

Abstract
In rheumatoid arthritis (RA) there are currently no useful indicators to predict a clinical response to tumour necrosis factor-α (TNF-α) blockade. The purpose of this study was to determine the role of peripheral blood cytokine profiling in differentiating between a good versus poor response to etanercept in RA. Peripheral blood samples were collected at baseline and at 3 months from 33 patients with active disease who were treated twice weekly by etanercept therapy. Responders are defined by the presence of three of four American College of Rheumatology criteria: ≥20% decrease in C-reactive protein (CRP), visual analogue score of disease activity, erythrocyte sedimentation rate and improvement of the disease activity score (28; four values) by ≥1·2 obtained at 3 months. Twelve cytokines were measured from serum collected on days 0 and 90 by proteomic array (protein biochip array, Investigator Evidence, Randox France), including interleukin (IL)-6, TNF-α, IL-1a, IL-1b, IL-2, IL-8, interferon-γ, IL-4, IL-10, monocyte chemoattractant protein (MCP)-1, epidermal growth factor (EGF) and vascular endothelium growth factor. Our results showed that high serum levels of MCP-1 and EGF were associated with a response to etanercept. In addition, the increase of two combined parameters CRP and EGF was predictive of a response to etanercept treatment at 3 months (sensitivity: 87·5% and specificity: 75%, accuracy: 84·4%). These findings suggest that cytokine profiling by proteomic analysis before treatment initiation may help to identify a responder patient to TNF-α blocking agents in RA.
Keywords: biomarkers, cytokine profiling, proteomics, rheumatoid arthritis, TNF-α antagonist
Introduction
Rheumatoid arthritis (RA) is a chronic, autoimmune, inflammatory polyarthritis that results in progressive joint damage and disability. Tumour necrosis factor (TNF)-α plays a key role in the associated pathological events and is considered as a therapeutic target. Indeed, TNF-α blocking agents (TBAs), such as infliximab, etanercept and adalimumab, have revolutionized the therapeutic care of methotrexate-resistent patients.
Various clinical trials with a TBA/methotrexate combination have shown efficacy in 60–80% of such patients [1–3]. TBAs reduce joint inflammation, retard joint damage and improve physical function [4,5]. None the less, 20–40% of the RA patients given a TBA/methotrexate combination do not respond to this treatment [1–3]. Moreover, TBAs may have side effects and are costly [6], and the efficacy of any given TBA in an individual patient is unpredictable [7,8]. For these reasons, a means of predicting responsiveness to a given TBA or other emerging biotherapies [such as inhibitors of the interleukin (IL)-1 or IL-6 pathways] would be most useful. Severals markers have been found to be informative in RA diagnosis and prognosis, such as C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), autoantibodies [rheumatoid factor (RF) and anti-cyclic citrullinated peptide antibodies] and bone proteins [9].
These parameters, however, have not been proved useful in predicting a response to TBAs. A major goal in RA today is to determine a profile at the initiation of the treatment, which could differentiate responders and non-responders.
This study was aimed at assessing the predictive response to etanercept by analysing a panel of cytokines with a proteomic approach in RA patients. In addition, it evaluated the feasibility and relevance of protein biochip array technology (PBAT) in a clinical application.
Materials and methods
Patients
A total of 33 patients were evaluated at Montpellier University Hospital and included in this study after informed consent had been obtained, all satisfying the American College of Rheumatology criteria revised in 1987 for RA diagnosis [10]. The criteria for patient eligibility were: RA and resistance to at least one disease modifying anti-rheumatic drug (DMARD) (methotrexate included). Exclusion criteria were: a history of severe or recurrent infectious disease; no contraception; pregnancy; a diagnosis of cancer; cardiac failure (stages III–IV of the New York Heart Association); and etanercept allergy. Every patient was treated with etanercept (ENBREL®), as recommended by the manufacturer and the French Drug Agency AFSSAPS (subcutaneous 25 mg two times per week). No DMARDs, including methotrexate, were associated with etanercept during the study period. Prior to the first dose of etanercept, disease activity score (DAS 28; four values), plasma CRP levels (determined by both immunological and biochemical analysis), patient assessment of disease activity (0–100 mm visual analogue scale), ESR, duration of morning stiffness, RF and dose of corticosteroid therapy (mg/day) were recorded. Patients were reassessed at 3 months. At this time, patients were categorized as responders if three of the following four criteria were achieved: a change of DAS28 ≥1·2, an improvement of 20% of patient assessment disease activity, of CRP, or of ESR. The study was approved by local ethical committee.
Proteomic analysis
Peripheral blood samples were collected at baseline and at 3 months from all patients. The 15-min centrifuge samples [550 g (relative centrifugal force)] were stored at minus 20°C. Twelve cytokines were measured in serum collected at both time-points by proteomic analysis (protein biochip array; Investigator Evidence, Randox, Mauguio, France) including IL-6, TNF-α, IL-1a, IL-1b, IL-2, IL-8, interferon (IFN)-γ, IL-4, IL-10, monocyte chemoattractant protein (MCP)-1, epidermal growth factor (EGF) and vascular growth factor (VEGF). Highly sensitive CRP (reagent Randox, Mauguio, France) was determined by latex-enhanced immunoturbidimetric method on a Olympus AU2700 biochemistry analyser (Rungis, Paris, France). The total intra-assay and total interassay variation coefficients for serum CRP were 3·3% and 2·6% respectively [11].
Statistical analysis
Values lower than the detection threshold were coded 0. The normality assumption of the variables in the two groups at the dates 0 and 90 was rejected. Twenty-four patients were classified as responders by these criteria and nine as non-responders. The variables were compared between responders and non-responders using the two-sided Kruskal–Wallis test. The link between the qualitative variables sex, previous biotherapy and RF was tested in each group by a χ2 test or Fisher's test. The change in each cytokine between day 0 and day 90 was tested by the paired Mann–Whitney U-test in each group. All the tests were implemented with r software (version 2.3.1). The significant level of P-values was fixed at 0·05 in the descriptive analysis. The loss of power induced by multiple tests in the cytokine analysis was corrected by the false discovery rate (FDR) approach [12]. Threshold P-values were given for each test. The relationship between a combination of variables (cytokines, CRP, RF, patient assessment of disease activity) at days 0 and 90 was explored using a logistic regression model. Logistic regression was implemented with sassoftware (version 8.1) with a forward procedure. The variables were introduced into the model if the P-value was < 0·05. The two variables retained in the model were EGF and CRP. The CRP variable tends to have a protective effect. However, it was not significant in the logistic regression (P-value = 0·24), because the small size of samples involved a loss of power. The EGF odds ratio could not be evaluated by the logistic regression; the contingency table between the response variable and EGF variable contained a zero value, and its P-value was 0·95. The variable EGF was stratified by the variable CRP. The hypothesis of homogeneity of EGF odds ratios across strata was not rejected. The Mantel–Haenszel estimator with a continuity correction was 0·067 and the 95% confidence interval was (0·007, 0·625).
Results
Rheumatoid arthritis patients and response to treatment (Table 1)
Table 1.
Demographic, clinical and biological data of rheumatoid arthritis (RA) patients at baseline.
| Parameters | Responders (n = 24) (mean ± s.d.) | Non-responders (n = 9) (mean ± s.d.) | P-value |
|---|---|---|---|
| Age (years) | 52·04 ± 12·8 | 50·56 ± 19·15 | 0·97† |
| Sex (men/women) | 5/19 | 2/7 | 0·93‡ |
| RA duration (years) | 14·57 ± 10·42 | 17·22 ± 13·59 | 0·47† |
| Prednisone (mg/day) | 13·69 ± 6·1 | 10·21 ± 5·27 | 0·13† |
| Previous DMARDs therapy (treatment number mean/patient) | 4·47 ± 1·77 | 3·75 ± 1·58 | 0·44† |
| Previous biotherapy (patient number) | 2 | 1 | 0·84‡ |
| Duration of morning stiffness (minutes) | 361·3 ± 515·7 | 240 ± 460·87 | 0·26† |
| DAS 28** (4 values) | 6·36 ± 1·31 | 5·3 ± 0·99 | 0·04*† |
| Rheumatoid factor (>30) | 68·18% | 88·88% | 0·21§ |
Demographic, clinical and biological data of RA patients at baseline were compared between responder and non-responder groups using the
two-sided Kruskal–Wallis test,
χ2 test or
Fisher's test.
The threshold P-value was 0·05;
28 joints were evaluated [for the disease activity score (DAS) 28 score]; DAS 28 score: a measure of RA activity score. DMARDs, disease modifying anti-rheumatic drugs; s.d., standard deviation.
The demographic, clinical and biological parameters for all patients at study initiation and at 3 months are shown in Table 1. Twenty-four patients were classified as responders and nine as non-responders. The average disease duration was 14·57 years for the responder group and 17·22 years for the non-responder group (P = 0·47). The number of prior DMARDs was 4·47 for the responder group and 3·75 for the non-responder group (P = 0·44). The high level of RA activity seen in all patients is consistent with their history of resistance to one or more DMARDs. Before treatment, however, CRP levels and disease activity (DAS28 score) were higher in the responder group [mean CRP at study initiation were 41·84 ± 30·25 mg/ml and 15·98 ± 17·14 mg/ml for the responder and non-responder groups respectively (P < 0·05)][mean DAS28 scores at study initiation were 6·36 and 5·3 for the responder and non-responder groups respectively (P < 0·05)]. Before treatment, demographic and clinical variables were not significantly different in responders compared with non-responders: sex (P = 0·93), age (P = 0·97), disease duration (P = 0·47), corticosteroid therapy (mg/day) (P = 0·13), number of previous DMARDs (P = 0·44), number who received previous biological therapies (P = 0·84) and RF (P = 0·28). By definition, the DAS28 score improved significantly at 3 months in responders (DAS28 = 6·36 ± 1·31 at day 0; DAS28 = 3·76 ± 1·79 at day 90), whereas it remained high in non-responders (DAS28 = 5·3 ± 0·99 at day 0 ; DAS28 = 4·76 ± 0·46 at day 90) (Table 1).
Cytokine profiling before etanercept treatment correlates with treatment responsiveness (Table 2; Fig. 1)
Table 2.
Comparison of cytokine between responders and non-responders at day 0 and in the non-responder group between day 0 and day 90.
| Cytokine analysis | |||||
|---|---|---|---|---|---|
| Parameters | Responders (n = 24) day 0 (mean ± s.d.) | Non-responders (n = 9) day 0 (mean ± s.d.) | Non-responders (n = 9) day 90 (mean ± s.d.) | p1-value* Responders day 0/non-responders day 0 | p2-value** Non-responders day 0/day 90 |
| IL-2 | 28·27 ± 38·14 | 75·59 ± 192·97 | 39 ± 58·44 | 0·21 | 1·00 |
| IL-4 | 11·14 ± 15·23 | 74·9 ± 217·38 | 16·38 ± 25·4 | 0·16 | 0·80 |
| IL-6 | 66·69 ± 84·94 | 69·75 ± 139·57 | 38·2 ± 37·31 | 0·10 | 0·93 |
| IL-8 | 34·02 ± 39·48 | 72·69 ± 191·23 | 56·96 ± 83·2 | 0·07 | 0·80 |
| IL-10 | 3·41 ± 4·83 | 1·53 ± 4·19 | 5·52 ± 9·24 | 0·06 | 0·21 |
| EGF | 144·18 ± 136·7 | 9·4 ± 25·15 | 61·79 ± 75·3 | 0·002* | 0·15 |
| VEGF | 228·23 ± 236·54 | 235·39 ± 662·89 | 247·35 ± 294·36 | 0·06 | 0·45 |
| IFN-γ | 22·95 ± 32·14 | 94·23 ± 251·63 | 28·99 ± 47·2 | 0·26 | 1·00 |
| TNF-α | 12·82 ± 16·7 | 88·51 ± 230·74 | 59·74 ± 89·81 | 0·14 | 0·80 |
| IL-1a | 8·86 ± 14·43 | 5·31 ± 14·34 | 15·86 ± 23·05 | 0·07 | 0·30 |
| IL-1b | 5·71 ± 9·91 | 3·39 ± 8·78 | 13 ± 22·37 | 0·08 | 0·21 |
| MCP-1 | 199·83 ± 146·09 | 29·08 ± 70·81 | 192·62 ± 175·08 | 0·005* | 0·05 |
Cytokine serum levels were compared: (1) at day 0 between responder and non-responder patients using the two-sided Kruskal–Wallis test.
The threshold p1-value corrected by the false discovery rate (FDR) approach was 0·008; (2) at days 0 and 90 in the non-responder group using the paired Mann–Whitney U-test.
The threshold p2-value corrected by the FDR approach was lower than 0·005, and any variable was retained. IL, interleukin; IFN, interferon; MCP, monocyte chemoattractant protein; TNF, tumour necrosis factor; EGF, epidermal growth factor; VEGF, vascular endothelium growth factor; s.d., standard deviation.
Fig. 1.
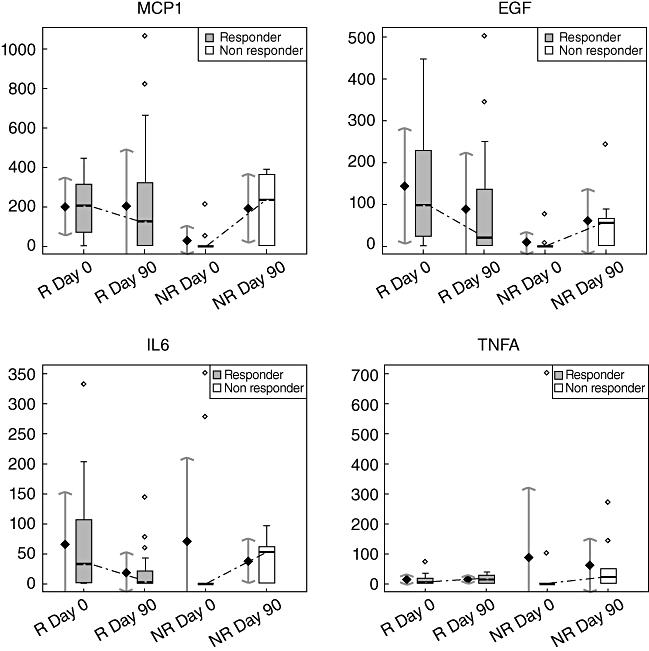
Cytokine level evolution during etanercept treatment in the responder (R) and non-responder (NR) groups. Box-plots are on the right and representations of the mean (±standard deviation intervals) on the left in each group at each time (MCP1, monocyte chemoattractant protein-1; EGF, epidermal growth factor; IL6, interleukin-6, TNFA, tumour necrosis factor-α).
Cytokine profiles were studied in all patients. We selected proinflammatory cytokines (IL-6, TNF-α, IL-1a, IL-1b, IL-2, IL-8, IFN-γ, MCP-1, EGF and VEGF) and anti-inflammatory cytokines (IL-4, IL-10). Some cytokine levels in serum at the initiation of etanercept treatment in RA may be used to predict a response to treatment at 3 months. Results of the comparison of cytokines between responders and non-responders at day 0 are presented in Table 2.
A high MCP1 serum level at day 0 was predictive of a good response (199·83 ± 146·09 in the responder group versus 29·08 ± 70·81; P = 0·01), and a high EGF serum level at day 0 was also predictive (144·18 ± 136·7 in the responder group versus 9·4 = 25·15; P = 0·002) (Fig. 1). MCP-1 and EGF serum levels at baseline may predict the treatment response to etanercept at 3 months. The other cytokine serum levels, particularly TNF-α and IL-1, were not statistically different at baseline between the responder and non-responder groups. This is consistent with earlier findings, suggesting that TNF-α and IL-1 serum levels at baseline may not be useful to predict response to TBA [13,14].
The area under the ROC curve (AUC) provides a measure of the cytokine's ability to discriminate the two patient groups (responders/non-responders). If the AUC lies between 0·8 and 0·9 then the cytokine is considered to be an excellent discriminate variable, which was the case for EGF and MCP-1 (AUC values were, respectively, 0·838 and 0·815) (data not shown).
Cytokine profiles were also compared in the responder and non-responder groups at day 90 (data not shown; see Supplementary material, Table S1). They showed no significant difference in cytokine serum levels. In particular, no significant differences in EGF and MCP-1 serum levels were found between the two groups at day 90, despite a significant difference at treatment initiation. Thus, at day 90, the cytokine profile did not discriminate between responders and non-responders.
Comparison of kinetic cytokine profiles at day 0 versus day 90 (Table 2; Fig. 1; Supplementary material Table S1)
Kinetics of the cytokine profiles were analysed at days 0 and 90 in the responder and non-responder groups. In the responder group, the serum IL-6 level decreased, although not significantly [IL-6 level: 66·69 ± 84·94 mg/ml at day 0, versus 18·11 ± 33·69 mg/ml at day 90 (P < 0·05)]; in contrast, IL-6 levels in the non-responder group increased during treatment, although not significantly (Table 2; Fig. 1). In the responder group, there was a tendency of IFN-γ levels to decrease [22·95 ± 32·14 mg/ml at day 0, versus 5·91 ± 12·05 mg/ml at day 90 (P = 0·06)]. In the non-responder group (results are presented in Table 2), MCP-1 serum levels increased from day 0 to day 90, although not significantly (29·08 ± 70·81 at day 0 versus 192·62 ± 175·08, P = 0·05). This suggests that the treatment, when it is not efficient, does not facilitate a decrease in the inflammatory process. No significant variations of growth factors (VEGF, EGF) or anti-inflammatory cytokines (IL-4, IL-10) levels were found in both groups. Even though all patients were treated with TBAs, their TNF-α, IL-1, and IL-8 levels did not decrease significantly (Fig. 1).
C-reactive protein serum level in RA patients treated with etanercept (Fig. 2)
Fig. 2.
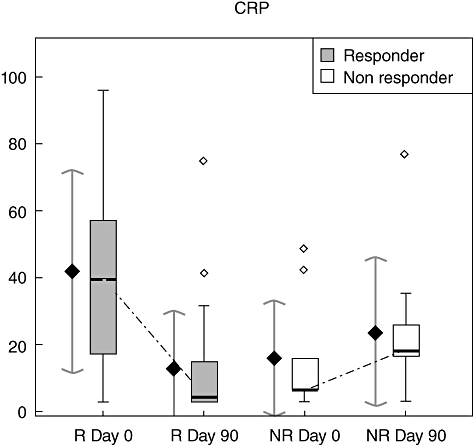
Cytokine level evolution during etanercept treatment in the responder and non-responder groups. Box-plots are on the right and representations of the mean (±standard deviation intervals) on the left in each group at each time (CRP, C-reactive protein).
At treatment initiation the CRP serum level was higher in the responder group, but not statistically significant (41·84 ± 30·25 mg/ml in the responder group versus 15·98 ± 17·14 mg/ml; P = 0·02) (Fig. 2). At day 90, comparison between the responder and non-responder groups showed no significant difference in CRP serum level [mean CRP levels at day 90 were 12·11 ± 16·54 and 13·26 ± 14·24 for the responder and non-responder groups respectively (P = 0·43)]. During etanercept treatment, the CRP level decreased significantly in the responder group [mean CRP levels were 41·84 ± 30·25 mg/ml at day 0, versus 13·07 ± 17·02 mg/ml at day 90 (P < 0·001; the threshold P-value corrected by FDR approach was 0·005)]. In contrast, in the non-responder group the CRP level increased during treatment, although not significantly (Fig. 2) [mean CRP in the non-responder group were 8·9 ± 13·36 mg/ml at day 0 and 13·26 ± 14·24 mg/ml at day 90 (P = 0·2)].
Biological parameters combination to identify responders to etanercept
We then performed a logistic regression analysis of all biological parameters at treatment initiation. The combination of the two parameters CRP and EGF was informative to identify responders and non-responders (sensitivity: 91·3% and specificity: 70%, accuracy: 85%). The combination of the above parameters levels could be used as a predictor of responsiveness to TBA with good sensitivity and specificity.
Discussion
Anti-TNF-α therapy does not benefit all patients. About 20–40% of RA patients are non-responders. In order to implement anti-TNF-α therapy in RA, it is important to identify the predictors for responsiveness to this treatment. The purpose of our study was to determine a cytokine profile at baseline predictive of response to etanercept at 3 months in RA.
Several studies have analysed predictive parameters. Genetic markers have been identified as polymorphisms in the TNF-α gene. In Korean RA patients, Kang et al. showed that the –857T allele of the TNF-α promoter, compared with the –857C allele, is associated with a good response to etanercept [15]. In Caucasian RA patients several authors have shown that the –308G allele of the TNF-α promoter, compared with the –308A allele, is associated with a better clinical response to anti-TNF-α therapy [16–20]. Demographic, clinical and radiological factors predicting response to anti-TNF-α therapy have also been studied. Disease duration, age of onset, baseline DAS 28 and baseline number of radiographic erosions were not shown to be correlated with anti-TNF-α therapy response in RA [21–23]. On the other hand, a low level of baseline disability (HAQ) was predictive of a response to anti-TNF-α therapy in RA [24,25]. Many laboratory parameters have been investigated. The available set of biological parameters used usually for RA diagnosis or prognosis [such as baseline CRP, human leucocyte antigen, RFs and anti-CCP] failed to predict a response to anti-TNF-α therapy [9,21,22,24,26–28]. However, Buch et al. showed that failure to suppress the CRP at week 2 identified the majority of patients who were non-responders at week 12 [27]. Therefore, to enable such a prediction, global approaches based on proteomics, genomics or transcriptomics have been considered recently [29–33].
The objectives of our study were to evaluate PBAT in a clinical application, and to determine a cytokine profile at baseline predictive of etanercept response at 3 months in RA. Three months of treatment was chosen as the end-point of our study, as recommended recently by international experts [34], because the objective of an efficient RA treatment is a rapid response. Two important points of clinical relevance arise from this study.
First, we evaluated the feasibility and the relevance of PBAT using a clinical approach in monitoring a treatment response in RA patients. PBAT is a promising and innovative technology. The chip system allows several tests to be performed simultaneously without dividing the original patient sample. In this study we used the technology Evidence® (Randox Laboratories, Crumlin, UK), which is a multi-analyte biochip array using a charge-coupled camera (CCD) and imaging system. The systems have been developed to allow simultaneous measurement of up to 25 analyses with a single drop of patient sample on a biochip [35]. A few studies have reported data concerning proteomics in a clinical approach. PBAT was evaluated for the diagnosis of acute coronary syndrome [36], and more recently for the prediction of mortality in haemodialysis patients by the use of cytokines [37].
Secondly, we identified a baseline cytokine profile predictive of a good response to etanercept at 3 months in RA. High serum levels of two proinflammatory cytokines, MCP-1 and EGF, were predictive of a TBA treatment response. Moreover, the combination of the two parameters CRP and EGF was informative to identify responders and non-responders with good sensitivity (91·3%), specificity (70%) and accuracy (85%).
Monocyte chemoattractant protein-1–3 are members of the CC family and may be induced by inflammatory cytokines [38,39]. TNF-α-dependent expression of MCP-1 has been described previously [33,40]. MCP-1 is regulated directly by the TNF-α/nuclear factor-κB pathway, whether in RA [41] or in another context [42,43]. Several research groups have detected MCP-1 in the synovial fluid of RA patients, and these concentrations were markedly higher than those seen in other rheumatic diseases, including osteoarthritis [40,44]. Similarly, treatment of RA patients with TBAs reduced MCP-1 expression significantly in the joint and tended to reduce the plasma concentrations of these chemokines [45,46], as did corticosteroids [47].
High amounts of EGF were detectable in the synovial fluid of RA patients and influenced the growth of rheumatoid synovial cells [48]. Klooster and colleagues showed that TNF-α and EGF have additive effects on chondrocyte function [49].
Several studies have examined the effect of TBA in RA on inflammation proteins, cytokines and growth factors [13,21,45,46]. Serum level decrease of several cytokines, such as IL-6, IL-8, MCP-1, VEGF, IL-1, regulated upon activation normal T cell expressed and secreted, intercellular adhesion molecule-1, E-selectin and vascular cell adhesion molecule-1, has been described after infliximab therapy in RA [14,45,46,50], but no parameters predicting a response to TBA have been identified to date.
Our data suggest that the combination of high CRP with high EGF is sufficiently sensitive and specific to identify responders. EGF and MCP-1 serum levels could also be useful predictive biological parameters to treatment response, which has not been shown previously. However, this result needs to be confirmed in a larger study in order to increase the number of patients in the non-responder group.
The circulating TNF-α levels have been shown not to correlate with a response to infliximab [21,22,51]. In our study, the analysis of other proinflammatory cytokine serum levels, such as TNF-α, IL-1a, IL-1b, IL-6, IL-2, IL-8, IFN-γ and VEGF, did not predict either TBA treatment response or levels of anti-inflammatory cytokines, such as IL-4 and IL-10. Even if TNF-α, IL-1a and IL-1b are key cytokines involved in the RA inflammatory process [52], their serum level assessment is not a useful tool to predict TBA response at treatment initiation or to follow the treatment.
Conclusion
This study has evaluated PBAT to identify responders in RA patients treated with etanercept, a soluble receptor TBA. We identified a responder cytokine profile at the start of the treatment: high serum levels of two proinflammatory cytokines (MCP-1 and EGF) and CRP.
The combined levels of a small set of 12 cytokines have provided, for the first time, a tool for the prediction of etanercept efficacy in patients with long-standing and very active RA. Future studies should identify additional cytokines whose profile changes correlate with responsiveness to other TBAs or biologics. Ultimately, we anticipate that a small series of parallel tests for drug-specific combinations of proteins, as quantified on proteomic biochip arrays, should allow physicians to select the most appropriate treatment for every RA patient, with the resultant beneficial eradication of the non-responder or moderate responder phenotypes. This study has evaluated the feasability and relevance of PBAT in predicting treatment responses to a TBA in RA.
Acknowledgments
We thank Christopher Payne (Department of Genome Sciences, University of Washington School of Medicine, Box 357730, 1705 Northeast Pacific Street, Seattle, WA 98195, USA) for his expert assistance in the re-reading of the manuscript. This work was supported in part by Wyeth for the protein biochip arrays.
Supplementary material
The following supplementary material is available for this article online:
Comparison of cytokine in the responder group between day 0 and day 90, and between responders and non-responders at day 90. Cytokine serum levels were compared: (1) at day 0 and day 90 in the responder group using the paired Mann–Whitney U-test. *The threshold p1-value corrected by the false discovery rate (FDR) approach was lower than 0·05, and any variable was retained; (2) at day 90 between responders and non-responders patients using the two-sided Kruskal–Wallis test. **The threshold p2-value corrected by the FDR approach was 0·008
This material is available as part of the online article from http://www.blackwell-synergy..com/doi/abs/10.1111/j.1365-2249.2008.03691.x
(This link will take you to the article abstract).
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Bathon JM, Martin RW, Fleischmann RM, et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N Engl J Med. 2000;343:1586–93. doi: 10.1056/NEJM200011303432201. [DOI] [PubMed] [Google Scholar]
- 2.Keystone EC, Kavanaugh AF, Sharp JT, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50:1400–11. doi: 10.1002/art.20217. [DOI] [PubMed] [Google Scholar]
- 3.Maini RN, Breedveld FC, Kalden JR, et al. Sustained improvement over two years in physical function, structural damage, and signs and symptoms among patients with rheumatoid arthritis treated with infliximab and methotrexate. Arthritis Rheum. 2004;50:1051–65. doi: 10.1002/art.20159. [DOI] [PubMed] [Google Scholar]
- 4.Klareskog L, van der Heijde D, de Jager JP, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363:675–81. doi: 10.1016/S0140-6736(04)15640-7. [DOI] [PubMed] [Google Scholar]
- 5.Smolen JS, Han C, Bala M, et al. Evidence of radiographic benefit of treatment with infliximab plus methotrexate in rheumatoid arthritis patients who had no clinical improvement: a detailed subanalysis of data from the anti-tumor necrosis factor trial in rheumatoid arthritis with concomitant therapy study. Arthritis Rheum. 2005;52:1020–30. doi: 10.1002/art.20982. [DOI] [PubMed] [Google Scholar]
- 6.den Broeder A, van de Putte L, Rau R, et al. A single dose, placebo controlled study of the fully human anti-tumor necrosis factor-alpha antibody adalimumab (D2E7) in patients with rheumatoid arthritis. J Rheumatol. 2002;29:2288–98. [PubMed] [Google Scholar]
- 7.Ang HT, Helfgott S. Do the clinical responses and complications following etanercept or infliximab therapy predict similar outcomes with the other tumor necrosis factor-alpha antagonists in patients with rheumatoid arthritis? J Rheumatol. 2003;30:2315–18. [PubMed] [Google Scholar]
- 8.Haraoui B, Keystone EC, Thorne JC, et al. Clinical outcomes of patients with rheumatoid arthritis after switching from infliximab to etanercept. J Rheumatol. 2004;31:2356–9. [PubMed] [Google Scholar]
- 9.Lequerre T, Jouen F, Brazier M, et al. Autoantibodies, metalloproteinases and bone markers in rheumatoid arthritis patients are unable to predict their responses to infliximab. Rheumatology (Oxf) 2007;46:446–53. doi: 10.1093/rheumatology/kel262. [DOI] [PubMed] [Google Scholar]
- 10.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 11.Dupuy AM, Badiou S, Descomps B, Cristol JP. Immunoturbidimetric determination of C-reactive protein (CRP) and high-sensitivity CRP on heparin plasma. Comparison with serum determination. Clin Chem Lab Med. 2003;41:948–9. doi: 10.1515/CCLM.2003.144. [DOI] [PubMed] [Google Scholar]
- 12.Benjamini Y, Hochberg Y. Controlling the false discovery rate – a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 13.Charles P, Elliott MJ, Davis D, et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J Immunol. 1999;163:1521–8. [PubMed] [Google Scholar]
- 14.Macias I, Garcia-Perez S, Ruiz-Tudela M, Medina F, Chozas N, Giron-Gonzalez JA. Modification of pro- and antiinflammatory cytokines and vascular-related molecules by tumor necrosis factor – a blockade in patients with rheumatoid arthritis. J Rheumatol. 2005;32:2102–8. [PubMed] [Google Scholar]
- 15.Kang CP, Lee KW, Yoo DH, Kang C, Bae SC. The influence of a polymorphism at position–857 of the tumour necrosis factor alpha gene on clinical response to etanercept therapy in rheumatoid arthritis. Rheumatology (Oxf) 2005;44:547–52. doi: 10.1093/rheumatology/keh550. [DOI] [PubMed] [Google Scholar]
- 16.de Vries N, Tak PP. The response to anti-TNF-α treatment: gene regulation at the bedside. Rheumatology (Oxf) 2005;44:705–7. doi: 10.1093/rheumatology/keh662. [DOI] [PubMed] [Google Scholar]
- 17.Lee YH, Rho YH, Choi SJ, Ji JD, Song GG. Associationof TNF-alpha -308 G/A polymorphism with responsiveness to TNF-alpha-blockers in rheumatoid arthritis: a meta-analysis. Rheumatol Int. 2006;27:157–61. doi: 10.1007/s00296-006-0175-7. [DOI] [PubMed] [Google Scholar]
- 18.Mugnier B, Balandraud N, Darque A, Roudier C, Roudier J, Reviron D. Polymorphism at position -308 of the tumor necrosis factor alpha gene influences outcome of infliximab therapy in rheumatoid arthritis. Arthritis Rheum. 2003;48:1849–52. doi: 10.1002/art.11168. [DOI] [PubMed] [Google Scholar]
- 19.Seitz M, Wirthmuller U, Moller B, Villiger PM. The -308 tumour necrosis factor-alpha gene polymorphism predicts therapeutic response to TNFalpha-blockers in rheumatoid arthritis and spondyloarthritis patients. Rheumatology (Oxf) 2007;46:93–6. doi: 10.1093/rheumatology/kel175. [DOI] [PubMed] [Google Scholar]
- 20.Guis S, Balandraud N, Bouvenot J, et al. Influence of -308 A/G polymorphism in the tumor necrosis factor alpha gene on etanercept treatment in rheumatoid arthritis. Arthritis Rheum. 2007;57:1426–30. doi: 10.1002/art.23092. [DOI] [PubMed] [Google Scholar]
- 21.Mugnier B, Roudier J. Factors predicting responsiveness to anti-TNFalpha therapy in patients with rheumatoid arthritis: using biotherapies rationally. Joint Bone Spine. 2004;71:91–4. doi: 10.1016/j.jbspin.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Maini R, St Breedveld EW, Clair F, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999;354:1932–9. doi: 10.1016/s0140-6736(99)05246-0. [DOI] [PubMed] [Google Scholar]
- 23.Wolbink GJ, Voskuyl A, Lems WF, et al. Relationship between serum trough infliximab levels, pretreatment C reactive, protein levels, and clinical response to infliximab treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64:704–7. doi: 10.1136/ard.2004.030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyrich KL, Watson KD, Silman AJ, Symmons DP. Predictors of response to anti-TNF-alpha therapy among patients with rheumatoid arthritis: results from the British Society for Rheumatology Biologics Register. Rheumatology (Oxf) 2006;45:1558–65. doi: 10.1093/rheumatology/kel149. [DOI] [PubMed] [Google Scholar]
- 25.Weaver AL, Lautzenheiser RL, Schiff MH, et al. Real-world effectiveness of select biologic and DMARD monotherapy and combination therapy in the treatment of rheumatoid arthritis: results from the RADIUS observational registry. Curr Med Res Opin. 2006;22:185–98. doi: 10.1185/030079905X65510. [DOI] [PubMed] [Google Scholar]
- 26.Lequerre T, Vittecoq O, Le Loet X. Comments about the editorial by Benedicte Mugnier and Jean Roudier entitled ‘Factors predicting responsiveness to anti-TNFalpha therapy in patients with rheumatoid arthritis: using biotherapies rationally’. Joint Bone Spine. 2005;72:346–7. doi: 10.1016/j.jbspin.2005.01.004. author reply 7–8. [DOI] [PubMed] [Google Scholar]
- 27.Buch MH, Seto Y, Bingham SJ, et al. C-reactive protein as a predictor of infliximab treatment outcome in patients with rheumatoid arthritis: defining subtypes of nonresponse and subsequent response to etanercept. Arthritis Rheum. 2005;52:42–8. doi: 10.1002/art.20711. [DOI] [PubMed] [Google Scholar]
- 28.Wolbink GJ, Groot E DE, Lems WF, Nurmohamed MT, Tak T, Dijmans BAC. Relationship between serum levels of infliximab, C-reactive protein and clinical response to infliximab patients with rheumatoid arthritis. Ann Rheum Dis. 2003;62(SI):98. doi: 10.1136/ard.2004.030452. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drynda S, Ringel B, Kekow M, et al. Proteome analysis reveals disease-associated marker proteins to differentiate RA patients from other inflammatory joint diseases with the potential to monitor anti-TNFalpha therapy. Pathol Res Pract. 2004;200:165–71. doi: 10.1016/j.prp.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 30.Jarvis JN, Centola M. Gene-expression profiling: time for clinical application? Lancet. 2005;365:199–200. doi: 10.1016/S0140-6736(05)17754-X. [DOI] [PubMed] [Google Scholar]
- 31.Bargnoux AS, Bonardet A, Chong G, et al. Evaluation of an immunoassay (Abbott-IMX Analyzer) allowing routine determination of sirolimus: comparison with LC-MS method. Transplant Proc. 2006;38:2352–3. doi: 10.1016/j.transproceed.2006.06.127. [DOI] [PubMed] [Google Scholar]
- 32.Dupuy AM, Lehmann S, Cristol JP. Protein biochip systems for the clinical laboratory. Clin Chem Lab Med. 2005;43:1291–302. doi: 10.1515/CCLM.2005.223. [DOI] [PubMed] [Google Scholar]
- 33.Lequerre T, Gauthier-Jauneau AC, Bansard C, et al. Gene profiling in white blood cells predicts infliximab responsiveness in rheumatoid arthritis. Arthritis Res Ther. 2006;8:R105. doi: 10.1186/ar1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furst DE, Breedveld FC, Kalden JR, et al. Updated consensus statement on biological agents for the treatment of rheumatic diseases, 2006. Ann Rheum Dis. 2006;65(Suppl 3):iii2–15. doi: 10.1136/ard.2006.061937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fitzgerald SP, Lamont JV, McConnell RI, Benchikh el O. Development of a high-throughput automated analyzer using biochip array technology. Clin Chem. 2005;51:1165–76. doi: 10.1373/clinchem.2005.049429. [DOI] [PubMed] [Google Scholar]
- 36.Serio F DI, Amodio G, Ruggieri E, et al. Proteomic approach to the diagnosis of acute coronary syndrome: preliminary results. Clin Chim Acta. 2005;357:226–35. doi: 10.1016/j.cccn.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 37.Badiou S, Cristol JP, Jaussent I, et al. Fine-tuning of the prediction of mortality in hemodialysis patients by use of cytokine proteomic determination. Clin J Am Soc Nephrol. 2008;3:423–30. doi: 10.2215/CJN.02010507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brieland JK, Flory CM, Jones ML, et al. Regulation of monocyte chemoattractant protein-1 gene expression and secretion in rat pulmonary alveolar macrophages by lipopolysaccharide, tumor necrosis factor-alpha, and interleukin-1 beta. Am J Respir Cell Mol Biol. 1995;12:104–9. doi: 10.1165/ajrcmb.12.1.7811465. [DOI] [PubMed] [Google Scholar]
- 39.DeForge LE, Kenney JS, Jones ML, Warren JS, Remick DG. Biphasic production of IL-8 in lipopolysaccharide (LPS)-stimulated human whole blood. Separation of LPS- and cytokine-stimulated components using anti-tumor necrosis factor and anti-IL-1 antibodies. J Immunol. 1992;148:2133–41. [PubMed] [Google Scholar]
- 40.Koch AE, Kunkel SL, Harlow LA, et al. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest. 1992;90:772–9. doi: 10.1172/JCI115950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taberner M, Scott KF, Weininger L, Mackay CR, Rolph MS. Overlapping gene expression profiles in rheumatoid fibroblast-like synoviocytes induced by the proinflammatory cytokines interleukin-1 beta and tumor necrosis factor. Inflamm Res. 2005;54:10–16. doi: 10.1007/s00011-004-1315-8. [DOI] [PubMed] [Google Scholar]
- 42.Tian B, Nowak DE, Jamaluddin M, Wang S, Brasier AR. Identification of direct genomic targets downstream of the nuclear factor-kappaB transcription factor mediating tumor necrosis factor signaling. J Biol Chem. 2005;280:17435–48. doi: 10.1074/jbc.M500437200. [DOI] [PubMed] [Google Scholar]
- 43.Zhou A, Scoggin S, Gaynor RB, Williams NS. Identification of NF-kappa B-regulated genes induced by TNFalpha utilizing expression profiling and RNA interference. Oncogene. 2003;22:2054–64. doi: 10.1038/sj.onc.1206262. [DOI] [PubMed] [Google Scholar]
- 44.Harigai M, Hara M, Yoshimura T, Leonard EJ, Inoue K, Kashiwazaki S. Monocyte chemoattractant protein-1 (MCP-1) in inflammatory joint diseases and its involvement in the cytokine network of rheumatoid synovium. Clin Immunol Immunopathol. 1993;69:83–91. doi: 10.1006/clin.1993.1153. [DOI] [PubMed] [Google Scholar]
- 45.Taylor PC, Peters AM, Paleolog E, et al. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43:38–47. doi: 10.1002/1529-0131(200001)43:1<38::AID-ANR6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 46.Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–96. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- 47.Haynes PP, Youssef DR, Triantafillou S, et al. Effects of pulse methylprednisolone on inflammatory mediators in peripheral blood, synovial fluid, and synovial membrane in rheumatoid arthritis. Arthritis Rheum. 1997;40:1400–8. doi: 10.1002/art.1780400807. [DOI] [PubMed] [Google Scholar]
- 48.Satoh K, Kikuchi S, Sekimata M, Kabuyama Y, Homma MK, Homma Y. Involvement of ErbB-2 in rheumatoid synovial cell growth. Arthritis Rheum. 2001;44:260–5. doi: 10.1002/1529-0131(200102)44:2<260::AID-ANR42>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 49.Klooster AR, Bernier SM. Tumor necrosis factor alpha and epidermal growth factor act additively to inhibit matrix gene expression by chondrocyte. Arthritis Res Ther. 2005;7:R127, 38. doi: 10.1186/ar1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klimiuk PA, Sierakowski S, Domyslawska I, Chwiecko J. Regulation of serum chemokines following infliximab therapy in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2006;24:529–33. [PubMed] [Google Scholar]
- 51.Hrycaj P, Korczowska I, Lacki JK. Infliximab in rheumatoid arthritis: can the response be predicted? Ann Rheum Dis. 2003;62(SI):403. Abstract. [Google Scholar]
- 52.Moller B, Villiger PM. Inhibition of IL-1, IL-6, and TNF-alpha in immune-mediated inflammatory diseases. Springer Semin Immunopathol. 2006 doi: 10.1007/s00281-006-0012-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of cytokine in the responder group between day 0 and day 90, and between responders and non-responders at day 90. Cytokine serum levels were compared: (1) at day 0 and day 90 in the responder group using the paired Mann–Whitney U-test. *The threshold p1-value corrected by the false discovery rate (FDR) approach was lower than 0·05, and any variable was retained; (2) at day 90 between responders and non-responders patients using the two-sided Kruskal–Wallis test. **The threshold p2-value corrected by the FDR approach was 0·008