

Abstract
Epilepsy is a complex disease with diverse clinical characteristics that preclude a singular mechanism. One way to gain insight into potential mechanisms is to reduce the features of epilepsy to its basic components: seizures, epileptogenesis, and the state of recurrent unprovoked seizures that defines epilepsy itself. A common way to explain seizures in a normal individual is that a disruption has occurred in the normal balance of excitation and inhibition. The fact that multiple mechanisms exist is not surprising given the varied ways the normal nervous system controls this balance. In contrast, understanding seizures in the brain of an individual with epilepsy is more difficult because seizures are typically superimposed on an altered nervous system. The different environment includes diverse changes, making mechanistic predictions a challenge. Understanding the mechanisms of seizures in an individual with epilepsy is also more complex than understanding the mechanisms of seizures in a normal individual because epilepsy is not necessarily a static condition but can continue to evolve over the lifespan. Using temporal lobe epilepsy as an example, it is clear that genes, developmental mechanisms, and neuronal plasticity play major roles in creating a state of underlying hyperexcitability. However, the critical control points for the emergence of chronic seizures in temporal lobe epilepsy, as well as their persistence, frequency, and severity, are questions that remain unresolved.
Introduction
Seizures and epilepsy have been documented since the earliest civilizations, before much was understood about the nervous system at all. Most individuals with epilepsy were thought to be possessed, and the word “seizure” is derived from that notion, implying that gods take hold or “seize” a person at the time a convulsion occurs. Fortunately, these conceptions about individuals with epilepsy have changed, and the current definition of a seizure has little religious connotation. A general definition for the word “seizure” is a period of abnormal, synchronous excitation of a neuronal population. Seizures typically last seconds or minutes but can be prolonged and continuous in the case of status epilepticus. Importantly, the clinical manifestations vary, and some seizures may not involve muscular contractions (convulsions) at all.
The difference between seizures and epilepsy is commonly confused. The two are not the same. Epilepsy is defined by a state of recurrent, spontaneous seizures. If one seizure occurs in an individual, it may not necessarily mean that they have epilepsy because the seizure may have been provoked and that individual may never have a seizure again. The concept of epileptogenesis refers to the development of the state of epilepsy. It refers to the sequence of events that converts the normal brain into one that can support a seizure. It is assumed that groups of neurons become hyperexcitable, poised to abnormally discharge.
Much of what we know about epilepsy emerged in the 1800s with the first evaluation of autopsy specimens from individuals with epilepsy. The seminal work of Bouchet and Cazauvieilh in 1825 [1], followed by Sommer [2] and other scientists decades later (for a review see Scharfman and Pedley [3]) suggested profound structural changes to the brain in patients with epilepsy. A new era in epileptology began with neurologists such as Hughlings Jackson [4,5] in the late 1800s providing suggestions for the ways seizures might occur. In the 1900s, the most important advances were the development of the electroencephalogram (EEG) and the first recordings of the EEG in patients with epilepsy by Gibbs et al. [6], Jasper et al. [7], and Penfield and Jasper [8]. In parallel, the breakthroughs in understanding the essential aspects of nerve cell function, from Hodgkin and Huxley [9] to others (for a review see Hille [10]), shaped a growing appreciation that epilepsy was a complex disorder that could best be understood through diverse approaches. Today, the combined efforts of clinical and basic research have demonstrated the wealth of potential mechanisms, facilitated by the emergence of the field of neuroscience.
Reviewing some of the basic principles in neurobiology can provide a framework to understand the mechanisms of seizures, epileptogenesis, and epilepsy. To understand how seizures can be initiated, some of the most fundamental aspects of nerve cell function are useful to review, such as electrical basis of the transmembrane potential and action potential. Mechanisms governing synaptic transmission can also provide insight. Finally, one of the most remarkable characteristics of the nervous system, its plasticity, is reflected in mechanisms underlying epileptogenesis. Thus, fundamental aspects of central nervous system (CNS) structure and function have provided a long list of potential mechanisms for seizures, epileptogenesis, and epilepsy. These ideas have been supported by information from genetic models of epilepsy, where mutations in the molecular components of nerve cell function have been shown to cause epilepsy in mice.
The Neurobiology of Seizures
Seizures can be caused by multiple mechanisms, and often they appear so diverse that one would suspect that no common theme applies. However, one principle that is often discussed is that seizures arise when there is a disruption of mechanisms that normally create a balance between excitation and inhibition. Thus, normally there are controls that keep neurons from excessive action potential discharge, but there are also mechanisms that facilitate neuronal firing so the nervous system can function appropriately. Disrupting the mechanisms that inhibit firing or promoting the mechanisms that facilitate excitation can lead to seizures. Conversely, disrupting the mechanisms that bring neurons close to their firing threshold, or enhancing the ways neurons are inhibited, usually prevents seizure activity.
Although the concept of a balance provides a useful model for mechanisms that can initiate seizures, it is daunting to consider the array of potential mechanisms. One approach that distills the long list into a more manageable form is to examine mechanisms at different “levels” of the nervous system: first as ions and membranes, then cells and circuits/synapses, and finally large-scale neuronal networks.
The electrical basis of nerve cell function
At the most fundamental level, the nervous system is a function of its ionic milieu, the chemical and electrical gradients that create the setting for electrical activity. Therefore, some of the most easily appreciated controls on excitability are the ways the nervous system maintains the ionic environment. An example is the electrical basis of resting membrane potential. Resting potential is set normally so that neurons are not constantly firing but are close enough to threshold so that it is still possible that they can discharge, given that action potential generation is essential to CNS function. The control of resting potential becomes critical to prevent excessive discharge that is typically associated with seizures.
Normally a high concentration of potassium exists inside a neuron and there is a high extracellular sodium concentration, as well as additional ions, leading to a net transmembrane potential of −60 mV [11]. If the balance is perturbed (eg, if potassium is elevated in the extracellular space), this can lead to depolarization that promotes abnormal activity in many ways [12]: terminals may depolarize, leading to transmitter release, and neurons may depolarize, leading to action potential discharge. Pumps are present in the plasma membrane to maintain the chemical and electrical gradients, such as the sodium-potassium ATPase, raising the possibility that an abnormality in these pumps could facilitate seizures. Indeed, blockade of the sodium-potassium ATPase can lead to seizure activity in experimental preparations [13], suggesting a role in epilepsy [14]. The sodium-potassium pump is very interesting because it does not develop in the rodent until several days after birth, and this may contribute to the greater risk of seizures in early life [15,16]. In addition to pumps, glia also provide important controls on extracellular ion concentration, which has led many to believe that glia are just as important as neurons in the regulation of seizure activity [17,18]. Thus, the control of the ionic environment provides many potential targets for novel anticonvulsants.
It is important to bear in mind that seizures, by themselves, can lead to the changes in the transmembrane gradients. For example, seizures are followed by a rise in extracellular potassium, a result of excess discharge. This can lead to a transient elevation in extracellular potassium that can further depolarize neurons. Thus, the transmembrane potential is a control point that, if perturbed, could elicit seizures and begin a “vicious” cycle, presumably controlled by many factors that maintain homeostasis, such as pumps and glia.
The ionic basis of the action potential is another example of a fundamental aspect of neurobiology that can suggest potential mechanisms of seizures. Neurons are designed to discharge because of an elegant orchestration of sodium and potassium channels that rely on chemical and ionic gradients across the cell membrane. Abnormalities in the sodium channel might lead to a decrease in threshold for an action potential if the method by which sodium channel activation is controlled changes (ie, sodium channels are activated at more negative resting potentials or sodium channel inactivation is impaired). Indeed, it has been shown that mutations in the subunits of the voltage-dependent sodium channels can lead to epilepsy. A specific syndrome, generalized epilepsy with febrile seizure plus, is caused by mutations in selected genes responsible for subunits of the voltage-dependent sodium channel [19]. The mutation does not block sodium channels, presumably because such a mutation would be lethal, but they modulate sodium channel function. This concept, that modulation—rather than essential function—is responsible for genetic epilepsies, has led to a greater interest in directing the development of new anticonvulsants at targets that are not essential to, but simply influence, CNS function.
Synaptic transmission
Research into seizures has gravitated to mechanisms associated with synaptic transmission because of its critical role in maintaining the balance between excitation and inhibition. As more research has identified the molecular mechanisms of synaptic transmission, it has become appreciated that defects in almost every step can lead to seizures.
Glutamatergic and γ-aminobutyric acid (GABA)-ergic transmission, as the major excitatory and inhibitory transmitters of the nervous system, respectively, have been examined in great detail. It is important to point out, however, that both glutamate and GABA may not have a simple, direct relationship to seizures. One reason is that desensitization of glutamate and GABA receptors can reduce effects, depending on the time-course of exposure. In addition, there are other reasons. GABA-ergic transmission can lead to depolarization rather than hyperpolarization if the gradients responsible for ion flow through GABA receptors are altered. For example, chloride is the major ion that carries current through GABAA receptors, and it usually hyperpolarizes neurons because chloride flows into the cell from the extracellular space. However, the K+Cl− co-transporters (KCCs) that are pivotal to the chloride gradient are not constant. In development, transporter expression changes, and this has led to evidence that one of the transporters, NKCC1, may explain seizure susceptibility early in life [20•].
The relationship of glutamate to excitation may not always be simple either. One reason is that glutamatergic synapses innervate both glutamatergic neurons and GABA-ergic neurons in many neuronal systems. Exposure to glutamate could have little net effect as a result, or glutamate may paradoxically increase inhibition of principal cells because the GABA-ergic neurons typically require less depolarization by glutamate to reach threshold. It is surprisingly difficult to predict how glutamatergic or GABA-ergic modulation will influence seizure generation in vivo, given these basic characteristics of glutamatergic and GABA-ergic transmission.
Synchronization
Excessive discharge alone does not necessarily cause a seizure. Synchronization of a network of neurons is involved. Therefore, how synchronization occurs becomes important to consider. There are many ways neurons can synchronize. In 1964, Matsumoto and Ajmone-Marsan [21] found that the electrographic events recorded at the cortical surface during seizures corresponded to paroxysmal depolarization shifts (PDS) of cortical pyramidal cells occurring synchronously. These studies led to efforts to understand how neurons begin to fire in concert when normally they do not.
Glutamatergic interconnections are one example of a mechanism that can lead to synchronization. Indeed, studies of the PDS suggested that the underlying mechanism was a “giant” excitatory postsynaptic potential [22], although it was debated widely at that time if this was the only cause. Thus, pyramidal cells of cortex are richly interconnected to one another by glutamatergic synapses. Gap junctions on cortical neurons are another mechanism for synchronization. Gap junctions allow a low-resistance pathway of current flow from one cell to another, so that coupled neurons are rapidly and effectively synchronized. It was thought that gap junctions were rare, so it was unlikely that they could play a major role, but further study led to the appreciation that even a few gap junctions may have a large impact on network function [23]. Another mechanism of synchronization involves, paradoxically, inhibition. Many GABA-ergic neurons that innervate cortical pyramidal cells, such as the cell type that controls somatic inhibition (the basket cell), make numerous connections to pyramidal cells in a local area. Therefore, discharge of a single interneuron can synchronously hyperpolarize a population of pyramidal cells. As GABA-ergic inhibition wanes, voltage-dependent currents of pyramidal cells become activated. These currents, such as T-type calcium channels and others, are relatively inactive at resting potential, but hyperpolarization relieves this inhibition. The result is a depolarization that is synchronous in a group of pyramidal cells [24].
Some of the changes that develop within the brain of individuals with epilepsy also promote synchronization. Such changes are of interest in themselves because they may be one of the reasons why the seizures are recurrent. These changes include growth of axon collaterals of excitatory neurons, typically those that use glutamate as a neurotransmitter and are principal cells. An example is the dentate gyrus granule cell of hippocampus. In animal models of epilepsy and in patients with intractable temporal lobe epilepsy (TLE), the axons of the granule cells develop new collaterals and the new collaterals extend for some distance. They do not necessarily terminate in the normal location but in a novel lamina, one that contains numerous granule cell dendrites. Electron microscopy has shown that the new collaterals innervate granule cell dendrites, potentially increasing recurrent excitatory circuits. Some argue that recurrent inhibition increases as well as recurrent excitation [25], but the fact remains that new synaptic excitatory circuits develop that are sparse or absent in the normal brain [26]. The resultant “synaptic reorganization” not only can support synchronization, potentially, but it also illustrates how the plasticity of the nervous system may contribute to epileptogenesis [27].
The Neurobiology of Epilepsy
Why consider that the neurobiology of seizures may be distinct from epilepsy? The premise is that the state of recurrent seizures is associated with underlying structural and functional abnormalities. Some of the alterations may be easily visualized and, therefore, compelling, but one could argue that even without evidence of structural changes, the molecular and subcellular components of the nervous system are not the same as a normal brain. Data from laboratory animals support this premise: when the brains of animals with recurrent seizures are examined, there are diverse changes in gene expression and function at the ultrastructural level. In light of this, the neurobiology of epilepsy is a different question than the neurobiology of a seizure.
Mechanisms of seizures in TLE
One of the types of epilepsy that can serve as a useful example is TLE. There are two fundamental questions to address if one is to explain seizure generation in TLE. First, what is the reason for the underlying hyperexcitability of the brain that characterizes the interictal period, the setting upon which a seizure is initiated? Second, what causes the intermittent, recurrent seizures?
It is important to consider the assumptions that have influenced the research to answer these questions because several assumptions may be misleading. For example, it is often assumed that all seizures in TLE are due to the same mechanism. If TLE is multifocal, that may not be the case. Even if there is a predominant focus, there might be more than one problem and, therefore, more than one mechanism. Indeed, in a patient with extremely frequent seizures, perhaps there are many underlying mechanisms, and this is why seizures are so common. Intractability to currently available anticonvulsants may reflect the fact that there are many mechanisms for seizure generation in a given patient, and if one is inhibited other mechanisms still exist.
What are some of the mechanisms that have been identified? In broad terms, there are 1) seizures due to genetic causes, 2) seizures due to developmental disorders or malformations, and 3) seizures that develop after a progression of changes in response to an insult or injury. None of these mechanisms are mutually exclusive, however [28]. Genes are likely to influence acquired epilepsy, and a cortical malformation may be present in those individuals who develop epilepsy after an insult or injury. On its own, the cortical abnormality may not generate seizures but it could influence the development of TLE. Genetic factors are reviewed elsewhere [19,29], as are the known types of developmental disorders that are likely to contribute [30–32]. The following text addresses current concepts related to the ways an initial insult or injury can lead to epilepsy.
Mechanisms of acquired epileptogenesis in TLE
In many individuals with TLE, an initial insult or injury leads to a period of time without evidence of overt seizures, and then recurrent seizures begin. These stages are schematically illustrated in Figure 1. It is important to point out that many aspects of the schematic are debatable. For example, one could challenge the idea that the initial insult, early in life, is the onset, because TLE may be influenced by genetics from the time of conception. It is also not clear if there really is very little seizure activity in the “latent” period between the initial insult or injury, so the use of the term “silent” period is no longer warranted. Whether the state of recurrent seizures represents that end of the progression and is, therefore, a static period is also debated.
Figure 1.
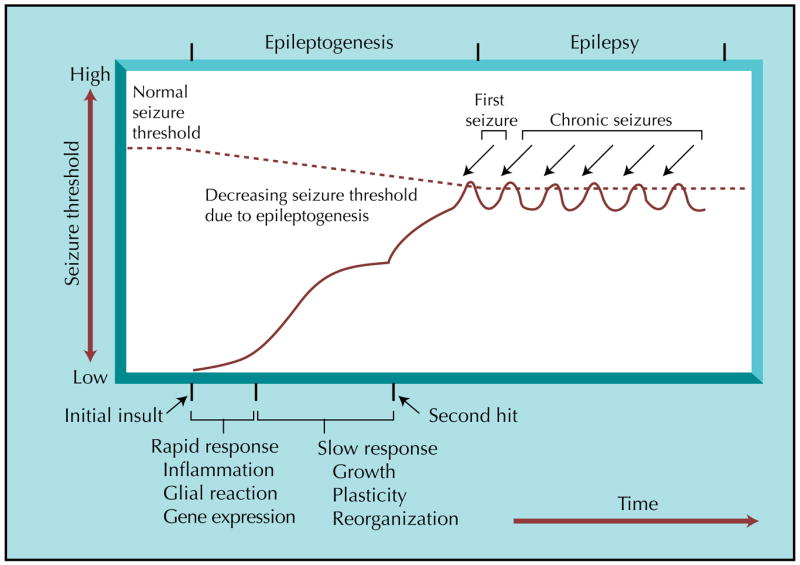
A timeline for epileptogenesis in temporal lobe epilepsy. Acquired epilepsy, using temporal lobe epilepsy as an example, can be simplified as three stages: an initial insult, followed by epileptogenesis, and ultimately ending in a state of recurrent, spontaneous seizures (epilepsy). The initial insult can take several forms and is followed by both rapid and slower progressive changes with rapid and slower durations. Rapid changes in animal models include neuronal excitation and calcium influx, triggering a cascade of events that include second messenger and immediate early gene responses, modifications to pre-existing proteins, and protein synthesis. Within days, there can be cell death and proliferation as well as inflammatory, glia, and vascular responses. Slower responses include growth (eg, axon outgrowth, synaptogenesis, angiogenesis), leading to synaptic reorganization, and this may in turn cause other changes. Over time, seizure threshold is lowered by a growing increase in excitability, and the risk of a seizure increases. These changes may be sufficient to cause epilepsy or may stall until a second “hit” occurs. The second hit could be environmental, or it could be due to time-dependent gene expression or co-morbidity. Therefore, genes, development, and the responses to the initial insult are likely to act together to result in a state of chronic seizures.
These considerations notwithstanding, the schematic represents an outline of a time-course upon which one can superimpose potential mechanisms. Thus, the fact that many individuals with TLE have experienced an event (typically early in life) that could be an initial insult is compelling. It suggests that an initial insult begins a process that ultimately leads to recurrent seizures. If it does not actually “begin” the process, at the very least it seems likely that it facilitates it. Examples include birth trauma, a febrile seizure, or infection such as encephalitis. However, the insult also may occur later, and a common example is a war injury as an adult that leads to TLE decades later. The fact that these events are injurious, leading to neuronal damage, and the fact that many patients with TLE have dramatic neuronal loss upon autopsy, have led many to conclude that damage to neurons is critical to the pathophysiology of TLE [33]. Furthermore, it appears that certain types of neurons are more vulnerable than others, such as hippocampal CA1 and the endfolium relative to the dentate gyrus and CA2 [3]. This has led to the assumption that the pattern of neuronal loss may also be important in TLE and the idea that protection of vulnerable neuronal populations could be advantageous. However, there is no clear proof to date that the selective loss of CA1 pyramidal cells, for example, is a key component of TLE.
The fact that a delay appears to occur between an initial insult and the first seizures has led many to suggest that a progression of changes in response to the initial insult is critical in epileptogenesis. However, the latent period can be very long, even decades in some patients. That observation has led some to suggest that additional environmental factors or “second hits” could be important in ultimately causing seizures to emerge [34].
These ideas, although reasonable and helpful in some ways, leave one with as many questions as answers. Why do some individuals with an initial insult never develop TLE? What occurs during the latent period? What are the mechanisms, exactly? Can they help explain why current anticonvulsants often fail in TLE?
Clues from animal models
In laboratory animals, the environment can be much better controlled and so can genetic factors. In addition, the lifespan is much shorter. Therefore, animal research provides opportunities to answer some of the questions listed previously.
Before evaluating data from laboratory animals, the models used in animals to simulate TLE require discussion. One reason is that they are not perfect, although they do allow a model of the process by which an initial insult leads to recurrent limbic seizures after a delay or latent period. The most common approach is to elicit status epilepticus in an adult male rat or mouse, using either a convulsant (pilocarpine, kainic acid) or electrical stimulation. Although prolonged status in the adult rodent typically leads to widespread damage to the brain much more than in TLE, administration of anticonvulsants soon after the onset of status or use of anesthesia [35,36] limits the damage, so that it resembles what is observed in humans, although not exactly. Additional models include the treatment of young rats with heat to initiate febrile seizures [37], whereas other laboratories use head trauma [38]. Importantly, data from these models have changed some previous conceptions about the nature of the initial insult and latent period and have identified potential mechanisms.
The perspective that a critical step is neuronal damage, presumably due to excitotoxicity, has been revised in light of the data from the febrile seizure model, where neuronal death does not appear necessary. However, there may simply be a difference in mechanisms associated with TLE after febrile seizures compared with TLE that follows an insult that occurs as an adult. Nevertheless, the febrile seizure model has provided important information about the events associated with febrile seizures and how they lead to epileptogenesis. For example, proinflammatory cytokines appear to be important [39•]. Gene expression studies corroborate a role of the immune system [40•].
A theme that appears constant across models is that the initial insult sets into motion a cascade of changes in gene expression that alters molecular and cellular constituents of neuronal (and glial) networks. Some of these are due to the upregulation of growth factors, as if the brain recapitulates developmental programs in response to the initial insult [41]. The distribution and expression of proteins that control the balance of excitation and inhibition dramatically changes. In addition, there are new synaptic connections that develop. There seem to be different examples from model to model, but the general theme is consistent. The fact that some changes involve complex growth of axons over long distances provides an explanation of why the latent period may require a long period of time, but this is still an unsettled question.
Although this discussion provides suggestions for the mechanisms of epileptogenesis in TLE, it does not explain the emergence of recurrent seizures at some point in the process (ie, what finally happens to initiate the first seizure). Presumably, enough changes occur to perturb the balance between excitation and inhibition so that even the normal range of brain activity can periodically reach seizure threshold.
What makes seizures occur repeatedly once they have begun? At the present time, there are two general answers. First, seizures involve a series of post-seizure sequelae that essentially prevent seizures for a period of time thereafter. When the post-seizure “depression” wanes, another seizure could occur, particularly if underlying hyperexcitability is such that even the normal range of activity in the CNS is able to reach seizure threshold. Evidence for this hypothesis comes from the recordings of the postictal period, when the EEG essentially becomes a flat line and behavioral immobility occurs. However, it is not a completely satisfying explanation because the EEG and behavior can recover long before another seizure occurs. One could argue that there are other aspects of the brain that must recover before another seizure can occur, but little evidence has been collected to support this idea.
Another explanation is that seizures are triggered by many aspects of the environment. For example, there are circadian changes in hormones, peptides, and other neuromodulators that may initiate seizures at certain times of the day. These ordinarily would not trigger seizures, but in a brain that has been altered so that there is an underlying state of increased excitability it is possible that they would. In humans, stressful life events appear to increase the risk of a seizure in some patients as do other external or environmental influences such as visual stimuli [42]. Changes inherent to normal life, such as the sleep-wake cycle, puberty, or aging, also influence seizures in individuals with epilepsy [43].
Conclusions
In summary, seizure generation in the normal brain has many potential mechanisms, and this is not surprising in light of the multitude of ways the CNS is designed to balance excitation and inhibition. In TLE, it seems reasonable to suggest that there are changes in the CNS that create a setting of increased excitability, and superimposed upon this are intermittent recurrent seizures. One could argue that the mechanisms that create the altered state of excitability reflect the mechanisms for normal growth and plasticity that are essential for limbic system function. Thus, the mechanisms that allow the CNS to develop into a complex structure and the mechanisms that provide plasticity, which are so important to its ability to function in a changing environment, do not come without risk—the risk of epileptogenesis. Although many of these issues are still unresolved, past years have witnessed substantial progress, largely resulting from the interplay between clinical and basic research in epilepsy.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Bouchet C, Cazauvieilh M. De L’epilpsie consideree dans ses raports avec l’alienation mentale. Recherche sur la nature et le siege de ces deux maladies. Arch Gen Med. 1925;9:510–542. [Google Scholar]
- 2.Sommer W. Erkrankung des Ammonshornes als aetiolgisches Moment der Epilepsie. Arch Psychiat Nervendrankh. 1880;10:631–675. [Google Scholar]
- 3.Scharfman HE, Pedley TA. Temporal lobe epilepsy. In: Gilman A, editor. The Neurobiology of Disease. New York: Academic Press; 2006. [Google Scholar]
- 4.Jackson JH. On a particular variety of epilepsy (“intellectual aura”), one case with symptoms of organic brain disease. Brain. 1988;11:179–207. [Google Scholar]
- 5.Jackson JH. Epileptic attacks in a patient who had symptoms pointing to gross organic disease of the right temporosphenoidal lobe. Brain. 1899;22:534–549. [Google Scholar]
- 6.Gibbs FA, Gibbs EL, Lennox WG. Epilepsy: a paroxysmal cerebral dysrhythmia. Brain. 1937;60:377–388. doi: 10.1016/s1525-5050(02)00050-1. [DOI] [PubMed] [Google Scholar]
- 7.Jasper HH, Pertuiset B, Flanigin H. EEG and cortical electrograms in patients with temporal lobe seizures. Arch Neurol Psychiatry. 1951;65:272–290. doi: 10.1001/archneurpsyc.1951.02320030009002. [DOI] [PubMed] [Google Scholar]
- 8.Penfield W, Jasper H. Epilepsy and the Functional Anatomy of the Human Brain. Boston: Little, Brown & Co; 1954. [Google Scholar]
- 9.Hodgkin AL, Huxley AF. Propagation of electrical signals along giant nerve fibers. Proc R Soc Lond B Biol Sci. 1952;140:177–183. doi: 10.1098/rspb.1952.0054. [DOI] [PubMed] [Google Scholar]
- 10.Hille B. Ion Channels of Excitable Membranes. New York: Sinauer; 2001. [Google Scholar]
- 11.McCormick DA, Huguenard JR. Electrophysiology of the Neuron: An Interactive Tutorial. Oxford: Oxford University Press; 1994. [Google Scholar]
- 12.Somjen GG. Ion regulation in the brain: implications for pathophysiology. Neuroscientist. 2002;8:254–267. doi: 10.1177/1073858402008003011. [DOI] [PubMed] [Google Scholar]
- 13.Vaillend C, Mason SE, Cuttle MF, Alger BE. Mechanisms of neuronal hyperexcitability caused by partial inhibition of Na+-K+-ATPases in the rat CA1 hippocampal region. J Neurophysiol. 2002;88:2963–2978. doi: 10.1152/jn.00244.2002. [DOI] [PubMed] [Google Scholar]
- 14.Grisar T, Guillaume D, Delgado-Escueta AV. Contribution of Na+,K(+)-ATPase to focal epilepsy: a brief review. Epilepsy Res. 1992;12:141–149. doi: 10.1016/0920-1211(92)90034-q. [DOI] [PubMed] [Google Scholar]
- 15.Haglund MM, Stahl WL, Kunkel DD, Schwartzkroin PA. Developmental and regional differences in the localization of Na, K-ATPase activity in the rabbit hippocampus. Brain Res. 1985;343:198–203. doi: 10.1016/0006-8993(85)91180-1. [DOI] [PubMed] [Google Scholar]
- 16.Fukuda A, Prince DA. Postnatal development of electrogenic sodium pump activity in rat hippocampal pyramidal neurons. Brain Res. 1992;65:101–114. doi: 10.1016/0165-3806(92)90013-m. [DOI] [PubMed] [Google Scholar]
- 17.Fellin T, Haydon PG. Do astrocytes contribute to excitation underlying seizures? Trends Mol Med. 2005;11:530–533. doi: 10.1016/j.molmed.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Duffy S, MacVicar BA. Modulation of neuronal excitability by astrocytes. Adv Neurol. 1999;79:573–581. [PubMed] [Google Scholar]
- 19.Meisler MH, Kearney J, Ottman R, Escayg A. Identification of epilepsy genes in human and mouse. Annu Rev Genet. 2001;35:567–588. doi: 10.1146/annurev.genet.35.102401.091142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20•.Dzhala VI, Talos DM, Sdrulla DA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. This study provided important new evidence for a role of NKCC1 in seizures that occur early in life, and a suggestion for a potential new anticonvulsant called bumetanide. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto R, Ajmone-Marsan C. Cortical cellular phenomena in experimental epilepsy: ictal manifestations. Exp Neurol. 1964;80:305–326. doi: 10.1016/0014-4886(64)90026-3. [DOI] [PubMed] [Google Scholar]
- 22.Brown TH, Johnston D. The synaptic nature of the paroxysmal depolarization shift in hippocampal neurons. Ann Neurol. 1984;16:S65–S71. doi: 10.1002/ana.410160711. [DOI] [PubMed] [Google Scholar]
- 23.Traub RD, Michelson-Law H, Bibbig AE, et al. Gap junctions, fast oscillations and the initiation of seizures. Adv Exp Med Biol. 2004;548:110–122. doi: 10.1007/978-1-4757-6376-8_9. [DOI] [PubMed] [Google Scholar]
- 24.Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P, et al. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1996;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- 25.Sloviter RS, Zappone CA, Harvey BD, Frotscher M. Kainic acid-induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyper-inhibition in chronically epileptic rats. J Comp Neurol. 2006;494:944–960. doi: 10.1002/cne.20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nadler JV. The recurrent mossy fiber pathway of the epileptic brain. Neurochem Res. 2003;28:1649–1658. doi: 10.1023/a:1026004904199. [DOI] [PubMed] [Google Scholar]
- 27.Sutula TP, Dudek FE. Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emergent property of a complex system. In: Scharfman HE, editor. The Dentate Gyrus: A Comprehensive Guide to Structure, Function, and Clinical Implications. Amsterdam: Elsevier; 2007. [DOI] [PubMed] [Google Scholar]
- 28.Lewis DV. Losing neurons: selective vulnerability and mesial temporal sclerosis. Epilepsia. 2005;46(Suppl 7):39–44. doi: 10.1111/j.1528-1167.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 29.Ottman R. Analysis of genetically complex epilepsies. Epilepsia. 2005;46(Suppl 10):7–14. doi: 10.1111/j.1528-1167.2005.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehericy S, Dormont D, Semah F, et al. Developmental abnormalities of the medial temporal lobe in patients with temporal lobe epilepsy. Am J Neuroradiol. 1995;16:617–626. [PMC free article] [PubMed] [Google Scholar]
- 31.Baulac M, DeGrissac M, Hasboun D, et al. Hippocampal developmental changes in patients with partial epilepsy: magnetic resonance imaging and clinical aspects. Ann Neurol. 1998;44:223–233. doi: 10.1002/ana.410440213. [DOI] [PubMed] [Google Scholar]
- 32.Ho SS, Kuzniecky RI, Gilliam F, et al. Temporal lobe developmental malformations and epilepsy: dual pathology and bilateral hippocampal abnormalities. Neurology. 1998;50:748–754. doi: 10.1212/wnl.50.3.748. [DOI] [PubMed] [Google Scholar]
- 33.Meldrum BS. Epileptic brain damage: a consequence and a cause of seizures. Neuropathol Appl Neurobiol. 1997;23:185–201. [PubMed] [Google Scholar]
- 34.Walker MC, White HS, Sander JW. Disease modification in partial epilepsy. Brain. 2002;125:1937–1950. doi: 10.1093/brain/awf203. [DOI] [PubMed] [Google Scholar]
- 35.Riban V, Bouilleret V, Pham-Le BT, et al. Evolution of hippocampal epileptic activity during the development of hippocampal sclerosis in a mouse model of temporal lobe epilepsy. Neuroscience. 2002;112:101–111. doi: 10.1016/s0306-4522(02)00064-7. [DOI] [PubMed] [Google Scholar]
- 36.Scharfman HE, Sollas AL, Smith KL, et al. Structural and functional asymmetry in the normal and epileptic rat dentate gyrus. J Comp Neurol. 2002;454:424–439. doi: 10.1002/cne.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bender RA, Dube C, Baram TZ. Febrile seizures and mechanisms of epileptogenesis: insights from an animal model. Adv Exp Med Biol. 2004;548:213–215. doi: 10.1007/978-1-4757-6376-8_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pitkanen A, McIntosh TK. Animal models of post-traumatic epilepsy. J Neurotrauma. 2006;23:241–261. doi: 10.1089/neu.2006.23.241. [DOI] [PubMed] [Google Scholar]
- 39•.Dube C, Vezzani A, Behrens M, et al. Interleukin-1beta contributes to the generation of experimental febrile seizures. Ann Neurol. 2005;57:152–155. doi: 10.1002/ana.20358. This study provided some of the first evidence that a critical element of febrile seizures is the proinflammatory cytokine interleukin-1β. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•.Lukasiuk K, Pitkanen A. Large-scale analysis of gene expression in epilepsy research: is synthesis already possible? Neurochem Res. 2004;29:1169–1178. doi: 10.1023/b:nere.0000023604.91584.6c. This article compares gene expression comprehensively across animal models to clarify the groups of gene families, by their function, that are implicated in different stages of epileptogenesis. [DOI] [PubMed] [Google Scholar]
- 41.Scharfman HE. Seizure-induced neurogenesis and its dependence on growth factors and cytokines. In: Binder DK, Scharfman HE, editors. Growth Factors and Epilepsy. Hauppague: Nova Sciences; 2005. [Google Scholar]
- 42.Guerrini R, Genton P. Epileptic syndromes and visually induced seizures. Epilepsia. 2004;45(Suppl 1):14–18. doi: 10.1111/j.0013-9580.2004.451011.x. [DOI] [PubMed] [Google Scholar]
- 43.Bazil CW. Sleep disturbances in epilepsy patients. Curr Neurol Neurosci Rep. 2005;5:297–298. doi: 10.1007/s11910-005-0074-4. [DOI] [PubMed] [Google Scholar]