoplast, RNA editing, RNA binding
1. Introduction
Trypanosomatids are causative agents of several devastating tropical diseases such as African sleeping sickness, Chagas’ disease and leishmaniasis. Available drugs are obsolete, difficult to administer and have many undesirable side-effects. Therefore, there is an ongoing effort to design new drugs against these parasites. From the pharmacological perspective, unique metabolic processes and protein complexes with a singular structure, composition and essential function are of particular interest. One such remarkable process is kinetoplast (k) RNA editing, an essential mechanism by which certain mitochondrial pre-mRNAs are post-transcriptionally modified via exclusive insertion and deletion of uridylate residues to otherwise encrypted transcripts. RNA editing is a requirement for translation of these mRNAs, as it creates start and stop codons, eliminates frameshifts and in some cases creates entire open reading frames (ORFs) (reviewed in references Lukeš et al., 2005; Simpson et al., 2004; Stuart et al., 2005). The RNA editing process appears to take place by a series of cut and paste steps and is carried out by a multi-protein complex termed the editosome (Panigrahi et al., 2003a, 2003b) or L-complex (Aphasizhev et al., 2003). Genetic information for kRNA editing is contained in short, mitochondrially-encoded transcripts, called guide (g) RNAs. Each gRNA transfers the genetic information for specific uridine insertions and/or deletions at multiple sites in the pre-mRNA through base pairing interactions (Blum et al., 1990; Sturm and Simpson, 1990; Hermann et al., 1997).
Evidence for the existence of gRNA-binding proteins in kinetoplastid mitochondria has been presented by several laboratories (Köller et al., 1994; Bringaud et al., 1995; Leegwater et al., 1995; Hayman and Read, 1999; Madison-Antenucci and Hajduk 2001). One of these proteins, identified by virtue of its specific cross-linking to the gRNA molecules, is the Trypanosoma brucei mitochondrial RNA binding protein (MRP) 1, originally called gBP21 (Köller et al., 1997). It has been shown that this protein has a high affinity for gRNAs (Köller et al., 1997) and promotes the annealing of gRNA and its cognate pre-mRNA in vitro (Lambert et al., 1999; Müller et al., 2001). Based on these findings, MRP1 was proposed to play an active role in the first step of kRNA editing, gRNA/pre-mRNA hybridization (Müller et al., 2001). The involvement of MRP1 in RNA editing was further supported by its association with the editosomal complex, albeit RNA mediated and perhaps transient (Allen et al., 1998; Aphasizhev et al., 2003).
Proteins orthologous to MRP1 were identified in Leishmania tarentolae (Lt28) and Crithidia fasciculata (gBP29) (Blom et al., 2001; Aphasizhev et al., 2003). Importantly, a second gRNA-binding protein, gBP27, was found in C. fasciculata that co-purified and co-immunoprecipitated with gBP29 (Blom et al., 2001). A similarly tight association was subsequently observed for the homologous proteins in L. tarentolae, in which Ltp28 and Ltp26, the gBP27 ortholog, form a 100-kDa hetero-tetrameric complex (Aphasizhev et al., 2003). The complex has been shown to bind RNA and promote annealing of gRNA to its cognate pre-mRNA (Aphasizhev et al., 2003), overlapping in function with MRP1 from T. brucei.
This finding indicated a possibility that MRP1 in T. brucei might be involved in a multi-protein complex in analogy to L. tarentolae (Aphasizhev et al., 2003) and C. fasciculata (Blom et al., 2001), which was later corroborated by identification of the orthologue of Ltp26 and gBP27, named MRP2 (Vondrušková et al., 2005). Silencing of MRP1 and MRP2 by RNA interference revealed a mutual dependence for stability, which further supported participation of both proteins in the formation of a single multi-protein complex (Vondrušková et al., 2005). In cells with down-regulated MRP1/MRP2 complex, the assembly and functionality of respiratory complexes were affected due to the disruption of RNA editing and/or stability of the transcripts encoding the mitochondrial-encoded subunits (Zíková et al., 2006). However, the exact function of the MRP1/MRP2 complex in kRNA editing and/or RNA stability has remained largely obscure. Recently, the crystal structure of the T. brucei MRP1/MRP2 complex reconstituted in E. coli with and without gRNA revealed a unique mechanism of RNA binding to the complex in a sequence non-specific manner (Schumacher et al., 2006). In the present study we have used natively isolated and recombinant T. brucei MRP1/MRP2 complexes to characterize their interactions with several mitochondrial gRNA and pre-mRNA molecules and to examine its RNA annealing activity towards various RNA species. These studies indicate that the T. brucei MRP1/MRP2 complex binds gRNAs and pre-mRNAs with almost the same affinity and can promote RNA annealing between gRNA/pre-mRNA pairs.
2. Materials and methods
2.1. Trypanosome culture and transfection
The whole ORF including the mitochondrial signal sequence of MRP1 and MRP2 was amplified by PCR and subcloned into the pLEW79-tandem affinity purification (TAP) vector (Panigrahi et al., 2003b). The T. brucei cell line 29-13 co-expressing the Tet repressor and T7 RNA polymerase was transfected with NotI-linearized pLew79-MRP1-TAP and pLew79-MRP2-TAP. Phleomycin-resistant clones were selected and checked for tightly tetracycline-regulated expression. Expression of MRP1-TAP or MRP2-TAP in the recombinant cell lines was induced for 48 h with 1µg/ml of tetracycline.
2.2. Isolation of native MRP1/MRP2 complex from T. brucei
The TAP protocol was adapted from the published method (Puig et al., 2001). The cell pellet from 2 × 1010 cells or the mitochondria-enriched fraction isolated by hypotonic lysis from 2 × 1011 cells was lysed on ice for 20 min in 20 ml of IPP buffer (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; 0.1% NP40) with 1% Triton X-100. Two tablets of complete proteinase inhibitors (Roche) were added directly to the extract. After centrifugation at 9,800 g for 30 min, the supernatant was incubated with 200 µl of IgG Sepharose FF (AP Biotech) for 2 h. After the wash, beads were incubated with tabacco etch virus (TEV) protease in the TEVCB buffer (10 mM Tris-HCl pH 8.0; 150 mM NaCl; 0.1% NP40; 0.5 mM EDTA; 1 mM DTT) for 2 h at 16°C. The TEV eluate was purified over calmodulin resin (Stratagene) in the CBB buffer (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; 0.1% NP40; 10 mM β-mercaptoethanol (ME); 1 mM Mg acetate; 1 mM imidazole; 2 mM CaCl2) or loaded onto 11 ml 10%–30% glycerol gradient and fractionated for 20 h at 4 °C and 168,000 g in a SW-40 rotor. Fractions of 750 µl were collected from the top of the gradient and selected fractions were pooled and purified over the calmodulin resin in the CBB buffer. MRP-tagged complexes were eluted with 4 × 250 µl of the CEB buffer (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; 0.1% NP40; 10 mM ME; 1 mM Mg acetate; 1 mM imidazole; 2 mM EGTA).
2.3. SDS-PAGE, Western blotting and blue native electrophoresis
Cell lysates and protein samples were fractionated by SDS-PAGE, blotted, probed with affinity purified polyclonal anti-MRP1 or anti-MRP2 antibodies and developed using the ECL system (Amersham) (Vondrušková et al., 2005). The monoclonal mouse antibodies against the editosomal proteins KREPA1, KREPA2, KREPA3, KREL2 (1:25) and the polyclonal rabbit antiserum against KRET1 (1:1000) (a kind gift from L. Simpson, UCLA) were used at dilutions shown in parenthesis. For blue native electrophoresis, 100 ng of the MRP1/MRP2 complex isolated from T. brucei or overexpressed in E. coli (see below) was incubated in buffer A (1% dodecyl maltoside; 750 mM aminocaproic acid; 0.5 % CBB-G; 50 mM Bis-Tris; 0.5 mM EDTA, pH 7.0) for 15 min on ice and loaded onto 4–16 % blue native gel (BN) (500 mM aminocaproic acid; 50 mM Bis-Tris, pH 7.0). The gel was run for 3 h at 250 V and 5 mA and silver stained (Invitrogen).
2.4. Electron microscopy and image analysis
The specimens were placed on glow-discharged carbon-coated copper grids and negatively stained with 2% uranyl acetate. Electron microscopy was performed with a Philips TEM 420 electron microscope using 80 kV at 60,000 × magnification. Micrographs were digitized with a pixel size corresponding to 0.51 nm at the specimen level. Image analysis was carried out using the SPIDER software (Frank et al., 1996) From 10 micrographs of the untreated MRP1/MRP2 complex preparation (Fig. 3A), 830 top-view projections were selected for the analysis, and 720 top-view projections from 15 micrographs of the RNase-treated complex (Fig. 3B), were selected. Next, these projections were rotationally and translationally aligned and processed by multivariate statistical analysis followed by classification (Harauz et al., 1988). For the final analysis, the best of the class members were summed using a cross-correlation coefficient of the alignment procedure as a quality parameter. The resolution of the images was calculated by using the Fourier ring correlation method (van Heel, 1987).
Fig. 3.
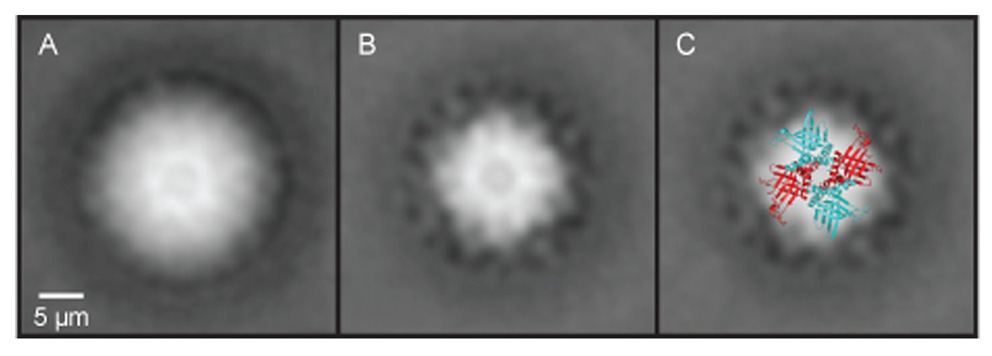
Transmission electron microscopy image reconstruction of native mitochondrial RNA-binding protein (MRP) 1/MRP2 complexes isolated from Trypnaosoma brucei. (A) View of the MRP1/MRP2 complex prior to the RNase treatment. Clearly visible in the structure is the central hole in the cavity of the MRP1/MRP2 heterotetramer complex. (B) View of the same complexes after the RNase treatment. Comparison of the RNase-treated and non-treated complexes indicates that the RNA does not bind in the central cavity but on the surface and/or the outside of the complex. (C) Superimposition of the crystal structure on an averaged electron microscopy picture of the MRP1/MRP2 complex.
2.5. Mass spectrometry analysis
One hundred µl of elution 2 from the calmodulin binding column was precipitated with 600 µl of acetone, and the samples were further denatured with 8 M urea/1 mM DTT, and treated overnight with 100 ng of trypsin. The resulting peptides were purified using RPC-18 magnetic beads (Dynabeads). In parallel, 30 µl of the elution 2 was separated by SDS-PAGE and the protein bands were visualized by Sypro Ruby Staining (Molecular Probes). All visible protein bands were excised from the gel and digested with trypsin in-gel as described elsewhere (Panigrahi et al., 2001). Peptides were identified using a Thermo Electron LCQ DECA XP Spectrometer or LTQ Linear Ion Trap Mass Spectrometer. The collision induced dissociation (CID) spectra were compared with the T. brucei protein database downloaded from GeneDB using TurboSequest software, and protein matches were determined using PeptideProphet and ProteinProphet (Keller et al., 2002; Nesvizhskii et al., 2003).
2.6. RNA sequences
A6U5 -5′ GGA AAG GUU AGG GGG AGG AGA GAA GAA AGG GAA AGU UGU GAU UUU UGG AGU UAU AGA AUA CUU ACC UGG CAU C 3′ gA6[14]16ΔG - 5′ GGA UAU ACU AUA ACU CCG AUA ACG AAU CAG AUU UUG ACA GUG AUA UGA UAA UUA UUU UUU UUU UUU UUU UU 3′ 5′CybUT - 5′ GGU UAU AAA UUU UAU AUA AAA GCG GAG AAA AAA GAA AGG GUC UUU UAA UGU CAG GUU GUU UAU AUA GAA U 3′ gCyb-558 - 5′ GGG AUU AAA AGA CAA UGU GAA UUU CUA GGU GAU AAA GGG AAU AAU UUU UUU UUU UUU UU 3′
2.7. Annealing and binding assays
All RNA molecules (A6U5, gA6[14], 5′CybUT, gCyb-558) were synthesized by in vitro transcription from a linear DNA template using T7 RNA polymerase following standard procedures. The synthesized RNAs were radioactively labeled using [γ-32P] ATP and T4 polynucleotide kinase and purified on an 8 M urea 15 % (w/v) polyacrylamide gel. The binding reaction contained 12.5 nM of one RNA reactant with the protein concentration varying from 0 to 1 μM. The annealing reaction contained 12.5 nM of both RNA reactants (pre-mRNA and gRNA), with the protein concentration ranging from 0 to 1 μM. All reactions were performed in a 20 μl-reaction containing 6 mM HEPES-KOH, pH 7.5; 50 mM KCl; 2.1 mM MgCl2; 0.1 mM EDTA and 0.5 mM DTT incubated for 30 min at 27 °C. After the incubation, the binding reaction was mixed with 6 × loading buffer (0.25% (w/v) bromophenol blue, 0.25% (w/v) xylene cyanol FF, 40% (w/v) sucrose in H2O) and immediately loaded onto a pre-cast 10 % TBE gels (Bio-Rad). The annealing reactions were stopped by the addition of 20 µg proteinase K, and further incubated for 15 min at 27 °C (Müller et al., 2001). Gels were run at 100 V for 2.5 h at 4 °C, fixed in 10% (v/v) isopropanol and 7% (v/v) acetic acid, dried and exposed in Phosphoimager cassettes. Band intensities were quantified using the ImageQuant TL v2003.02 program, and the Prizm3 (GraphPad) software package was used for all nonlinear regression curve fitting and statistical analyses.
2.8. Purification of the recombinant MRP1 and MRP2 proteins and the MRP1/MRP2 complex
The MRP1 and MRP2 proteins were overexpressed and purified as described elsewhere (Vondrušková et al., 2005). To obtain sufficient amounts of the MRP1/MRP2 complex, the MRP1 gene fragment encoding residues 20 to 176 (lacking the N-terminal mito-targeting sequence and the C-terminal region found to cause insolubility) and the MRP2 gene fragment encoding residues 30 to 224 (lacking the N-terminal mito-targeting sequence) were co-expressed in the pET-Duet-1 co-expression vector in E. coli. Although a significant fraction of the complex was found in inclusion bodies, 2–5 mg of soluble, pure MRP1/MRP2 complex was obtained per liter of bacterial culture.
3. Results
3.1. MRP1 and MRP2 are conserved in trypanosomatids
To date, MRP1 and MRP2 homologues have only been identified in T. brucei, L. tarentolae and C. fasciculata (Blom et al., 2001; Aphasizhev et al., 2003; Vondrušková et al., 2005). Using the T. brucei MRP1 and MRP2 sequences, we searched the databases and found additional homologues of both proteins in the genomes of Leishmania infantum, Leishmania major, Trypanosoma cruzi and Trypanosova vivax (Supplementary Fig. S1). These alignments revealed a somewhat higher level of similarity than estimated previously from the narrower dataset. While the N-terminal 28 (Trypanosoma spp.) to 52 amino acids (C. fasciculata) of MRP1 lack any conservation, most of the central part of the gene (~140 residues) is highly conserved. The C-terminus is again highly variable (Supplementary Fig. S1A). The conservation follows a rather different pattern in MRP2, with three highly conserved blocks 25 to 42 amino acids long, one occupying the most C-terminal region, being separated by highly variable regions (Supplementary Fig. S1B). With the exception of marginal similarity with bacterial ribosomal protein L1, the MRP proteins have no significant sequence similarity with other known proteins.
3.2. Isolation of the MRP1/MRP2 complex from T. brucei procyclics
To identify interaction partners of MRP1 and MRP2, both proteins containing the mitochondrial targeting sequence and the C-terminal TAP-tag have been expressed in T. brucei procyclic cells under the control of a tetracycline-inducible promoter. The TAP-tagged MRP2 complex was purified from lysed 2 × 1010 cells, and clarified whole cell lysate was incubated with IgG-Sepharose beads. The MRP2 protein attached to a calmodulin binding peptide (MRP2-CBP) was released by incubation with TEV protease and further purified via a calmodulin binding column. The purification was checked by Western blot analysis using a specific anti-MRP2 antibody (Fig. 1A). SyproRuby staining of the elution 2 revealed three major gel bands (Fig. 1Ba), all of which were analyzed by LC-MS/MS, as well as a protein mixture from the same elution. Data obtained from the MS analysis revealed that the ~30kDa band is the tagged MRP2 protein and the ~20kDa band contains both the MRP1 and MRP2 proteins, which co-migrate after the mitochondrial signal sequence is cleaved. This result suggests that the TAP-tag attached to the MRP2 protein does not compromise its capacity to be efficiently incorporated into the MRP1/MRP2 complex. The 50 kDa band was identified as α and β tubulin with 31 and 43 unique peptides, respectively. Moreover, cytosolic ribosomal proteins, TEF-1 elongation factor, hsp70, glycerol kinase, and a few other proteins were also rarely present.
Fig. 1.
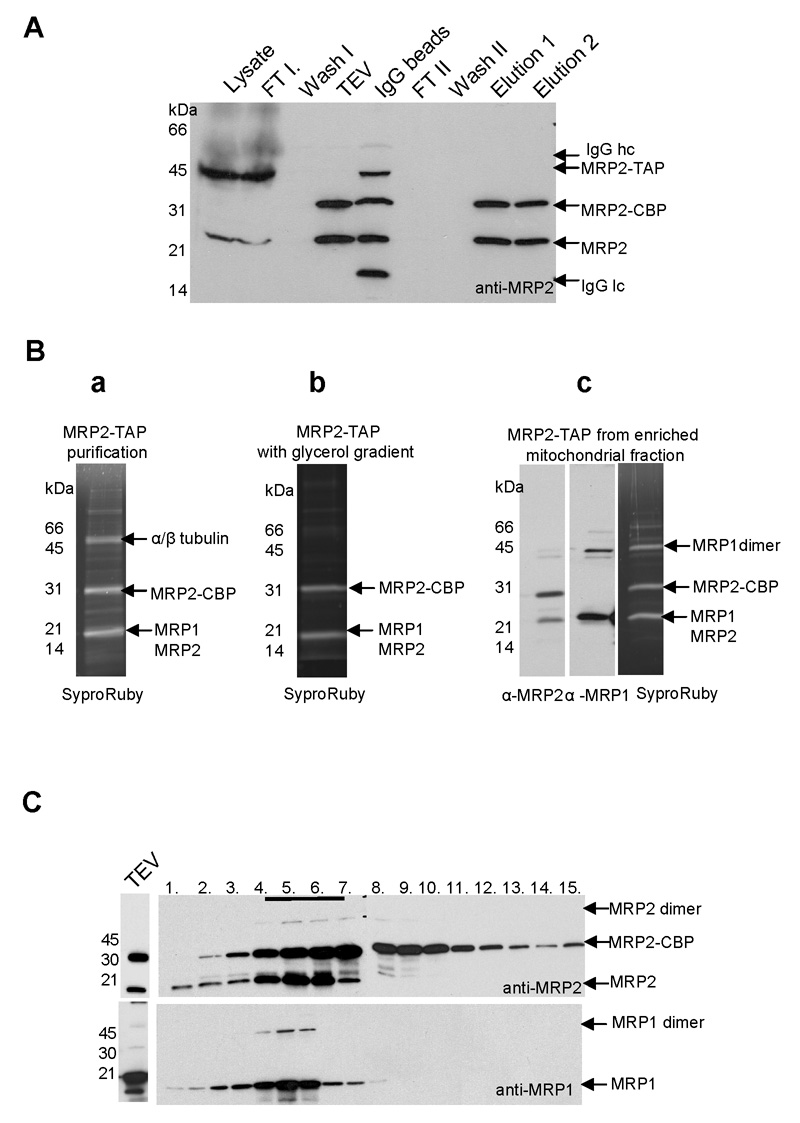
Purification of the mitochondrial RNA-binding protein (MRP) 1/MRP2 complex by tandem affinity purification (TAP) chromatography. (A) The TAP-tagged MRP2 complex was purified from a clarified whole cell lysate, which was incubated with IgG-Sepharose beads. The MRP2 protein attached to a calmodulin-binding peptide (MRP2-CBP) was released by incubation with the tabacco etch virus (TEV) protease and further purified via calmodulin-affinity chromatography and EGTA release. The purification was checked by Western blot analysis with the specific anti-MRP2 antibody; clarified cell lysate, flow through from the IgG column (FT I), first wash (Wash I), TEV eluate (TEV), IgG beads after TEV elution (IgG beads), flow through from the calmodulin-binding column (FT II), second wash (Wash II) and EGTA elutions fractions 1 and 2. Arrows indicate the location of TAP-tagged MRP2 (MRP2-TAP), MRP2-CBP, endogenous MRP2 and heavy (IgG hc) and light chains (IgG lc) of IgG molecule visible in IgG beads sample. (B) Sypro Ruby staining of calmodulin elution fraction 2 from three different MRP2-TAP tag purifications from: the whole cell lysate (a); the whole cell lysate with a glycerol gradient step of MRP2-TAP TEV eluate followed by calmodulin column with EGTA release (b); the mitochondria-enriched fraction including glycerol gradient step. The calmodulin elution fraction 2 was also analyzed by Western blot analysis using anti-MRP1 and anti-MRP2 antibodies (c). All visible bands were subjected to LC-MS/MS and proteins identified in each band are indicated by arrows. (C) Fractionation of the MRP2-TAP TEV eluates on a 10–30% glycerol gradient. Fractions were collected from the top of the gradients. Aliquots from all fractions were analyzed by SDS-PAGE probed with anti-MRP2 (a) and anti-MRP1 antibodies (b). Fractions 4–7, which were positive for endogenous MRP2 and MRP1 and tagged MRP2-CBP, were further subjected to calmodulin affinity column.
The TEV eluate was fractionated using a 10–30% glycerol gradient and fractions were screened with the anti-MRP2 and anti-MRP1 antibodies. Western blot analysis revealed that both wild type MRP2 and MRP2-CBP proteins co-sediment with wild type MRP1. Interestingly, some dimers under non-reducing SDS-PAGE condition, which are resistant to 10% SDS, were observed (Fig. 1C). Glycerol gradient fractions 4 through 7 that contained most of the MRP1 and MRP2 signal were pooled and the MRP1/MRP2 complex was purified by a second affinity step. A Sypro Ruby stained SDS PAGE gel, onto which the eluate of the second purification step has been loaded, revealed only two bands (Fig. 1Bb), which were subsequently subjected to LC-MS/MS analysis and identified as MRP1 and MRP2 with 15 and 24 unique peptides, respectively. Although still present, tubulin, cytosolic ribosomal protein and a few other proteins were identified only with a few peptides as apparent contaminants of the TAP purification. One of these putative contaminants, a 42 kDa protein of unknown function (Tb927.4.1300), was tagged with an (HA)3 tag and expressed in T. brucei procyclics. Using a monoclonal antibody against this tag, Tb927.4.1300 was immunolocalized to the cytoplasm, confirming its spurious association with the MRP1/MRP2 complex (data not shown).
To obtain additional evidence about the composition of the MRP1/MRP2 complex, the TAP-tagged MRP2 was further purified from a clarified mitochondria-enriched fraction isolated by hypotonic lysis from 2 × 1011 cells. A glycerol gradient step was also included and sedimentation of the MRP1/MRP2 complex was similar as shown in Fig. 1C. Sypro Ruby-stained SDS PAGE gel revealed three major bands, which were subsequently subjected to LC-MS/MS analysis. In all analyzed samples both MRP1 and MRP2 were equally frequent (35 and 34 unique peptides, respectively). As judged from its mobility in the gel, Western analysis and MS analysis, the high molecular weight band is predominantly a homodimer of the MRP1 protein with a small amount of MRP2 homodimer and/or heterodimer (Fig. 1Bc). Results of several TAP tag purifications were identical, including the presence of a SDS-resistant dimer of the MRPs.
In order to detect putative RNA-sensitive interactions, the same purification protocol was performed with the addition of RNase A (0.1 mg/ml) during the first step. Glycerol gradient sedimentation, elution profile and MS data were very similar to those obtained without the RNase treatment (data not shown). In another experiment, the TAP tag was attached to MRP1 and complexes containing tagged MRP1 were isolated from the T. brucei mitochondria. The same purification profile as for the MRP2-TAP tagged complex was obtained (data not shown).
Since the Leishmania orthologues of MRP1 and MRP2, Ltp26 and Ltp28, were shown to interact with the RNA ligase-containing L-complex, editosome and RET1 in an unstable manner (Aphasizhev et al., 2003), we undertook analysis of these interactions in T. brucei. After the first purification step, glycerol gradient fractions of the TEV eluate (Fig. 1C) were subjected to adenylation assays in order to detect editosomal ligases, but these assays failed to detect any activity. Next, Western analysis using monoclonal antibodies against the core editosomal proteins (KREPA1, KREPA2, KREPA3 and KREL2) and polyclonal antibody against RET1 were performed in order to detect any possible association with the editosome. Again, the results were negative, regardless of whether or not RNase A treatment has been performed during the first purification step (data not shown).
3.3. Escherichia coli-reconstituted versus natively isolated T. brucei MRP1/MRP2 complexes
The MRP1 and MRP2 genes were cloned in tandem into the pETDuet-1 system. Co-expression of both proteins in E. coli produced a soluble MRP1/MRP2 complex that was readily purified via Ni-NTA chromatography. This reconstituted complex, as well as the MRP1/MRP2 complex purified from T. brucei, were used for native size determination on 4–16% BN gel electrophoresis and visualized by silver staining. Regardless of the way the complexes were isolated, they form equally sized high molecular weight complexes of 100 kDa, 200 kDa and 400 kDa, corresponding to the predominant heterotetramer and its putative multimers (Figs. 2A, B).
Fig. 2.
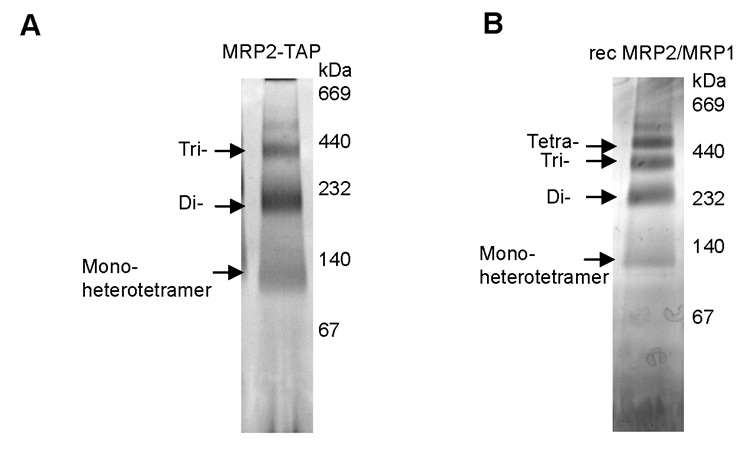
Blue native electrophoresis of the mitochondrial RNA-binding protein (MRP) 1/MRP2 complexes isolated from Trypanosoma brucei and Escherichia coli. The native T. brucei MRP2-tandem affinity purification (TAP) complex (A) and the recombinant E. coli MRP1/MRP2 complex (B) were separated on a 4–16% Blue native gel and silver stained (lane 1). The native high molecular weight marker (Sigma) was used to determine the size of molecular complexes.
3.4. Electron microscopy of the MRP1/MRP2 complex isolated from T. brucei
The structures of the natively isolated MRP1/MRP2 complex, with and without the RNase A treatment, were investigated using transmission electron microscopy (TEM) (see Materials and Methods). Both preparations contained dispersed particles of uniform size and shape and were almost free of contaminants. Particles extracted from micrographs were separately aligned, treated with multivariate statistical analysis and classified. The most representative class averages of both RNase A untreated and treated MRP1/MRP2 complexes are shown (Fig. 3). Although no symmetry was imposed during the image analysis, the resulting reconstructed structures clearly revealed a pseudo-C4 structure containing a central hole (Figs. 3A,B) with dimensions essentially identical to the crystal structures of the MRP1/MRP2 complex reconstituted from proteins overexpressed in E. coli (Schumacher et al., 2006).
Notably, the overall structure of the T. brucei MRP1/MRP2 complex with the non-occluded central hole was retained in the presence of RNA, indicating that the RNA molecules do not bind in the central hydrophilic cavity. Instead, the extra density observed in the presence of RNA surrounds the edges of the tetrameric structure, which corresponds to RNA binding at the β-sheet face of the complex and/or the outside loop regions as seen in the crystal structure with gRNA (Schumacher et al., 2006). Superimposition of the crystal structure on an averaged TEM picture of the MRP1/MRP2 complex illustrates the coherence between the natively purified MRP1/MRP2 complex and its E. coli-derived version (Fig. 3C).
3.5. Binding activities of the MRP1 and MRP2 proteins, and the MRP1/MRP2 complex
Since MRP1 was previously shown to be a high affinity gRNA-binding protein (Lambert et al., 1999), we first tested whether its binding partner, MRP2, has a similar property. The gRNA gA6[14] (70 nucleotides long) was synthesized by in vitro transcription and [32P]-end labeled. Binding by MRP2 in a native polyacrylamide gel results in a slower electrophoretic mobility of the labeled gRNA that is bound in a concentration-dependent fashion, with relatively high concentrations of the protein being required for efficient binding (Fig. 4A). The presence of a single RNA binding site in each MRP protein, which binds RNA with the same efficacy, was demonstrated by side-by-side binding assays performed with separately overexpressed and purified MRP1 and MRP2 proteins (Fig. 4B).
Fig. 4.
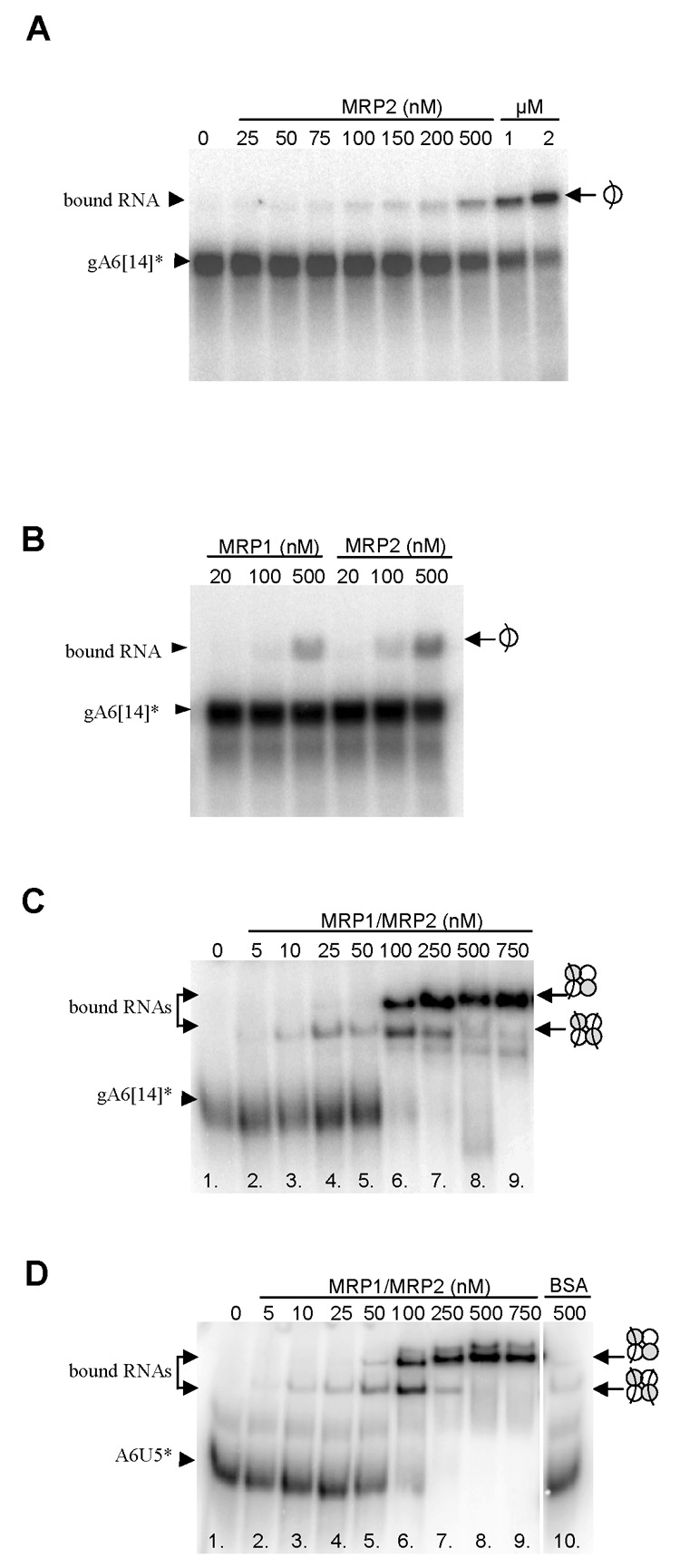
Binding activities of the mitochondrial RNA-binding protein (MRP) 1 and MRP2, and the MRP1/MRP2 complex. The icons to the left depict the RNA-protein interaction. (A) Autoradiogram of a representative binding experiment with the recombinant MRP2 protein and radiolabeled gA6[14]. The protein concentrations are indicated. The icon to the right indicates one RNA molecule bound to one MRP2 protein. (B) Autoradiogram of a side-by-side binding experiment with the recombinant MRP1 (lanes 1–3) and MRP2 proteins (lanes 4–6) and radiolabeled gA6[14]. The protein concentrations are indicated.(C) Autoradiogram of a binding experiment with radiolabeled gA6[14]. The protein concentrations are indicated. The icon to the right indicates one or two RNA molecules bound to one MRP1/MRP2 complex. (D) Autoradiogram of a binding experiment with radiolabeled A6U5 at the MRP1/MRP2 complex concentrations indicated (lines 1–9). Control binding reaction was set up with 500 nM BSA (instead of MRP1/MRP2 complex).
Further experiments were performed with the MRP1/MRP2 complex reconstituted in E. coli. First, we tested whether the complex distinguishes between gRNAs and pre-mRNAs. As shown in Figs. 4C and 4D, labeled A6U5, pre-edited mRNA (74 nucleotides long) covering the first editing site of the T. brucei ATPase 6 mRNA, and its cognate gRNA gA6[14] were bound with equal efficiency. Nevertheless, the binding pattern differed significantly from that observed for the solitary MRP1 (Köller et al., 1997; Müller et al., 2002) and MRP2 proteins (Fig. 4A). At low protein concentrations (10 to 50 nM) and RNA in excess, a single shifted band was present (Fig. 4C; lanes 3, 4 and 5). However, in excess of the protein (100 and 250 nM) over RNA, a second shifted band appeared at the intensity depending on protein concentration (Fig. 4C; lanes 6 – 9).
The observed pattern can be explained by two alternative models. The first postulates that the lower band represents single RNA molecule bound to one heterotetrameric MRP1/MRP2 complex, whereas the upper band represents single RNA molecule bound to one di-heterotetrameric complex, a form observed in native gel (Fig. 2). However, since the crystal structure revealed two identical RNA binding sites related by the C2 symmetry (Schumacher et al., 2006), a second explanation postulating that the lower and upper bands correspond to the MRP1/MRP2 complexes with two and one RNA molecules bound, respectively, appears to be more plausible. As a result of the increased charge, the MRP1/MRP2 complex fully loaded with two RNA molecules has a higher electrophoretic mobility (Figs. 4C and D). While the enthalpic contribution to the free energy of binding would be identical for both binding sites, the symmetry of the MRP1/MRP2 complex increases the entropy for RNA binding to the first degree. Once the first RNA molecule is bound, the symmetry of the MRP1/MRP2 complex is lost and binding to the second degree causes lowering of entropy and is thus less probable. Therefore, at high protein concentrations, the competition would result in each MRP1/MRP2 complex ending up with only a single RNA molecule bound. No binding activity was detected by the addition of BSA (Fig. 4D, line 10).
The apparent dissociation constants (Kda) for binding gRNAs (gA6[14] and gCyb-558) and pre-mRNAs (A6U5 and 5′CybUT) were determined for the natively purified and recombinant MRP1/MRP2 complexes under native PAGE conditions. Binding of the recombinant complex to gRNA and/or pre-mRNA molecules is approximately the same, with the apparent Kda between 140–190 nM (Table 1).
Table 1.
Binding of the native and recombinant mitochondrial RNA-binding proteins (MRP) 1/MRP2 complexes to selected guide RNA and pre-mRNA molecules in apparent Kda values.
| Protein | A6U5 | gA6[14] | CybUT | gCyb-558 |
|---|---|---|---|---|
| Native complex | 117.8 | 111.7 | N/D | N/D |
| Recombinant complex | 147.3 | 143.3 | 140 | 190.3 |
N/D - values not determined
3.6. Annealing activities of the MRP1/MRP2 complex
To test whether the MRP1/MRP2 complex promotes annealing of complementary RNA molecules, as was previously shown for MRP1 alone (Müller et al., 2001), control experiments were performed using the natively purified and recombinant complexes and labeled gA6[14] or A6U5 RNAs. In an initial experiment, a range of concentrations of the MRP1/MRP2 complex was tested for annealing activity of A6U5 pre-mRNA with its labeled cognate gRNA (Figs. 5A and B). Under the reaction conditions used (see Materials and Methods), up to 13% of the input RNA was spontaneously converted into the annealed product in the absence of the MRP1/MRP2 complex (Figs. 5A, lane 2 and 5B, lanes 1,4). In its presence, up to 79% of the input RNA was converted into the annealed product (Figs. 5A, lanes 3–9 and 5B, lanes 2,3,5). The amount of the pre-mRNA/gRNA hybrid peaked at 200 nM of the protein and decreased at its high concentrations (600–1,000 nM) (Fig. 5A, lanes 10–14), presumably because under these conditions annealing was overcome by the binding activity. With all RNA bound by the MRP1/MRP2 complex, no free RNA was available for hybridization, an explanation further supported by the prevailing species with a single bound RNA under conditions shown in lane 15 (Fig. 5A).
Fig. 5.
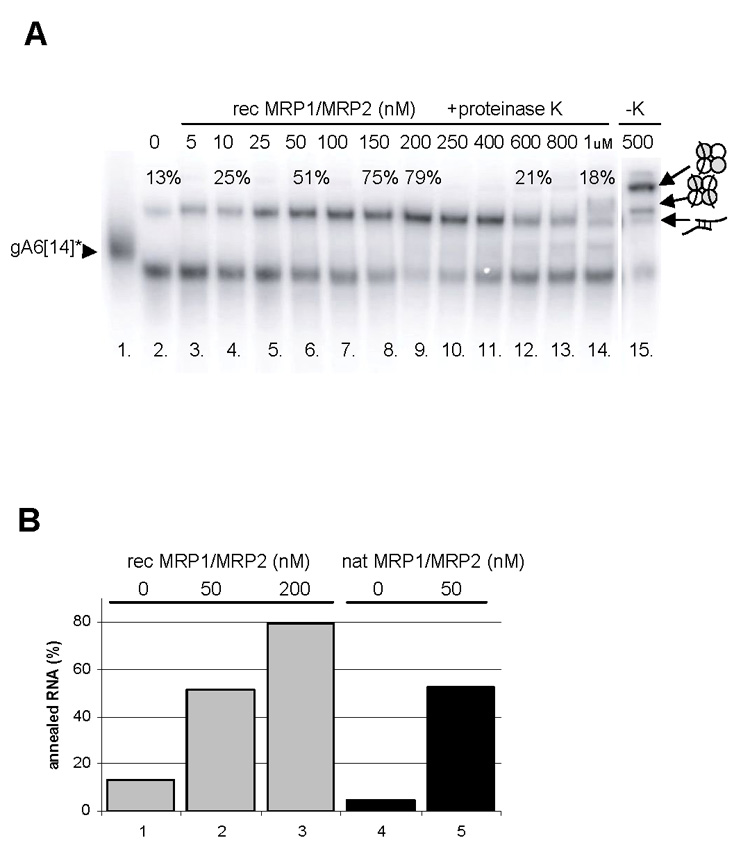
Annealing activities of the mitochondrial RNA-binding protein (MRP) 1/MRP2 complex. (A) Autoradiogram of a representative annealing experiment with the recombinant MRP1/MRP2 complex and radiolabeled gA6[14] and A6U5 pre-mRNA. The protein concentrations are indicated. Reactions were stopped by the addition of 20 mg/ml proteinase K (lanes 3–14), or the proteinase K treatment was omitted (lane 15). Free, annealed and bound RNAs are shown by arrowheads to the left. The percentage of annealed RNAs is indicated. The icons to the right depict the RNA-protein interactions. (B) Stimulation of annealing by the recombinant (grey columns) and natively purified MRP1/MRP2 complexes (black columns). Bars are derived from the densitometric analysis of several experiments as described in (A). The level of RNA-RNA “self-annealing” in the absence of the MRP1/MRP2 complex is shown in lanes 1 and 4. The reaction was performed with 50 nM (lanes 2 and 5) and 100 nM (lane 3) of the complex.
Using the prediction program for possible hybridization sites for RNA molecules we found out that duplex formation between the same RNA molecules or between the non-cognate RNA molecules may occur (Supplementary Figs. S2A and 2B). To test this prediction, we performed an experiment with different combinations of gRNAs and pre-mRNAs (the above described pair, and 5′CybUT [70 nucleotides long], a pre-edited mRNA corresponding to the editing domain of cytochrome B, and its cognate gRNA gCyb-558 [59 nucleotides long]) added to the MRP1/MRP2 complex (Fig. 6).
Fig. 6.
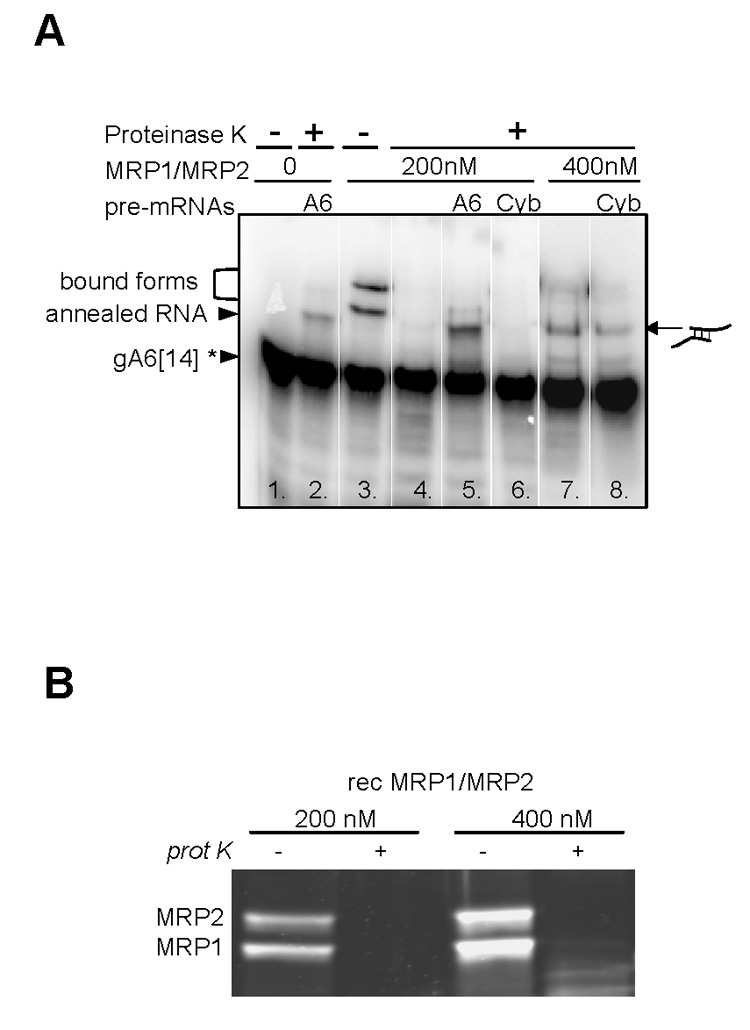
Specific and non-specific annealing activities of the mitochondrial RNA-binding protein (MRP) 1/MRP2 complex. (A) Autoradiogram of a representative annealing experiment with 200 nM and 400 nM of the recombinant MRP1/MRP2 complex, radiolabeled gA6[14], and non-labeled pre-mRNA A6U5 or CybUT. Free, annealed and bound RNAs are shown by arrowheads to the left and the icon to the right depicts the RNA-RNA interaction. (B) SDS-PAGE and Sypro Ruby staining of the MRP1/MRP2 complex at 200 nM and 400 nM concentration before and after proteinase K treatment.
A control reaction containing gA6[14] and 200 nM of the MRP1/MRP2 complex shows that the higher bands represent RNA molecules bound to the complex (Fig. 6A, lane 3), since after the proteinase K treatment these bands disappear (Fig. 6A, lane 4). Thus in the reaction containing gA6[14] and its cognate pre-edited mRNA A6U5, the higher band most likely corresponds to the gRNA/pre-mRNA duplex (Fig. 6A, lane 5). The same size band representing gRNA/pre-mRNA duplex was observed in the presence of both RNA molecules but in the absence of the MRP1/MRP2 complex (Fig. 6, lane 2).The annealing of the non-cognate gA6[14] and/or CybUT RNA molecules was not observed under these conditions (Fig. 6A, lane 6). However, at higher protein concentration (400 nM) and upon the addition of proteinase K, a higher band appeared in the reaction containing gA6[14] alone or with CybUT pre-mRNA (Fig. 6A, lanes 7 and 8). These higher bands might represent a duplex of two gA6[14] molecules when only gA6[14] was added to the reaction, and/or a sub-population of heterogeneous hybrid duplex of the gA6[14] and CybUT RNAs when reaction contains gA6[14] and non-cognate CybUT RNAs. An alternative explanation of the higher bands in lanes 7 and 8 (Fig. 6A) holds that these bands represent different intramolecular structures of RNA promoted by the MRP1/MRP2 complex. Such an explanation, however, contradicts the predicted thermodynamic stabilities of lowest energy secondary structures for individual gRNAs and mRNAs and their hybrids (Supplementary Fig. S2B) (Yu et al., 2006; Leung and Koslowsky, 2001). The same pattern was observed with the labeled pre-edited A6U5 RNA used in the same experimental setup (data not shown). In order to verify that the described shifted band is indeed the RNA-RNA annealed product and not an RNA-protein complex, we show a Sypro Ruby-stained SDS PAGE gel of the reactions containing 200 nM or 400 nM of the MRP1/MRP2 complex before and after proteinase K treatment. Obviously, the protein complex was completely eliminated by the treatment (Fig. 6B).
4. Discussion
Extensive mass spectrometry analysis of the MRP1/MRP2 complex from the mitochondrion of T. brucei procyclics, natively purified by TAP-tagging of either of its subunits, strongly indicated that this RNA binding complex consists solely of the MRP1 and MRP2 proteins arranged in a heterotetramer. By its composition confined to MRP1 and MRP2, the T. brucei complex differs from the L. tarentolae Ltp26/Ltp28 complex that was reported to contain substochiometric amounts of the AP1, AP2 and AP3 proteins (Aphasizhev et al., 2003). Interestingly, when the T. brucei homologue of the L. tarentolae AP-1 was used as bait for the TAP-tag purification, the purified protein complex contained TbAP-1 and TbAP-2 along with a few proteins of unknown function, but not the MRP1 or MRP2 proteins (H. Hashimi and J. Lukeš, unpublished data). The MRP1 protein and the Ltp26/Ltp28 complex were shown to interact with the editosomal activities in a transient and RNA-sensitive manner and under low salt conditions (Allen et al., 1998; Aphasizhev et al., 2003), while no such interactions have been reported for the C. fasciculata gBP27/gBP29 complex (Blom et al., 2001). Despite numerous attempts, we were unable to show any of these interactions for the MRP1/MRP2 complex in T. brucei procyclics. Blue native electrophoresis of the MRP1/MRP2 complex indicated that a 100 kDa heterotetramer is capable of forming higher molecular weight complexes probably by oligomerization of the heterotetramer. Three major complexes of approximate size 100, 200 and 400 kDa are formed under native conditions. Similar high molecular weight complexes were reported from C. fasciculata (Blom et al., 2001) and L. tarentolae (Aphasizhev et al., 2003).
An implication from the TEM analysis is that the MRP1/MRP2 complex, which was natively purified from T. brucei, forms a pseudo-C4 symmetrical structure. Although the TEM resolution does not allow distinction between the MRP1 and MRP2 subunits, we were able to superimpose the crystal structure upon an averaged TEM picture of the complex. This clearly illustrates the coherence between the genuine T. brucei MRP1/MRP2 complex and its version reconstituted in E. coli. The TEM analysis also revealed that the overall architecture of the complex does not change upon RNA binding. The natively purified MRP1/MRP2 complex with bound RNA has an altered structure at the edges of the tetramer, whereas the prominent central hole remains unchanged. These findings are in perfect agreement with the crystal structure (Schumacher et al., 2006) and put it into a biologically relevant perspective.
The capacity of the T. brucei MRP1 protein to bind RNA molecules (Müller et al., 2001) was demonstrated here also for the MRP2 protein and the MRP1/MRP2 complex natively purified from T. brucei. Since each free MRP protein binds a single RNA molecule (Köller et al., 1997), one would expect that the heterotetrameric MRP1/MRP2 complex will bind four RNA molecules. However, our binding experiments confirmed the results obtained by crystallography, namely that there are only two RNA binding sites in the complex. Combined, these data showed that the MRP1/MRP2 complex binds in a different mode than the MRP1 and MRP2 proteins alone.
As was shown by crystallography, the gRNA stem/loop II base is anchored to the basic surface of the MRP1/MRP2 complex, while the stem/loop I is unfolded and its bases are exposed to the solvent (Schumacher et al., 2006). We propose that the complex destabilizes the RNA of low thermodynamic stability converting them to an annealing-active conformation and thus functions as an RNA matchmaker. Whereas the annealing activity of the native and recombinant MRP1/MRP2 complex was detectable at protein concentration as low as 10 nM, its peak was observed at 100 to 250 nM, causing the conversion of 70 to 80% of the added RNA into the double stranded form. Comparable annealing activities have been described for the Ltp26/Ltp28 complex of L. tarentolae (Aphasizhev et al., 2003).
It is generally accepted that the first step in assisted hybridization of gRNA to pre-mRNA is gRNA binding by the MRP1/MPR2 complex that recognizes the archetypal secondary structure of gRNA, namely the stem-loop II domain (Hermann et al., 1997;Allen et al., 1998;Schumacher et al., 2006). However, our binding data indicate that the MRP1/MPR2 complex binds gRNA and pre-mRNA with almost the same affinity, which might imply that assisted hybridization can be initiated by either of these species. This speculation is further supported by chemical and enzymatic probing and secondary structure prediction of 5‘ CybUT and A6U5 pre-mRNAs revealing that, in analogy to gRNAs, pre-mRNAs adopt a higher order structure (J. Kopečná and L. Trantírek, unpublished data).
The initiation of the editing process assumes formation of an RNA hybrid between pre-mRNA and its cognate gRNA, an event thought to involve several steps. Based on the available data (Müller et al., 2001, 2002; Aphasizhev et al., 2003; Schumacher et al., 2006; this study), we propose that during the first step, by binding to positively charged surface of the MRP1/MRP2 complex (Fig. 7; A), the gRNA or pre-mRNA is converted to an annealing-active conformation (Fig. 7; B). At the same time, repulsion between the negatively charged bound RNA and approaching RNA is lowered, and the heteroaromatic bases of the bound molecule are aligned to base pair with the incoming RNA. This would lower the energy required to reach the transition state of hybridization reaction represented by base-pairing of pre-mRNA with the MRP1/MRP2 complex-bound gRNA (Fig. 7C). The catalytic cycle is most likely completed by the entropically driven release of the RNA hybrid from the complex (Fig. 7D).
Fig. 7.
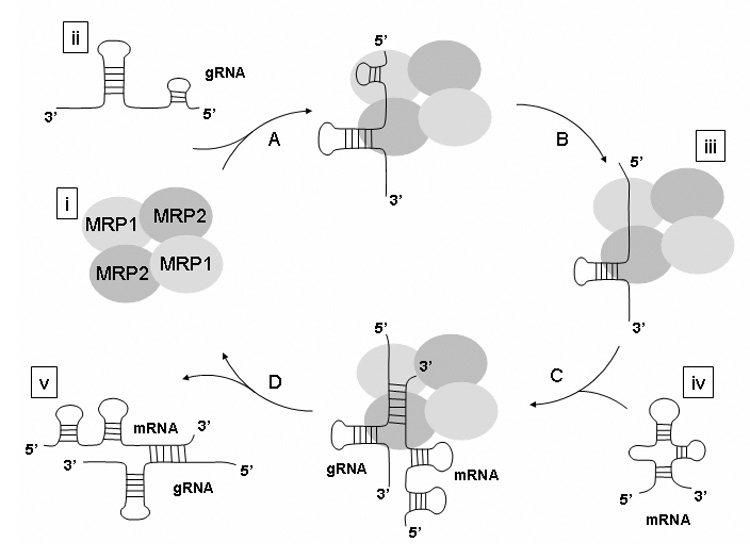
Proposed mechanism of catalyzed annealing of two RNA molecules by the mitochondrial RNA-binding protein (MRP) 1/MRP2 complex. The mechanism involves high affinity binding of the MRP1/MRP2 complex to RNA and stabilization of a bound RNA in an annealing-competent conformation. Two separate guide (g) RNA binding determinants were proposed for the MRP1/MRP2 complex; a binding pocket for the stem/loop II and for the stem/loop I (the anchor sequence). The crystal structure revealed that the base of the stem/loop II is bound by one site (MRP2), while the anchor sequence is bound by a second binding platform (MRP1) (A). The MRP1/MRP2-stem/loop II interaction would then disrupt the stem/loop I-stem/loop II interaction, tethering the thermodynamically labile anchor sequence (Schumacher et al., 2006). The end result of MRP1/ MRP2-gRNA binding is the presentation of the anchor sequence in an unfolded state with the bases exposed to the solvent in a conformation suitable for hybridization with cognate pre-mRNA (B). At the same time, repulsion between the negatively charged bound RNA and approaching RNA is lowered, and the heteroaromatic bases of the bound molecule are aligned to base pair with the incoming RNA. (C). The catalytic cycle is most likely completed by the entropically driven release of the RNA hybrid from the complex (D). Schematization of the components is based on the crystal structure of in vivo reconstituted MRP1/MRP2 complex in Escherichia coli (Schumacher et al., 2006) [i], gRNA secondary structure derived from the enzymatic probing data (Hermann and Read, 1999) [ii], co-crystalization of the MRP1/MRP2 complex with gRNA (Schumacher et al., 2006) [iii], secondary structure of the A6U5 pre-mRNA predicted using the program mfold (Mathews et al., 1999; Zuker, 2003) [iv], and secondary structure of the gRNA/pre-mRNA hybrid derived from the enzymatic and chemical probing data (Leung and Koslowsky, 2001) [v]. To be compatible with experimental data, the proposed scenario postulates the initiation of the hybridization process by either gRNA or pre-mRNA binding to the MRP1/MRP2 complex.
Predictions of possible hybridization sites for several RNA molecules provide a possible explanation for the formation of non-cognate RNA hybrids observed in vitro. They revealed that annealing between the same or non-cognate RNA molecules might occur, with the formation of the cognate pairs not always being a thermodynamically favorable process (Supplementary Fig. S2A, B). The small differences in free energies of dimer formation between cognate and non-cognate RNA pairs suggest that enzymatic catalysis of the hybridization process is required to prevent extensive accumulation of the non-translatable RNA hybrids, which would adversely affect the efficiency of editing. Since these predictions are based on the primary sequences and do not consider the formation of stable secondary structure elements, and assume non-specific mechanism of the MRP1/MRP2 complex-assisted annealing, one has to keep in mind, however, that these predictions provide only a raw qualitative estimate.
Although the MRP1/MRP2 complex is also able to catalyze the annealing of non-cognate RNA pairs in vitro, all available data indicate that the complex-assisted formation of the cognate gRNA/pre-mRNA pair is a preferred reaction route. The product specificity of the MRP1/MRP2 complex can be explained by contacts between the protein, and both single-and double-stranded regions of the gRNA (Schumacher et al., 2006). In this way, the protein orientates the conserved stem-loop II of gRNA that may represent a sterical and charge hindrance for the incoming RNA molecules (Fig. 7) and thus functions as a factor discriminating among different gRNA and pre-mRNA species (Hermann et al., 1997; Allen et al., 1998).
While this finding is in accordance with the proposed mechanism of assisted RNA hybridization via non-specific reduction of the negative charge of the RNA backbone (Schumacher et al., 2006), it also implies that along with the cognate duplexes, spuriously mismatched gRNA/pre-mRNA hybrids might occur at some rate. The formation of non-cognate RNA pairs may be responsible for the frequently observed “misediting by misguiding” (Sturm et al., 1992; Arts et al., 1993; Maslov et al., 1994). This unexpected flexibility and seemingly mistake-prone mechanism may in fact generate protein diversity, the first case of which has recently been detected (Ochsenreiter and Hajduk, 2006). At this point we can only speculate whether this is the reason why the extremely complex and apparently superfluous form of RNA editing, as it is found in the kinetoplastid mitochondrion, was not eliminated during the long evolutionary history of these flagellates.
Supplementary Material
Supplementary Fig. S1. Amino acid sequence alignment of the mitochondrial RNA-binding proteins (MRP) 1 (A) and MRP2 (B) from trypanosomatid flagellates. “*” indicates fully conserved residues “:” indicates strongly conserved residues and “.” indicates weakly conserved residues according to ClustalX. Putative N-terminal targeting sequences according to MitoProt are in italics and underlined.
Supplementary Fig. S2. Features of the RNA molecules used. (A) Sequence aligments of the guide (g) RNA and/or mRNA substrate pairs and the schematic representation of predicted hybridization sites between two RNA molecules. The complementarity between the RNA molecules is shown with : indicating G:U base pairing and | indicating the Watson-Crick base pairing. (B) Free energies of hybrid structures of two RNA molecules predicted by the program RNAcofold.
Acknowledgements
We acknowledge the help of Aswini Panigrahi with mass spectrometry. This work was supported by the Grant Agency of the Czech Republic 204/06/1558 (to J.L.) and 204/04/P191 (to L.T.), the Ministry of Education of the Czech Republic (LC07032, 1K04011, MSM6007665810 and 2B06129) and the National Institutes of Health grants 5R03TW6445 (to K.S. and J.L.) and AI14102 (to K.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Note: Supplementary data associated with this article
References
- Allen TE, Heidmann S, Reed R, Myler PJ, Göringer HU, Stuart KD. Association of guide RNA binding protein gBP21 with active RNA editing complexes in Trypanosoma brucei. Mol. Cell. Biol. 1998;18:6014–6022. doi: 10.1128/mcb.18.10.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizhev R, Aphasizheva I, Nelson RE, Simpson L. A 100-kD complex of two RNA-binding proteins from mitochondria of Leishmania tarentolae catalyzes RNA annealing and interacts with several RNA editing components. RNA. 2003;9:62–76. doi: 10.1261/rna.2134303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizhev R, Aphasizheva I, Nelson RE, Gao G, Simpson AM, Kang X, Falick AM, Sbicego S, Simpson L. Isolation of a U-insertion/deletion editing complex from Leishmania tarentolae mitochondria. EMBO J. 2003;22:913–924. doi: 10.1093/emboj/cdg083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts GJ, van der Spek H, Speijer D, van den Burg J, van Steeg H, Sloof P, Benne R. Implications of novel guide RNA features for the mechanism of RNA editing in Crithidia fasciculata. EMBO J. 1993;12:1523–1532. doi: 10.1002/j.1460-2075.1993.tb05796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom DM, Van den Berg M, Breek CKD, Speijer D, Muijsers AO, Benne R. Cloning and characterization of two guide RNA-binding proteins from mitochondria of Crithidia fasciculata: gBP27, a novel protein, and gBP29, the orthologue of Trypanosoma brucei gBP21. Nucl. Acids Res. 2001;29:2950–2962. doi: 10.1093/nar/29.14.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum B, Bakalara N, Simpson L. A model for RNA editing in kinetoplastid mitochondria: "Guide" RNA molecules transcribed from maxicircle DNA provide the edited information. Cell. 1990;60:189–198. doi: 10.1016/0092-8674(90)90735-w. [DOI] [PubMed] [Google Scholar]
- Bringaud F, Peris M, Zen KH, Simpson L. Characterization of two nuclear-encoded protein components of mitochondrial ribonucleoprotein complexes from Leishmania tarentolae. Mol. Biochem. Parasitol. 1995;71:65–79. doi: 10.1016/0166-6851(95)00023-t. [DOI] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Harauz G, Boekema E, van Heel M. Statistical image analysis of electron micrographs of ribosomal subunits. Methods Enzymol. 1988;164:35–49. doi: 10.1016/s0076-6879(88)64033-x. [DOI] [PubMed] [Google Scholar]
- Hayman ML, Read LK. Trypanosoma brucei RBP16 is a mitochondrial Y- box family protein with guide RNA binding activity. J. Biol. Chem. 1999;274:12067–12074. doi: 10.1074/jbc.274.17.12067. [DOI] [PubMed] [Google Scholar]
- Hermann T, Schmid B, Heumann H, Göringer HU. A three-dimensional working model for a guide RNA from Trypanoma brucei. Nucl. Acids Res. 1997;25:2311–2318. doi: 10.1093/nar/25.12.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Purvine S, Nesvizhskii AI, Stolyar S, Goodlett DR, Kolker E. Experimental protein mixture for validating tandem mass spectral analysis. OMICS. 2002;6:207–212. doi: 10.1089/153623102760092805. [DOI] [PubMed] [Google Scholar]
- Köller J, Nörskau G, Paul AS, Stuart K, Göringer HU. Different Trypanosoma brucei guide RNA molecules associate with an identical complement of mitochondrial proteins in vitro. Nucl. Acids Res. 1994;22:1988–1995. doi: 10.1093/nar/22.11.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köller J, Müller U, Schmid B, Missel A, Kruft V, Stuart K, Göringer HU. Trypanosoma brucei gBP21: an arginine-rich mitochondrial protein that binds to guide RNA with high affinity. J. Biol. Chem. 1997;272:3749–3757. doi: 10.1074/jbc.272.6.3749. [DOI] [PubMed] [Google Scholar]
- Lambert L, Müller UF, Souza AE, Göringer HU. The involvement of gRNA-binding protein gBP21 in RNA editing-an in vitro and in vivo analysis. Nucl. Acids Res. 1999;27:1429–1436. doi: 10.1093/nar/27.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leegwater P, Speijer D, Benne R. Identification by UV cross-linking of oligo(U)-binding proteins in mitochondria of the insect trypanosomatid Crithidia fasciculata. Eur. J. Biochem. 1995;227:780–786. doi: 10.1111/j.1432-1033.1995.tb20201.x. [DOI] [PubMed] [Google Scholar]
- Leung SS, D. J. Koslowsky DJ. Interactions of mRNAs and gRNAs involved in trypanosome mitochondrial RNA editing: Structure probing of an mRNA bound to its cognate gRNA. RNA. 2001;7:1803–1816. [PMC free article] [PubMed] [Google Scholar]
- Lukeš J, Hashimi H, Zíková A. Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr. Genet. 2005;48:277–299. doi: 10.1007/s00294-005-0027-0. [DOI] [PubMed] [Google Scholar]
- Madison-Antenucci S, Hajduk S. RNA editing-associated protein 1 is an RNA binding protein with specificity for preedited mRNA. Mol. Cell. 2001;7:879–886. doi: 10.1016/s1097-2765(01)00231-3. [DOI] [PubMed] [Google Scholar]
- Maslov DA, Thiemann O, Simpson L. Editing and misediting of transcripts of the kinetoplast maxicircle G5 (ND3) cryptogene in an old laboratory strain of Leishmania tarentolae. Mol. Biochem. Parasitol. 1994;68:155–159. doi: 10.1016/0166-6851(94)00160-x. [DOI] [PubMed] [Google Scholar]
- Mathews DH, Sabina H, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Müller UF, H. U. Göringer HU. Mechanism of the gBP21-mediated RNA/RNA annealing reaction: matchmaking and charge reduction. Nucl. Acids Res. 2002;30:447–455. doi: 10.1093/nar/30.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller UF, Lambert L, Göringer HU. Annealing of RNA editing substrates facilitated by guide RNA-binding protein gBP21. EMBO J. 2001;20:1394–1404. doi: 10.1093/emboj/20.6.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Ochsenreiter T, Hajduk SL. Alternative editing of cytochrome c oxidase III mRNA in trypanosome mitochondria generates protein diversity. EMBO Rep. 2006;7:1128–1133. doi: 10.1038/sj.embor.7400817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Schnaufer A, Carmean N, Igo RP, Jr, Gygi SP, Ernst NL, Palazzo SS, Weston DS, Aebersold R, Salavati R, Stuart KD. Four related proteins of the Trypanosoma brucei RNA editing complex. Mol. Cell. Biol. 2001;21:6833–6840. doi: 10.1128/MCB.21.20.6833-6840.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Schnaufer A, Ernst NL, Wang B, Carmean N, Salavati R, Stuart K. Identification of novel components of Trypanosoma brucei editosomes. RNA. 2003a;9:484–492. doi: 10.1261/rna.2194603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Allen TE, Haynes PA, Gygi SP, Stuart K. Mass Spectrometric Analysis of the Editosome and Other Multiprotein Complexes in Trypanosoma brucei. J. Am. Soc. Mass. Spectrom. 2003b;14:728–735. doi: 10.1016/S1044-0305(03)00126-0. [DOI] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Schumacher MA, Karamooz E, Zíková A, Trantírek L, Lukeš J. Crystal structures of Trypanosoma brucei MRP1/MRP2 guide-RNA-binding complex reveals RNA matchmaking mechanism. Cell. 2006;126:701–711. doi: 10.1016/j.cell.2006.06.047. [DOI] [PubMed] [Google Scholar]
- Schuster P, Fontana W, Stadler PF, Hofacker IL. From sequences to shapes and back - a case-study in RNA secondary structure. Proc. Biol. Sci. 1994;255:279–284. doi: 10.1098/rspb.1994.0040. [DOI] [PubMed] [Google Scholar]
- Simpson L, Aphasizhev R, Gao G, Kang X. Mitochondrial proteins and complexes in Leishmania and Trypanosoma involved in U-insertion/deletion RNA editing. RNA. 2004;10:159–170. doi: 10.1261/rna.5170704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK. Complex management: RNA editing in trypanosomes. Trends Biochem. Sci. 2005;30:97–105. doi: 10.1016/j.tibs.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Sturm NR, Simpson L. Kinetoplast DNA minicircles encode guide RNAs for editing of cytochrome oxidase subunit III mRNA. Cell. 1990;61:879–884. doi: 10.1016/0092-8674(90)90198-n. [DOI] [PubMed] [Google Scholar]
- Sturm NR, Maslov D, Blum B, Simpson L. Generation of unexpected editing patterns in Leishmania tarentolae mitochondrial mRNAs: misediting produced by misguiding. Cell. 1992;70:469–476. doi: 10.1016/0092-8674(92)90171-8. [DOI] [PubMed] [Google Scholar]
- van Heel M. Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy. 1987;21:111–123. doi: 10.1016/0304-3991(87)90078-7. [DOI] [PubMed] [Google Scholar]
- Vondrušková E, Van den Burg J, Z#x000ED;ková A, Ernst NL, Stuart K, Benne R, Lukeš J. RNA interference analyses suggest a transcript-specific regulatory role for mitochondrial RNA-binding proteins MRP1 and MRP2 in RNA editing and other RNA processing in Trypanosoma brucei. J. Biol. Chem. 2005;280:2429–2438. doi: 10.1074/jbc.M405933200. [DOI] [PubMed] [Google Scholar]
- Yu LE, Koslowski DJ. Interactions of mRNAs and gRNAs involved in trypanosome mitochondrial RNA editing: structure probing of a gRNA bound to its cognate mRNA. RNA. 2006;12:1050–1060. doi: 10.1261/rna.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zíková A, Horáková E, Jirků M, Dunajčíková P, Lukeš J. The effect of down-regulation of mitochondrial RNA-binding proteins MRP1 and MRP2 on respiratory complexes in procyclic Trypanosoma brucei. Mol. Biochem. Parasitol. 2006;149:65–73. doi: 10.1016/j.molbiopara.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. S1. Amino acid sequence alignment of the mitochondrial RNA-binding proteins (MRP) 1 (A) and MRP2 (B) from trypanosomatid flagellates. “*” indicates fully conserved residues “:” indicates strongly conserved residues and “.” indicates weakly conserved residues according to ClustalX. Putative N-terminal targeting sequences according to MitoProt are in italics and underlined.
Supplementary Fig. S2. Features of the RNA molecules used. (A) Sequence aligments of the guide (g) RNA and/or mRNA substrate pairs and the schematic representation of predicted hybridization sites between two RNA molecules. The complementarity between the RNA molecules is shown with : indicating G:U base pairing and | indicating the Watson-Crick base pairing. (B) Free energies of hybrid structures of two RNA molecules predicted by the program RNAcofold.