

Abstract
Immunodeficiency affects over half of all patients with ataxia telangiectasia (A-T) and when present can contribute significantly to morbidity and mortality. A retrospective review of clinical history, immunological findings, ataxia telangiectasia mutated (ATM) enzyme activity and ATM mutation type was conducted on 80 consecutive patients attending the National Clinic for Ataxia Telangiectasia, Nottingham, UK between 1994 and 2006. The aim was to characterize the immunodeficiency in A-T and determine its relationship to the ATM mutations present. Sixty-one patients had mutations resulting in complete loss of ATM kinase activity (group A) and 19 patients had leaky splice or missense mutations resulting in residual kinase activity (group B). There was a significantly higher proportion of patients with recurrent sinopulmonary infections in group A compared with group B (31 of 61 versus four of 19 P = 0·03) and a greater need for prophylactic antibiotics (30 of 61 versus one of 19 P = 0·001). Comparing group A with group B patients, 25 of 46 had undetectable/low immunoglobulin A (IgA) levels compared with none of 19; T cell lymphopenia was found in 28 of 56 compared with one of 18 and B cell lymphopenia in 35 of 55 compared with four of 18 patients (P = 0·00004, 0·001 and 0·003 respectively). Low IgG2 subclass levels and low levels of antibodies to pneumococcal polysaccharide were more common in group A than group B (16 of 27 versus one of 11 P = 0·01; 34/43 versus six of 17 P = 0·002) patients. Ig replacement therapy was required in 10 (12·5%) of the whole cohort, all in group A. In conclusion, A-T patients with no ATM kinase activity had a markedly more severe immunological phenotype than those expressing low levels of ATM activity.
Keywords: ataxia telangiectasia, ATM, immunodeficiency, mutation type
Introduction
The gene for ataxia telangiectasia (A-T) coding for ataxia telangiectasia mutated (ATM), a protein kinase, was identified in 1995 on the long arm of chromosome 11 [1]. A-T has an autosomal recessive mode of inheritance and usually presents with progressive cerebellar ataxia in early life. Other features include characteristic oculocutaneous telangiectasiae, increased susceptibility to cancers, particularly lymphomas and leukaemias, and immunodeficiency.
Immunodeficiency does not affect all A-T patients; some have no excess of infections and normal immunological investigations. When present, the immunodeficiency may affect the humoral immune system, cellular immune system or both. Typically, low immunoglobulin (Ig)A, low IgG2, defective polysaccharide antibody responses and lymphopenia, especially of the naive CD4 cells, are described [2–7]. Patients with immunodeficiency often suffer an excess of bacterial sinopulmonary infections; common warts are relatively common but opportunistic infections, such as pneumocystis pneumonia, are very rare [2], possibly because of relatively preserved functional T cell responses [8].
Ataxia telangiectasia patients show bi-allelic mutations in the ATM gene, a large gene encoding a 370 kDa protein kinase with major functions in the cellular response to DNA damage. These responses include phosphorylation of targets mediating control of cell cycle checkpoints, repair of DNA double-strand breaks and apoptosis [9]. Classical cases have two truncating mutations, resulting in an absence of functional protein kinase. Some A-T patients have a milder neurological presentation and/or a slower rate of neurodegeneration. These patients have been shown to carry either leaky splice site [10,11] or missense mutations [12], resulting in expression of some ATM with functional kinase activity. The degree of retained ATM activity correlates with preservation of neurological function [12]. Other milder, later-onset phenotypes associated with mutations allowing some functional ATM expression have also been described [12–14]. In these studies some of the patients with mutations allowing some functional protein expression did not show immunodeficiency, but larger-scale studies looking at genotype–phenotype correlation in relation to immunodeficiency have not been reported. Confirmation of such a correlation would allow clinical care measures for the prevention of infection to be focused on the subgroup of A-T patients with no functional protein expression. It might also be of relevance to future potential gene therapy strategies.
We investigated how the heterogeneity in immunodeficiency in A-T patients is related to the types of ATM mutations carried in 80 consecutive patients attending the UK National Ataxia Telangiectasia Clinic.
Methods
The clinical notes and immunology results of 80 A-T patients attending the UK National Ataxia Telangiectasia Clinic between October 1994 and June 2006 were reviewed and analysed. This clinic, held in Nottingham, is a transitional multi-disciplinary clinic in which children, adolescents and adults are seen by a combination of paediatric and adult clinicians.
The results of molecular studies already performed on the patients were available. These included data on the type of mutation in the ATM gene, ATM protein level and kinase activity as well as the degree of radiation sensitivity. The analyses were performed as described previously [10,12,15,16]. On the basis of these results, patients were divided into those with no functional ATM kinase activity (group A) and those with some functional ATM kinase (group B). The clinical history and immunological data were analysed in the two groups.
Clinical
A clinical history of susceptibility to infections was taken as more than three sinopulmonary infections per winter season requiring antibiotics or a severe/atypical response to a single infection.
Immunological analysis
Immunoglobulin G, IgA and IgM were measured by nephelometry (Behring II nephelometer, Siemens Healthcare Diagnostics, Deerfield, IL, USA). The limit of detection for IgA was 0·07 g/l. IgG subclasses were measured by radial immunodiffusion. Specific antibodies to pneumovax II were measured by enzyme-linked immunosorbent assay. Lymphocyte markers were measured by flow cytometry (Coulter EPICS XL-MCL, Beckman Coulter, Fullerton, CA, USA). T cell markers included CD3, CD4 and CD8; CD19 was used for B cells and CD16/56 for natural killer (NK) cells.
Normal ranges for Igs and Ig subclasses were based on those in the Protein Reference Unit Handbook[17]. Centile ranges (2·5th−97·5th) were calculated assuming normal distributions and reported low or high levels of Ig classes or subclasses were those falling outside these limits. Normal ranges for lymphocyte numbers and subpopulations were from published data [18], as were ranges for pneumococcal antibodies [19].
Statistical analysis
The data were analysed using spss with χ2 tests or Fisher's exact test if more than 20% of categories had an expected count of less than 5. The Mann–Whitney U-test was used to compare the ages of the two groups.
Ethics
The study was approved by Nottingham Local Research Ethics Committee 1. All patients or parents/guardians gave consent to participate in this study.
Results
Data were available on 80 patients, with a median age of 13 years (range 2–47); 43 were males.
Mutation type and ATM kinase activity
Forty patients were shown to have no ATM protein expression by virtue of carrying two truncating mutations. A further 19 patients expressed non-functional ATM protein, as a consequence of a mutation in the kinase domain or an exon deletion. Two patients, who were siblings, showed absent ATM activity with very radiosensitive cells but without identified mutations. This gave a total of 61 A-T patients, with a median age of 10 years (range 2–30) without ATM kinase activity (group A).
In contrast, 12 patients were shown to have the leaky splice mutation IVS 40–1050 A > G on one or both alleles, allowing production of a small amount of normal ATM kinase, and seven patients had a missense mutation on one or both alleles resulting in expression of some ATM protein which was shown to have kinase activity. This gave a total of 19 patients in this group with some retained kinase activity (median age 27; range 6–47 years) (group B).
There was a significant difference in age between the two groups (P < 0·00001).
The main clinical history and immunological findings in the two groups are summarized in Table 1.
Table 1.
Clinical and immunological parameters in relation to mutation type.
| Group A | Group B | P-value* | |
|---|---|---|---|
| Recurrent sinopulmonary infections | 31/61 | 4/19 | 0·033 |
| Ig therapy | 10/61 | 0/19 | 0·107 |
| Prophylactic antibiotics | 30/61 | 1/19 | 0·001 |
| IgG low or replaced | 12/56 | 0/19 | 0·03 |
| Low or undetectable IgA | 25/46† | 0/19 | 0·00004 |
| Low IgG2 | 16/27 | 1/11 | 0·01 |
| Low pneumococcal antibodies (23-valent) | 34/43 | 6/17 | 0·002 |
| Low total lymphocyte | 24/57 | 0/18 | 0·001 |
| Count | |||
| Low T cell count | 28/56 | 1/18 | 0·001 |
| Low CD4 count | 23/55 | 0/18 | 0·001 |
| Low CD8 count | 25/55 | 0/18 | 0·0003 |
| Low B cell count | 35/55 | 4/18 | 0·003 |
Denominator numbers vary, as not all the patients had all the tests performed. Group A patients are those with no ataxia telangiectasia mutated (ATM) kinase activity and group B patients have mutations resulting in some residual ATM kinase activity.
P-values refer to differences between group A and group B.
Of the 25 abnormal patients 22 had undetectable levels. IgA, immunoglobulin A; IgG, immunoglobulin G.
Clinical
Recommendations for prophylaxis (antibiotics or Ig replacement) were made by the clinic physicians, but the final decision on such treatment was left to the local treating clinician. Regular antibiotic prophylaxis, when used, was usually with cotrimoxazole prior to 2001 and with azithromycin after that date. Ig therapy was used for those with low total IgG levels or with specific antibody deficiency (poor responses to vaccine antigens) in whom prophylactic antibiotics failed to control recurrent respiratory infections.
Overall, 35 of 80 patients had a history of recurrent sinopulmonary infections. This included 31 of 61 patients in group A and four of 19 in group B (P = 0·033). All 10 patients (12·5% of the whole cohort) who required Ig replacement therapy and all but one of the patients receiving prophylactic antibiotics (including eight of the patients on Ig replacement therapy) were in group A.
Immunological data
Excluding patients on Ig replacement, 46 of 51 group A and 19 of 21 group B patients had Ig levels measured. Two patients in group A had significantly reduced total IgG levels and one had raised IgG. A total of 12 of 56 patients in group A therefore had either reduced IgG or were on Ig replacement therapy, while the levels were normal in group B patients.
All the patients with low serum IgA were in group A. In 22 of 25 of these patients the levels were undetectable. Raised levels were found in four group A patients, with one group B patient showing a marginally raised level (Fig. 1).
Fig. 1.
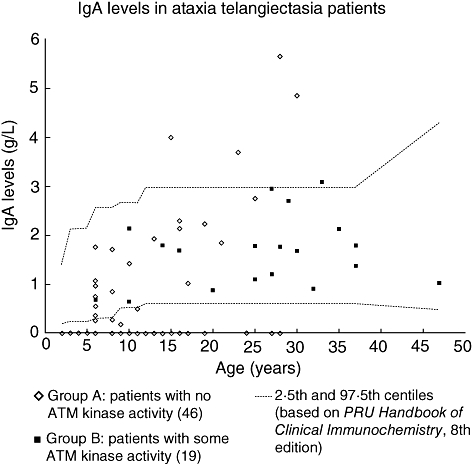
Serum immunoglobulin A (IgA) levels in ataxia telangiectasia patients. IgA levels in 46 group A patients [with no ataxia telangiectasia mutated (ATM) kinase activity] and 19 group B patients (some residual ATM kinase activity). Limit of detection for IgA was 0·07 g/l. Age-related normal reference ranges are taken from Protein Reference Unit Handbook of Clinical Immunochemistry, 8th edition [17].
IgM was raised in seven of 46 group A and in four of 19 group B patients (P = not significant). Only one patient had reduced IgM. None of those with high IgM levels had low IgG levels and three of them (all in group A) had IgA deficiency.
Deficiency of IgG2 was found significantly more commonly in group A than group B. The levels are shown in Fig. 2. There were no significant differences in IgG1 and IgG3 levels between the two groups (data not shown). There were significantly more patients with low pneumococcal antibodies in group A than in group B.
Fig. 2.
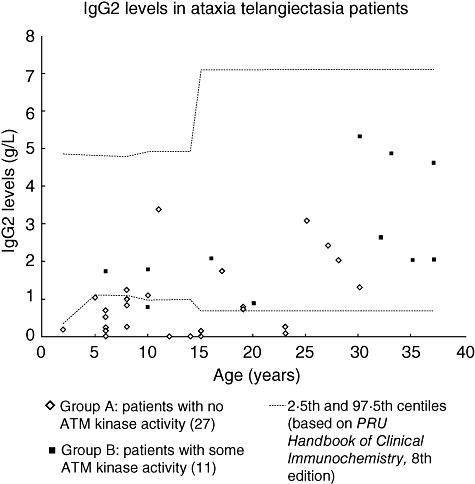
Serum immunoglobulin G2 (IgG2) levels in ataxia telangiectasia patients. IgG2 levels in 27 group A patients [with no ataxia telangiectasia mutated (ATM) kinase activity] and 11 group B patients (some residual ATM kinase activity). Age-related normal reference ranges are taken from Protein Reference Unit Handbook of Clinical Immunochemistry, 8th edition [17].
As seen from Table 1, the findings of overall lymphopenia and of low numbers of T, B, CD4 or CD8 cells were found almost exclusively in group A patients. Of the lymphopenic patients, 17 of 24 had both low T and B cell numbers. T and B lymphocyte counts are shown in Figs 3 and 4 respectively. NK cell numbers, evaluated in 56 group A and 18 group B patients, were mostly normal, with one patient in each of the groups having low numbers and three patients in each group having high numbers.
Fig. 3.
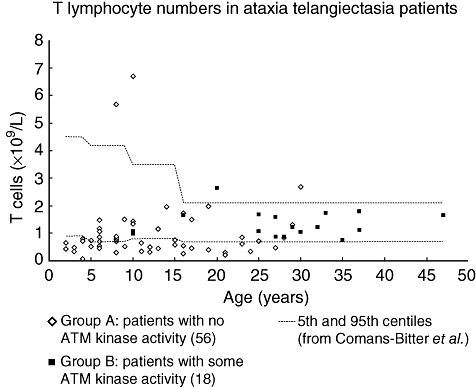
T lymphocyte counts in ataxia telangiectasia patients. Absolute numbers in 56 group A patients [with no ataxia telangiectasia mutated (ATM) kinase activity] and 18 group B patients (some residual ATM kinase activity). Age-related normal reference ranges are taken from Comans-Bitter et al. [18].
Fig. 4.
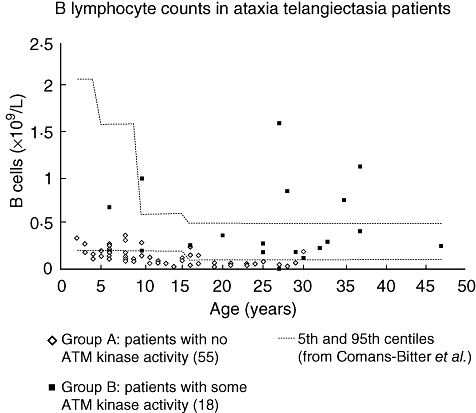
B lymphocyte counts in ataxia telangiectasia patients. Absolute numbers in 55 group A patients [with no ataxia telangiectasia mutated (ATM) kinase activity] and 18 group B patients (some residual ATM kinase activity). Age-related normal reference ranges are taken from Comans-Bitter et al. [18].
Of the 28 of 29 patients with T cell lymphopenia who had further subsets evaluated, 15 had both low CD4 and CD8 counts, while five each had either a low CD4 or CD8 count. In three patients, including the only one in group B with T cell lymphopenia, the CD4 and CD8 counts were just within the normal range.
There was no correlation between particular abnormal immunological findings (including Ig classes and subclasses, pneumococcal antibodies and T, B and NK lymphocyte counts) and a history of recurrent sinopulmonary infections.
Discussion
The findings in this study confirm those of others, with just over half of the patients exhibiting features of immunodeficiency. While a previous study did suggest a correlation between mutation type and the presence of immunodeficiency [20], this study is the first to be adequately designed to compare the clinical and immunological phenotype between those with genotypes causing complete loss of ATM function and those with genotypes associated with residual ATM function. It has been shown that those patients with complete absence of enzyme activity are much more likely to show clinical and/or immunological features of immunodeficiency than those with residual activity. For two of the most common immunological markers of immunodeficiency in A-T patients, IgA deficiency and lymphopenia (affecting T and B cells but not affecting NK cells), those patients with abnormalities were found exclusively in the group with no ATM activity. Similarly, the need for Ig replacement therapy (a decision taken on clinical rather than molecular grounds) was exclusively in the same group of patients. The findings with regard to pneumococcal antibody levels are difficult to interpret, because the measurements were taken randomly and not in relation to pneumococcal vaccination and there was uncertainty, in this retrospective study, about the numbers who had received pneumococcal immunization and its timing. Furthermore, the difference between the two groups may have been exaggerated by the fact that group A patients were significantly younger than group B patients and pneumococcal antibody levels tend to be lower in very young children. Nevertheless, the overall incidence of deficiency was similar to that described by others [7]. Twenty-four of the patients (including 13 from group A) are participants in a separate ongoing study of the immunogenicity of conjugate pneumococcal vaccine. Full data from this study will be reported elsewhere. Preliminary results indicate that all patients in group B and 9/13 in group A achieved protective antibody levels, suggesting that this vaccine may be able at least partially to overcome the defect in polysaccharide antibody responses.
These findings suggest that even a low level of ATM kinase activity (5%) is sufficient to permit relatively normal development of immunological function. Although the patients in group B (with ATM kinase activity) were significantly older than those in group A (without activity), for a number of reasons it is unlikely that this would explain the differences seen. A previous study has shown that immune function remains stable over time in A-T patients [2]. Data from normal children show that lymphocyte counts normally decrease with age, which would mitigate towards group B patients having lower, not higher, lymphocyte counts [18]. Although partial IgA deficiency in young children without A-T may improve with time, complete IgA deficiency does not usually do so [21] and most of the IgA-deficient patients seen were in the complete category. Furthermore, there was no trend towards higher IgA levels or lymphocyte counts with age.
A number of possible mechanisms have been proposed to explain the immunodeficiency in A-T. It was observed several years ago that many A-T patients had small thymuses with poorly developed Hassal's corpuscles on histology [22].
Low thymic output has been demonstrated more recently in A-T patients by measurement of TRECS (T cell recombination excision circles), markers of recent thymic emigrants [23]. Low thymic output could be one explanation for the T cell lymphopenia, and in particular the low numbers of naive CD4 cells described in A-T patients [3]. In this study TRECS were not measured, and there were insufficient cases in which naive and memory populations were measured to draw meaningful conclusions.
Ataxia telangiectasia mutated is also involved in stabilizing DNA double-stranded breaks in response to radiation damage and during VDJ recombination [24]. This is relevant to the generation of diversity of T and B cell antigen receptors. T cell receptor Vβ spectratyping studies confirm reduced antigen-receptor diversity and oligoclonal expansions [22].The same role for ATM in VDJ rearrangement is also relevant to B cell class-switching and, in addition, ATM is known to have a role Ig class-switch recombination (CSR) [25]. It is striking that the constant heavy chain genes of Ig isotypes most likely to be reduced in A-T, Cα and Cã2 are situated near the end of the constant region's reading frame and that most of the Vβs commonly under-represented in A-T patients are located in the portion of the T cell receptor β chain variable locus more distal from the diversity-joining-constant genes cluster [23]. This would be in keeping with the suggestion that V(D)J rearrangements involving excision of long intervening sequences may be impaired preferentially in A-T. The role of ATM in CSR might explain the raised IgM in 17% of the patients in this study, although it is possible that other factors such as recent infections caused increased IgM levels. If the mechanism of IgG and IgA deficiency were a defect in class-switching it might be expected that IgM would be raised in the patients with these deficiencies. High IgM levels occurred in both groups and there was no association between this finding and low IgG or IgA levels. It should be noted that in X-linked hyper-IgM syndrome, a classical example of a CSR defect, elevated IgM levels were only found in about one-half of the patients [26].
In certain circumstances ATM may protect against apoptosis [27]. It has been speculated that an increased sensitivity to oxidative stress in A-T may lead to premature cell death, preferentially affecting naive T cells [4]. Naive (CD45 RA) T cells, particularly CD4-positive T cells, are reduced in A-T [3,4,23]. The number of patients in this study tested for naive/memory T cell phenotypes was too small to allow any meaningful comparison between the groups.
It is likely that the immune defects in A-T are multi-factorial, with both reduction of T and B cell numbers (reduced thymic output, increased apoptosis) and abnormal lymphocyte function (restricted antigen receptor repertoires, abnormal B cell class-switching) contributing to the clinical phenotype. In this study the patients with humoral deficiency did not necessarily have lymphopenia and vice versa; indeed, four of seven of the patients with humoral deficiency severe enough to require Ig replacement therapy who had lymphocyte counts measured had normal lymphocyte counts. This suggests that the humoral immunodeficiency and the lymphopenia may be due to different processes. It is unclear why, in the complete absence of ATM, not all patients develop clinical features of immunodeficiency. It is also not clear why some patients with marked laboratory abnormalities did not have an excess of infections. This discrepancy between clinical and laboratory phenotype was also found in another large series [2]. It is possible that differences in innate immune mechanisms may contribute to this finding. Recent work has suggested that ATM has a role in signalling pathways relevant to the innate immune system [28].
The incidence of malignant disease in this series was too small to draw any conclusions as to whether there is a genotype–phenotype correlation in relation to this complication. A larger international study would be required to address this point.
The results of this study show that molecular techniques to characterize the genetic defect can be used to predict which patients require close monitoring and preventative measures to reduce morbidity from infection. However, it should be remembered, when managing A-T patients, that as the neurological deficit progresses they may become more prone to recurrent chest infections because of aspiration occurring, irrespective of the presence of immunodeficiency. The findings may also be relevant to potential future strategies for gene therapy or gene manipulation techniques [29]. Such strategies could potentially improve immune function, even if only low levels of ATM function were restored.
Conclusions
Ataxia telangiectasia patients with mutations leading to no expression of ATM kinase activity have a more severe immunological phenotype than those able to express small amounts of functional protein. This is relevant to their clinical management and also to future strategies for correcting the gene defect.
Acknowledgments
We thank the Ataxia Telangiectasia Society of the UK for their support of the National Ataxia Telangiectasia Clinic and also Cancer Research–UK for support (A. M. R. T., P. J. B.). None of the authors have any competing interests to declare in the context of this publication.
References
- 1.Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 2.Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, et al. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr. 2004;144:505–11. doi: 10.1016/j.jpeds.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 3.Paganelli R, Scala E, Scarselli E, et al. Selective deficiency of CD4+/CD45RA+ lymphocytes in patients with ataxia-telangiectasia. J Clin Immunol. 1992;12:84–91. doi: 10.1007/BF00918137. [DOI] [PubMed] [Google Scholar]
- 4.Schubert R, Reichenbach J, Zielen S. Deficiencies in CD4+ and CD8+ T cell subsets in ataxia telangiectasia. Clin Exp Immunol. 2002;129:125–32. doi: 10.1046/j.1365-2249.2002.01830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanal O, Ozaltin F, Tezcan I, et al. Serum IgD concentrations in patients with ataxia telangiectasia and with selective IgA deficiency. Int Arch Allergy Immunol. 1998;116:246. doi: 10.1159/000023951. [DOI] [PubMed] [Google Scholar]
- 6.Oxelius V-A, Berkel AI, Hanson LA, et al. IgG2 deficiency in ataxia-telangiectasia. N Engl J Med. 1982;306:515–7. doi: 10.1056/NEJM198203043060905. [DOI] [PubMed] [Google Scholar]
- 7.Sanal O, Ersoy F, Yel L, et al. Impaired IgG antibody production to pneumococcal polysaccharides in patients with ataxia telangiectasia. J Clin Immunol. 1999;19:326–34. doi: 10.1023/a:1020599810261. [DOI] [PubMed] [Google Scholar]
- 8.Pashankar F, Singhal V, Akabogu I, Gatti RA, Goldman FD. Intact T cell responses in ataxia telangiectasia. Clin Immunol. 2006;120:156–62. doi: 10.1016/j.clim.2006.04.568. [DOI] [PubMed] [Google Scholar]
- 9.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 10.Sutton IJ, Last JI, Ritchie SJ, et al. Adult-onset ataxia telangiectasia due to ATM 5762ins137 mutation homozygosity. Ann Neurol. 2004;55:891–5. doi: 10.1002/ana.20139. [DOI] [PubMed] [Google Scholar]
- 11.McConville C, Stankovic T, Byrd PJ, et al. Mutations associated with variant phenotypes in ataxia-telangiectasia. Am J Hum Genet. 1996;59:320–30. [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart GS, Last JI, Stankovic T, et al. Residual ataxia telangiectasia mutated protein function in cells from ataxia telangiectasia patients, with 5762ins137 and 7271T—>G mutations, showing a less severe phenotype. J Biol Chem. 2001;276:30133–41. doi: 10.1074/jbc.M103160200. [DOI] [PubMed] [Google Scholar]
- 13.Gilad S, Chessa L, Khosravi R, et al. Genotype–phenotype relationships in ataxia-telangiectasia and variants. Am J Hum Genet. 1998;62:551–61. doi: 10.1086/301755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saviozzi S, Saluto A, Taylor AM, et al. A late onset variant of ataxia-telangiectasia with a compound heterozygous genotype, A8030G/7481insA. J Med Genet. 2002;39:57–61. doi: 10.1136/jmg.39.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor AMR, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol. 2005;58:1009–15. doi: 10.1136/jcp.2005.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson D, Duedal S, Kirner J, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–22. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- 17.Milford-Ward A, Sheldon J, Ramsbottom A, Wild G. Protein reference unit handbook of clinical immunochemistry. 8th edn. Rotherham: PRU Publications; 2004. [Google Scholar]
- 18.Comans-Bitter WM, de Groot R, van den Beemd R, et al. Immunophenotyping of blood lymphocytes in childhood: reference values for lymphocyte subpopulations. J Pediatr. 1997;130:388–93. doi: 10.1016/s0022-3476(97)70200-2. [DOI] [PubMed] [Google Scholar]
- 19.Misbah SA, Griffiths H, Mitchell T, et al. Antipolysaccharide antibodies in 450 children with otitis media. Clin Exp Immunol. 1997;109:67–72. doi: 10.1046/j.1365-2249.1997.4291322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stray-Pedersen A, Jónsson T, Heiberg A, et al. The impact of an early truncating founder ATM mutation on immunoglobulins, specific antibodies and lymphocyte populations in ataxia-telangiectasia patients and their parents. Clin Exp Immunol. 2004;137:179–86. doi: 10.1111/j.1365-2249.2004.02492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberton DM, Colgan T, Ferrante A, et al. IgG subclass concentrations in absolute, partial and transient IgA deficiency in childhood. Pediatr Infect Dis J. 1990;9:S41–5. [PubMed] [Google Scholar]
- 22.Peterson RD, Kelly WD, Good RA. Ataxia–telangiectasia. Its association with a defective thymus, immunological-deficiency disease, and malignancy. Lancet. 1964;1:1189–93. doi: 10.1016/s0140-6736(64)91209-7. [DOI] [PubMed] [Google Scholar]
- 23.Giovannetti A, Mazzetta F, Caprini E, et al. Skewed T-cell receptor repertoire, decreased thymic output, and predominance of terminally differentiated T cells in ataxia telangiectasia. Blood. 2002;100:4082–9. doi: 10.1182/blood-2002-03-0976. [DOI] [PubMed] [Google Scholar]
- 24.Bredemyer AL, Sharma GG, Huang CY, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–70. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- 25.Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC. ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med. 2004;200:1103–10. doi: 10.1084/jem.20041162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levy J, Espanol-Boren T, Thomas C, et al. Clinical spectrum of hyper-IgM syndrome. J Pediatr. 1997;131:47–54. doi: 10.1016/s0022-3476(97)70123-9. [DOI] [PubMed] [Google Scholar]
- 27.Hotti A, Järvinen K, Siivola P, et al. Caspases and mitochondria in c-Myc-induced apoptosis: identification of ATM as a new target of caspases. Oncogene. 2000;19:2354–62. doi: 10.1038/sj.onc.1203567. [DOI] [PubMed] [Google Scholar]
- 28.Wu ZH, Shi Y, Tibetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signalling in response to genotoxic stimuli. Science. 2006;311:1141–6. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- 29.Du L, Pollard JM, Gatti RA. Correction of prototypic ATM splicing mutations and aberrant ATM function with antisense morpholino oligonucleotides. Proc Natl Acad Sci USA. 2007;104:6007–12. doi: 10.1073/pnas.0608616104. [DOI] [PMC free article] [PubMed] [Google Scholar]