

Abstract
Primary neonatal immune responses to infection or vaccines are weak when compared with those of adults. In addition, memory responses of neonatally primed animals may be absent, weak or T helper type 2 (Th2)-biased. Respiratory syncytial virus (RSV) is an important pathogen of human infants and infection during the neonatal period has been linked to the development of asthma in later life. Here we report that acute intranasal infection of neonatal mice with RSV induces significant RSV-specific antibody and CD8 T cell responses. These responses were boosted after RSV rechallenge during adulthood, demonstrating the establishment of memory after neonatal priming. Primary infection during neonatal life was associated, following rechallenge, with limited viral replication in the lung. Recall responses of both spleen and lymph node cells from neonatally primed and adult-primed mice were associated with interferon-γ secretion, indicative of a Th1-type response. However, interleukin (IL)-4 and IL-5 secretion were enhanced only in spleen and lymph node cells from neonatally primed mice. Rechallenge of neonatally primed mice was also associated with increased concentrations of chemokines monocyte chemoattractant protein-1, macrophage inflammatory protein-1α and regulated upon activation normal T cell expressed and secreted in the lung. These may play a role in the enhanced inflammatory cell recruitment and immunopathology induced following RSV reinfection. Our results demonstrate therefore that immunity to RSV can be established during neonatal life and, importantly, that the quality of the subsequent response is dependent upon the age of first infection.
Keywords: antibody responses, neonatal, spleen/lymph nodes
Introduction
The immune system of murine neonates is developmentally immature and responses generated during this period are typically weak, delayed and often poorly protective. This immaturity makes neonatal animals highly susceptible to infection. The relative immunodeficiency of neonatal mice was demonstrated originally in the late 1950s, when it was shown that during the period around birth mice were easily made tolerant to the transplantation of semi-allogeneic (F1) cells [1]. Both quantitative and qualitative factors contribute to this neonatal immunodeficiency. Neonatal animals have relatively low numbers of immune cells including B cells, T cells and antigen-presenting cells (APC), and the populations of B and T cells are a mixture of cells derived from both adult and fetal haematopoietic precursors [2]. In addition, functional differences have been observed between adult and neonatal immune cells (reviewed in [3]). It has been observed frequently that the CD4 T cell responses made by neonatal mice are biased towards those of the T helper 2 (Th2) type (for reviews see [2] and [4]) and that neonatal animals are defective in their ability to generate effective Th1-mediated responses, and this polarization results in impaired cellular immune responses. Adaptive CD4 T cell responses can be described as being of Th1 or Th2 types based on cytokine secretion profile: Th2 responses, characterized by the secretion of interleukin (IL)-4, IL-5 and IL-10, are required for effective humoral immunity and are implicated in allergic responses, whereas Th1 responses, which involve typically the production of interferon (IFN)-γ, are important for cell-mediated immune responses. Hence the reported Th2 bias in neonates is of particular consequence when considering protective immunity to intracellular pathogens, including some viruses, that depend on effective Th1 responses. Although it has been demonstrated that neonatal animals can make Th1 responses in the presence of strong Th1-promoting agents (reviewed in [5,6]), there is a considerable amount of in vitro data that supports the hypothesis that exposure to antigen during the neonatal period leads frequently to Th2-biased secondary responses to the same antigen, including alloantigens [7], conventional vaccines and live or attenuated virus [8,9].
In recent years, the idea that neonatal mice are tolerized more easily or are hyporesponsive has been challenged by a growing number of workers who have demonstrated that, under certain conditions, neonatal animals are competent to generate protective immune responses, including those of the Th1 type. Thus it is possible to induce mature adult-like neonatal cytotoxic T lymphocyte (CTL) responses against alloantigens [10,11], viruses [12] and conventional protein antigens [13], using appropriate Th1-inducing adjuvants, DNA vaccines [9], manipulations of dose or in the presence of adult APC.
Respiratory syncytial virus (RSV) is the major cause of lower respiratory tract infection in infancy and early childhood. Severe lower respiratory tract infection and bronchiolitis occurs in approximately 5% of infant infections and bronchiolitis appears to be largely a result of immune hyperresponsiveness [14]. RSV-induced bronchiolitis has also been associated with the predisposition to develop asthma and wheeze during later life, although the evidence for this comes largely from epidemiological studies [15], and a causal relationship has not been proven. It has been suggested that a Th2-biased memory response following neonatal exposure to RSV may influence or direct lung immune responses to unrelated antigens to an allergic phenotype in later life (for review see [16]). In support of this hypothesis, RSV infection of neonatal mice was shown to establish the subsequent pattern of Th cell responses, with neonatally primed mice having a greater percentage of IL-4-expressing CD4+ cells in the lung and enhanced lymphocyte and granulocyte recruitment to the lung, following secondary RSV challenge [17].
In the mouse model of RSV infection a selective recruitment of inflammatory cells is observed, driven presumably by chemotactic gradients. There is extensive evidence for the involvement of chemokines in various inflammatory responses induced in the lung and RSV has been shown to induce the up-regulation of chemokine production, including monocyte chemoattractant protein (MCP)-1, regulated upon activation normal T cell expressed and secreted (RANTES) and macrophage inflammatory protein (MIP)-1α in both mouse models [18,19] and human studies [20].
Here we show that mice primed at 7 days of age with live RSV made enhanced antibody and CD8 T cell responses when rechallenged with RSV as adults. The secondary Th cell response mounted in both spleen and lymph nodes following neonatal priming showed strong production of IFN-γ, but was also associated with the production of measurable quantities of Th2 cytokines IL-4 and IL-5 not found after adult priming. In addition, we report increased concentrations of inflammatory chemokines MCP-1, MIP-1α, RANTES and of tumour necrosis factor (TNF) in the lungs of neonatally primed mice compared with concentrations found in unprimed mice or mice primed as adults.
Materials and method
Mice and RSV
Adult BALB/c mice were obtained from Charles River Laboratories (UK) Ltd (Tranent, East Lothian, UK). Breeding females were monitored during gestation and date of birth of litters was recorded as day 0. RSV-A2 strain was grown in Vero cells and viral titre in cell-free supernatants determined by plaque assay. Adult or 7-day-old (neonatal) mice were infected intranasally with 3·75 × 104 plaque-forming units (pfu) per g body weight: adult mice were anaesthetized lightly by intraperitoneal injection of ketamine/xylazine and infected intranasally with RSV in 25 μl volume and neonatal mice were infected, without anaesthesia, with RSV in 5 μl volume. Mice were reinfected as adults, as described above, 7 weeks after primary infection. All animal studies were carried out in accordance with UK Home Office regulations for animal care and use.
Respiratory syncytial virus-specific antibody enzyme-linked immunosorbent assay
Sucrose density gradient purified RSV was coated onto Nunc-Immuno Maxisorp plates (Nunc, Roskilde, Denmark) at a predetermined concentration. RSV-specific serum antibody was detected with alkaline phosphatase-conjugated goat anti-mouse immunoglobulin M (IgM), IgG, IgG1 or IgG2a-specific antibodies (Southern Biotechnology Associates, Birmingham, AL, USA) and developed with the Bio-Rad alkaline phosphatase substrate kit (Bio-Rad, Hemel Hempstead, UK). Titres (end-point) are expressed as the reciprocal of the highest dilution giving an optical density value greater than three times the value of the lower plateau of the dilution curve.
Tissue collection and cell preparation
At specified times following infection, mice were terminally anaesthetized, exsanguinated via axillary vessels and serum was collected. The right lung from each mouse was snap-frozen in liquid nitrogen and stored at −80°C prior to viral titre and inflammatory cytokine/chemokine analysis. Left lungs, spleens and mediastinal lymph nodes were harvested from mice and stored in complete RPMI-1640 (Gibco Invitrogen, Paisley, UK) supplemented with 10% heat-inactivatedfetal calf serum (FCS), 25 mM HEPES buffer, 5 × 10−5 M 2-mercaptoethanol, 1% non-essential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin and 1 mM sodium pyruvate, on ice until cell suspensions were prepared by pressing gently between frosted slides. RBC were lysed and cells were washed and resuspended in complete RPMI-1640 (as above).
Enzyme-linked immunospot
Ninety-six-well filtration plates (MultiScreenTM-IP; Millipore, Watford, UK) were coated with 0·75 μg/well purified rat anti-mouse IFN-γ (clone R4-6A2) or 0·75 μg/well anti-mouse IL-4 (clone 11B11). Coated plates were washed and blocked with RPMI-1640 containing 10% FCS before the addition of cells. For detection of RSV-specific IFN-γ-secreting CD8 T cells, serial dilutions of responder cells (in duplicate) in complete RPMI-1640 were added to wells, followed by stimulation with RSV matrix protein (M2)82−90 class I peptide at 10 μg/ml and mouse rIL-2 (Biosource International, Nivelles, Belgium) at 5 U/ml. Cultures were incubated for 16 h at 37°C in 5% CO2. For detection of RSV-specific IL-4-secreting T cells, serial dilutions of responder cells were incubated with CD4- and CD8-depleted syngeneic adult splenocytes which had been infected with RSV (0·1 pfu/cell) and subjected to 3000 rad using a caesium irradiator (RSV-infected splenocytes). Control wells contained stimulator cells alone or responder cells alone ± IL-2. Cultures were incubated for 16 h as above. After incubation, plates were washed with phosphate-buffered saline (PBS), followed by incubation with biotinylated rat anti-mouse IFN-γ (clone XMG1·2) or rat anti-mouse IL-4 (clone BVD4-1D11) monoclonal antibody (mAb as appropriate), diluted in PBS [all antibodies used for enzyme-linked immunospot (ELISPOT) were obtained from BD Biosciences Pharmingen, Cowley, Oxfordshire, UK; unless specified otherwise]. Biotinylated antibodies were detected by the addition of alkaline phosphatase-conjugated goat anti-biotin antibodies (Vector Laboratories Ltd, Orton Southgate, UK), and plates were developed by the addition of alkaline phosphatase substrate (Bio-Rad). Plates were dried and spots counted. RSV-specific spots were determined by subtracting spots in control wells containing responder cells plus medium/IL-2 alone from the number of spots in wells containing responder cells plus peptide/IL-2 (for IFN-γ ELISPOT) and by subtracting spots in wells containing responder cells plus medium from spots in wells containing responder cells plus RSV-infected stimulator cells (IL-4 ELISPOT). Irradiated RSV-infected splenocytes produced no IL-4.
Flow cytometry and tetramer analysis
For analysis of RSV-specific CD8+ T cells, lung cells prepared as described above were resuspended in PBS, and incubated in the dark for 30 min at room temperature simultaneously with the phycoerythrin (PE)-conjugated RSV major histocompatibility complex class I tetramer H-2 kd/PE containing the RSV matrix protein (M2)82−90 peptide (iTAgTM Beckman Coulter, Villepinte, France) and with fluorescein isothiocyanate-conjugated anti-mouse CD8 (clone 53-6·7; BD Biosciences Pharmingen). Cells were washed with PBS and fixed in PBS−0·5% formalin prior to analysis. Flow cytometry was performed on a dual laser fluorescence activated cell sorter (FACSCalibur) and analysed using WinMDI software. Analysis was performed on cells gated in a viable lymphocyte gate (10 000 events per sample).
In vitro restimulation for Th1/Th2 cytokine analysis
Spleen or mediastinal lymph node cells (3 × 106/ml) were stimulated with CD4 and CD8 depleted syngeneic adult splenocytes (3 × 106/ml) which had been infected with RSV (0·1 pfu/cell), and subjected to 3000 rad using a caesium irradiator. Control responder cells were set up with medium alone. Cell cultures were set up in 48-well tissue culture plates and incubated for 72 h, after which supernatant was collected and stored at −80°C prior to analysis of Th1 and Th2 cytokines. Supernatants were diluted 1 : 1 or 1 : 4 with PBS and analysed using the BDTM cytometric bead array (CBA) Th1/Th2 cytokine kit (BD Biosciences).
Lung virus titration
Individual lungs, snap-frozen previously in liquid nitrogen, were homogenized on ice in 500 μl of serum-free RPMI-1640. Samples were centrifuged at 2000 g for 5 min at 4°C and 200 μl of serial dilutions of homogenate were added to BSC1 monolayers, plated previously in 24-well tissue culture plates. Cultures were incubated for 2 h at 33°C in 5% CO2 before the addition of complete RPMI-1640–10%FCS/2% methylcellulose. After 5 days' incubation at 33°C in 5% CO2, plates were fixed in 80% methanol, blocked with PBS−5% skimmed milk powder for 1 h at 37°C, after which virus-specific plaques were detected by incubation with a cocktail of mouse anti-RSV mAb (clones 131-2A, 131-2G, 130-H12, 130-5F, 130-8F), followed by alkaline phosphatase-conjugated anti-mouse IgG and alkaline phosphatase substrate buffer (Bio-Rad). A pretitred RSV stock solution was used as positive control.
Assay for inflammatory cytokines
Supernatants from the above lung homogenates were stored at −80°C prior to analysis of inflammatory cytokines. Supernatants were diluted 1 : 1 with PBS and analysed using the BDTM CBA mouse inflammation kit (BD Biosciences). RANTES, MIP-1α and eotaxin in 1 : 5 diluted lung homogenates were analysed using enzyme-linked immunosorbent assay (ELISA) kits (R&D DuoSet®, Oxon, Abingdon, UK) according to the manufacturer's instructions.
Statistical analysis
Data from experimental groups were compared using Student's t-test or Mann–Whitney rank sum test. Statistical significance was determined by P < 0·05.
Results
Neonatal infection with RSV primes for an enhanced response on secondary challenge
To evaluate the capacity of neonatal mice to make antibody responses to primary RSV infection, 7-day-old mice were infected intranasally with RSV and RSV-specific serum antibodies were measured by ELISA at various times after infection. Following infection, the serum of neonatal mice contained low but detectable amounts of RSV-specific IgM and IgG antibodies (Fig. 1a), with amounts of antibodies of both isotypes lower than those measured in the sera of mice infected as adults. In addition, the kinetics of the IgG responses were different in adults and neonates. In adult mice the peak of response was at approximately day 14, whereas in neonatal mice the peak was not seen until 28 days after infection. Comparative analysis of the IgG2a and IgG1 subclasses, selected as markers of Th1 and Th2 responses, respectively, demonstrated that for both adult and neonatal infection the predominant RSV-specific antibody isotype produced at 3 weeks after infection was IgG2a (Fig. 1b). IgG2a was also the predominant anti-RSV IgG isotype produced at 1, 2 and 4 weeks after infection in both adults and neonates (data not shown).
Fig. 1.
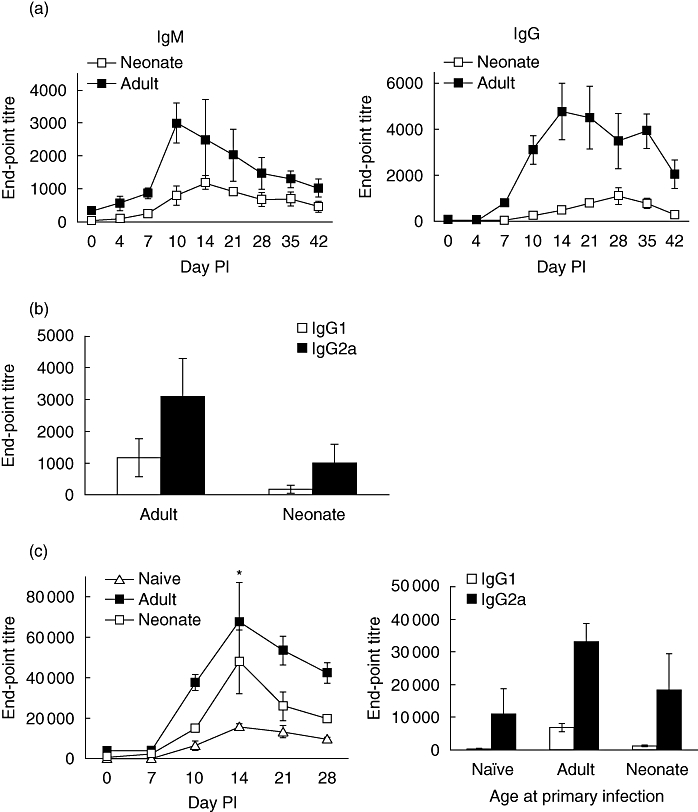
Primary respiratory syncytial virus (RSV) infection induces weak antibody responses in neonatal mice, but primes for enhanced responses following subsequent RSV infection. Mice were infected with RSV as adults (adult) or neonates (neonate) and RSV-specific IgM and IgG were quantified by enzyme-linked immunosorbent assay (ELISA) (a). Anti-RSV serum IgG1 and IgG2a responses were measured 21 days after primary adult or neonatal infection (b). Seven weeks after primary infection of adult or neonatal mice, animals were re-challenged with RSV (c). A third group of mice, previously uninfected (naive), were infected similarly with the same dose of RSV. RSV-specific IgG was quantified by ELISA at indicated time-points after infection, and RSV-specific IgG1 and IgG2a were quantified at 21 days after infection. Values represent mean values from a minimum of five mice ± standard deviation. Differences between unprimed adult and neonatally primed groups are shown; *P < 0·05 by Mann–Whitney rank sum test. The data shown are representative of two independent experiments.
Adult and 7-day-old mice were then infected with RSV and reinfected 7 weeks later to compare the memory responses of both groups. Figure 1c demonstrates that despite the weak antibody response of neonatal mice to primary infection, on secondary challenge, neonatally primed mice made an enhanced IgG response that was more rapid and of significantly greater magnitude than responses induced in adult mice during a primary infection (P < 0·05 at day 14). As for primary responses, it was also observed that the secondary anti-RSV antibody response was dominated by IgG2a antibodies 3 weeks after secondary challenge.
Appearance of RSV-specific CD8+ T cells following neonatal infection
To determine whether neonatal mice mount a primary CD8 response in the lung, 7-day-old mice were infected intranasally with RSV. Groups of five mice per time-point were killed at 4, 7, 10 and 14 days following infection and lungs from individual mice were analysed. The presence of RSV-specific CD8+ T cells was determined by flow cytometry using an RSV matrix protein (M2)82−90 class I tetramer. As shown in Fig. 2a, RSV-specific CD8+ cells first became detectable in the lungs of neonatal mice between 7 and 10 days after infection. At day 10 approximately 12% of CD8+ T cells in the lung were tetramer-positive, but this percentage had declined by day 14. To determine whether RSV infection during neonatal life induced memory in the CD8 T cell compartment, mice primed as adults or neonates were rechallenged 7 weeks later with RSV, and RSV-specific cells identified by tetramer analysis. Figure 2b shows that on day 0, less than 1% of CD8+ T cells in lungs of unprimed and neonatally primed mice were M2-specific, and a small population was present in lungs of adult-primed mice (2·5%). When cells were isolated from lungs of mice at an early time-point, 4 days after primary infection (naive group), less than 2% of lung CD8+ T cells were M2-specific. In contrast, after rechallenge, a dramatic increase in the percentage of M2-specific CD8+ T cells was observed in the lungs of both neonatally primed and adult-primed mice (25% and 37% respectively). On day 7 after rechallenge, an equal percentage of M2-specific CD8+ cells were present in lungs of neonatally primed and adult-primed mice, whereas approximately half this percentage were present in the lungs of unprimed mice.
Fig. 2.
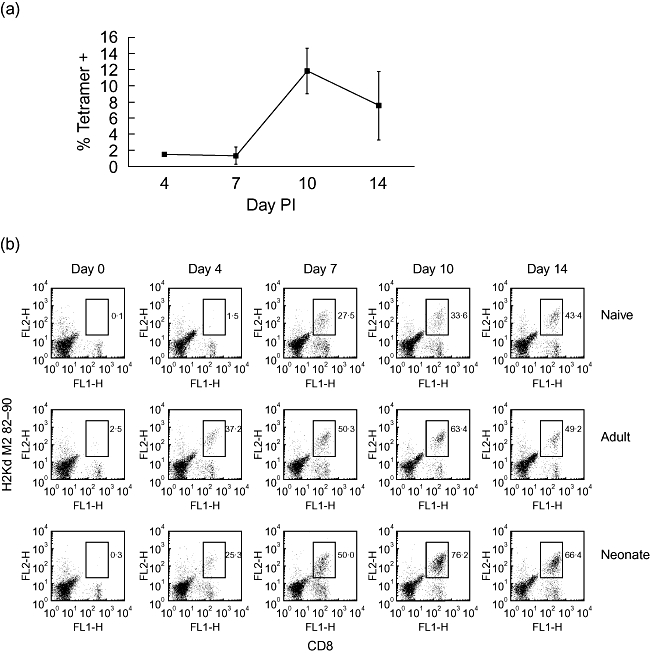
Effective boosting of respiratory syncytial virus (RSV)-specific CD8 T cells in lungs of mice primed as neonates and rechallenged as adults. Analysis of the neonatal primary response is shown in (a); mice were infected with RSV at 7 days of age and at the times indicated post-infection lungs were disrupted and cells analysed for expression of CD8 and RSV peptide-specific T cell receptor and expressed as tetramer-positive cells as a percentage of CD8+ cells. Values represent the mean values from at least four mice per group ± standard deviation. Mice primed as adults or neonates were rechallenged with RSV during adult life, 7 weeks post-priming. Mice previously naive were challenged once with RSV. At the times indicated post-secondary challenge with RSV (or primary challenge for the naive group), lungs were disrupted and tetramer and CD8 analysis was performed as above. Fluorescence activated cell sorter plots represent pooled lungs from at least four mice per group, and frequencies of tetramer-positive cells as a percentage of CD8+ T cell numbers are shown (b). The data shown are representative of two independent experiments.
Induction of anti-RSV IFN-γ-producing CD8 T cells in neonatal mice
Neonatal mice have been shown to make adult-like CTL and IFN-γ responses to viruses under appropriate circumstances [12]. To determine the ability of mice to mount a CD8 T cell response to RSV infection we isolated lung and spleen cells from adult or neonatal mice at various time-points after intranasal infection and examined IFN-γ secretion. Cells were tested by ELISPOT for IFN-γ production in response to stimulation with an RSV-specific matrix protein (M2)82−90 peptide. Primary intranasal infection of 7-day-old mice with RSV led to the induction of RSV peptide-specific IFN-γ-secreting cells in both lung (Fig. 3a) and spleen (Fig. 3b), although the numbers were low compared with those induced during primary infection of adult mice (Fig. 3c and d open symbols). The peak of the neonatal and adult primary response was observed at 10 days post-infection in both lung and spleen. To determine the capacity of RSV to induce CD8 T cell memory, mice primed as adults or neonates were rechallenged 7 weeks later and IFN-γ responses were determined. Four days after primary infection (naive group), IFN-γ responses of lung and spleen cells (Fig. 3c and d respectively) were barely detectable. In contrast, secondary challenge of mice primed either as adults or neonates revealed a significantly boosted response in the lung. Importantly, for neonatally primed mice, this is despite the low numbers of virus-specific IFN-γ-secreting cells generated during the primary response. At 4 days after rechallenge there were no significant differences in responses between neonatally primed and adult-primed mice in either lung (P = 0·314) or spleen (P = 0·280). For all groups, the peak of the lung response was observed at 10 days and declined thereafter.
Fig. 3.
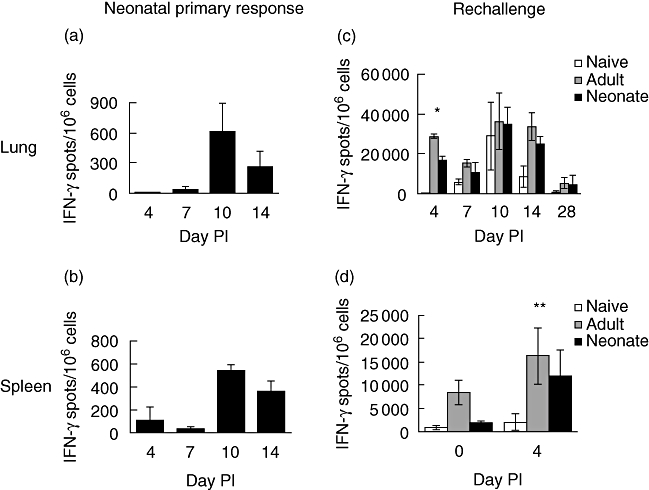
Effective boosting of respiratory syncytial virus (RSV)-specific interferon (IFN)-γ-secreting CD8 T cells in lungs and spleens of mice primed as neonates. Analysis of the neonatal primary response in the lung (a) and spleen (b) is shown. Mice primed as adults or neonates were rechallenged with RSV during adult life 7 weeks post-priming (c and d). Mice previously naive were challenged once with RSV. At the times indicated post-secondary challenge with RSV (or primary challenge for the naive group, and neonatal primary response), RSV peptide-specific IFN-γ-secreting cells in lung (a and c) and spleen (b and d) were detected by enzyme-linked immunospot assay (ELISPOT). Values represent means of at least five mice ± standard deviation. Insignificant differences between adult-primed and neonatally primed groups are shown for day 4; *P = 0·314 (lung) and **P = 0·280 (spleen) by Student's t-test. The data shown are representative of two independent experiments.
Induction of Th memory response in neonatally primed mice
The adaptive responses of neonatal mice are reported to have an inherent bias towards theTh2 type both during the primary response and on secondary challenge in later life. Therefore we examined the cytokine profile of virus-specific responders from both spleen and the draining lymph node (mediastinal) of the lung during the primary adult response and during rechallenge of adult-primed or neonatally primed mice. Seven days after primary infection (naive group) or rechallenge, splenocytes or lymph node cells were stimulated for 72 h with RSV-infected splenocytes or medium alone and supernatant collected and cytokines analysed by CBA assay (for IFN-γ and IL-5). Alternatively, because of the difficulties in detecting soluble IL-4 by ELISA, cells were stimulated for 16 h with RSV-infected splenocytes or medium and IL-4-secreting cells enumerated by ELISPOT. Figure 4a shows substantial IFN-γ secretion by spleen cells and draining lymph node cells from neonatally primed mice, equivalent to secretion by cells from unprimed adult RSV-infected mice. However, this was significantly less (P < 0·001) than that secreted by spleen or lymph node cells from adult-primed mice. The Th2 cytokine IL-5 was found to be secreted in significantly greater amounts by cells from neonatally primed mice than by T cells from adult-primed mice, and this was the case for both spleen (P < 0·05) and lymph node cells (P < 0·01). Furthermore, an identical pattern of IL-5 secretion was observed following restimulation in culture of positively selected CD4+ T cells (data not shown). ELISPOT analysis, 7 days after rechallenge, demonstrated that IL-4 responders, like cells secreting IL-5, were present at a significantly greater frequency in both spleen (P < 0·05) and lymph node (P < 0·01) of neonatally primed mice than were present in adult-primed mice (Fig. 4c). IL-4 may be secreted by cells other than T cells, but we found an identical pattern of IL-4 secretion following restimulation in culture of positively selected CD4+ cells (data not shown). Very low levels of IFN-γ and IL-5 secretion were detected in control cultures in the absence of RSV-infected splenocytes (Fig. 4b). Thus, rechallenge of mice with RSV following neonatal priming led to a robust IFN-γ response equivalent to the response observed in mice undergoing a primary infection. However, neonatal infection with RSV enhanced significantly the production of Th2 cytokines IL-4 and IL-5 by T cells in both the local lymph nodes and the spleen that were not seen in unprimed mice or mice primed as adults.
Fig. 4.
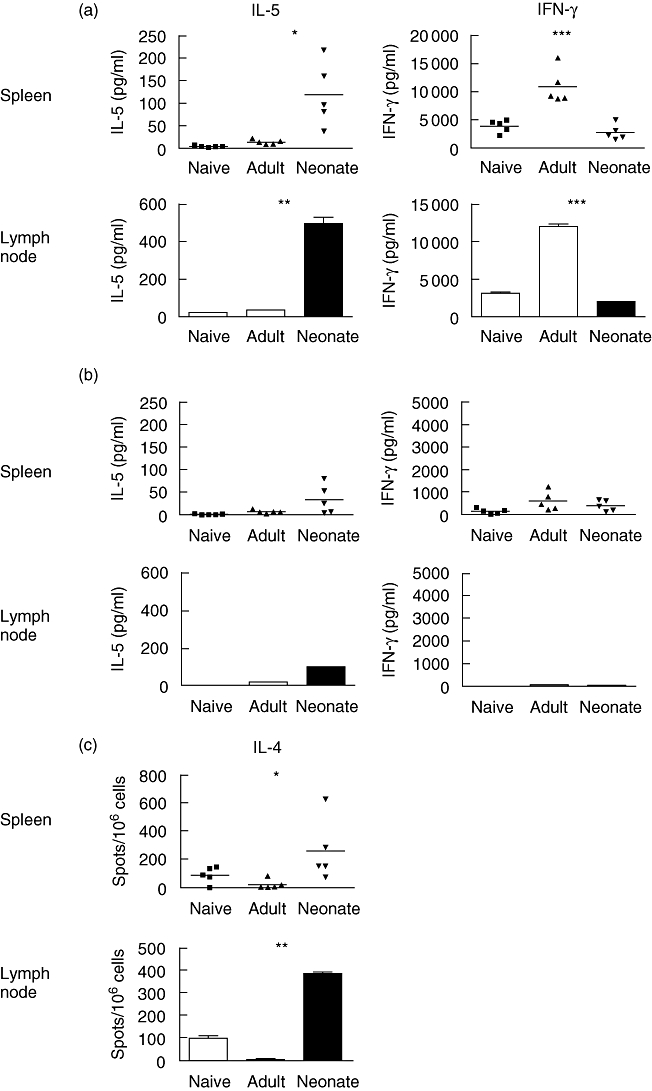
High-level interleukin (IL)-5 production is evident only in neonatally primed lymph node and spleen following respiratory syncytial virus (RSV) infection during adult life. Mice primed as adults or neonates were rechallenged with RSV during adult life 7 weeks post-priming. Mice previously naive were challenged once with RSV. Spleens and lymph node were harvested 7 days after secondary challenge (or primary challenge for the naive group). Spleen or lymph node cells were then restimulated in culture with RSV-infected syngeneic, irradiated splenocytes (a) or with medium alone (b) for 72 h. The IL-5 and interferon-γ content of cell culture supernatants (pg/ml) was assessed. IL-4 secreting cells were detected by enzyme-linked immunospot assay (ELISPOT) and expressed as spots/106 cells (c). Results for individual mice are shown for spleen cell cultures, and for pooled lymph node cultures (from at least four mice per group) and each bar represents the average ± standard deviation values from triplicate enzyme-linked immunosorbent assay wells. Differences between adult- and neonatally primed groups are shown; *P < 0·05 and **P < 0·01 and ***P < 0·001 by Student's t-test. The data shown are representative of two independent experiments.
Neonatal infection with RSV protects against infection during adult life
To determine whether infection of 7-day-old mice protected against or limited subsequent RSV infection during adult life, we infected groups of adult or neonatal mice and determined viral titre in lungs following rechallenge. Viral titre was analysed by plaque assay 4 days after rechallenge (or primary infection in the naive group), as pilot experiments indicated this to be the peak of viral replication in lungs of mice during primary infection (data not shown). Live virus was detected in lungs of mice undergoing a primary infection (Fig. 5), but could not be detected in the lungs of either adult- or neonatally primed mice following rechallenge. It appears therefore that the memory response generated in both adult-primed and neonatally primed mice was sufficient to induce effective protection against subsequent RSV infection.
Fig. 5.
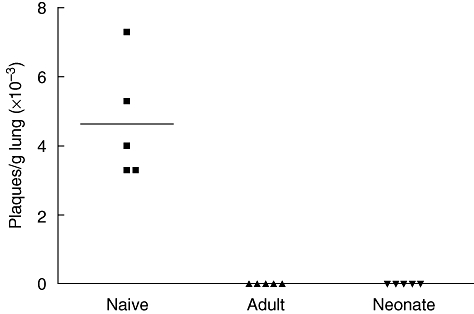
Neonatal infection with respiratory syncytial virus (RSV) protects against secondary challenge. Mice primed as adults or neonates were rechallenged with RSV during adult life, 7 weeks post-priming. Mice previously naive were challenged once with RSV. Lungs were harvested 4 days after secondary challenge (or primary challenge for naïve group) and viral titre determined by plaque assay and expressed as plaques per gram of lung. Data are expressed for individual mice and means shown.
Enhanced concentrations of inflammatory cytokines and chemokines in lungs of neonatally primed mice
Severe inflammation is partially responsible for RSV-induced pathology in humans, and in a mouse model it has been shown previously that secondary challenge of neonatally primed mice leads to enhanced inflammatory cell recruitment [17]. To identify possible mediators of enhanced inflammation in lungs of neonatally primed mice we analysed cytokines and chemokines (some of which have been shown previously to be involved in RSV-mediated pathology) in lung homogenates 4 days after rechallenge. Following infection, concentrations of inflammatory chemokines were lowest in the lungs of unprimed and adult-primed mice (Fig. 6). Concentrations of MCP-1 (P < 0·05), TNF (P < 0·001), MIP-1α (P < 0·01) and RANTES (P < 0·01) were all significantly higher in the lungs of neonatally primed mice than the concentrations found in lungs of adult-primed mice. We found no significant difference in the concentrations of eotaxin or IL-6 in lungs from the three groups of mice.
Fig. 6.
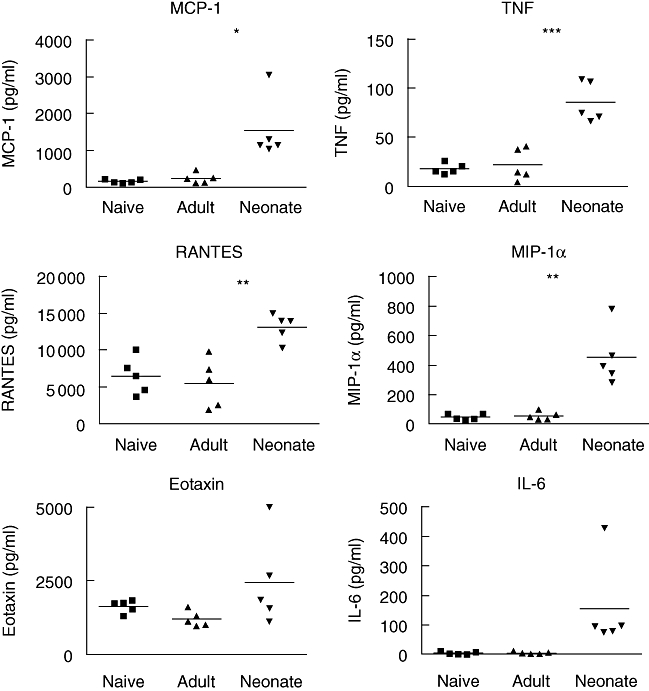
High level of inflammatory cytokines and chemokines in lungs of neonatally primed mice following secondary challenge with respiratory syncytial virus (RSV) during adult life. Mice primed as adults or neonates were rechallenged with RSV during adult life 7 weeks post-priming. Mice previously naive were challenged once with RSV. Lungs were harvested 4 days after secondary challenge (or primary challenge for the naive group) and concentration of cytokines and chemokines in supernatants were determined by cytometric bead array assay or enzyme-linked immunosorbent assay and expressed as pg/ml of supernatant. Differences between adult- and neonatally primed groups are shown; *P < 0·05, **P < 0·01 and ***P < 0·001 by Student's t-test. Data are expressed for individual mice. The data shown are representative of two independent experiments.
Discussion
Neonatal mice and humans are more susceptible to infection than adults, and many studies have sought to address the mechanisms underlying this vulnerability. These studies have described differences in terms of both cell number and function for T cells, B cells and APC in murine neonates. Although many studies have suggested that neonates are immunodeficient, it has become apparent more recently that, under appropriate circumstances, neonatal mice are competent to make effective immune responses against both infectious agents and vaccine antigens.
RSV is a pathogen of great clinical importance, being the major cause of viral bronchiolitis in infancy and early childhood, and infection with RSV has been implicated in the subsequent development of asthma and atopy in later life [15]. It is important, therefore, to develop a better understanding of early life responses to this virus. In adult BALB/c mice, primary infection with RSV produces a mild illness. However, mice exposed to RSV during neonatal life suffer more severe disease when reinfected in adulthood than do unprimed animals [17,21]. This observation suggests that neonates do mount immune responses to RSV and that these somehow modify subsequent responses in adult life. In this study we have examined the primary antibody and T cell responses of neonatal mice to intranasal infection with RSV. We have also examined the effect of neonatal infection on the quality and magnitude of response to subsequent RSV infection in adulthood. Our findings indicate that neonatal responses to RSV are weaker than those of adults, but despite this deficiency, neonatally infected animals are primed to mount significantly improved T and B cell responses on re-encounter with the virus in adult life. Furthermore, we show that neonatal priming drives a marked deviation of the subsequent Th cell response leading to the secretion of significant amounts of Th2 cytokines in addition to IFN-γ.
Many studies have shown that neonatal animals make poor antibody responses to T dependent antigens. Although humans can make IgG responses in utero to infectious agents, it has been observed frequently that IgG responses are relatively weak during the first year of life. Similarly, vaccination of murine neonates elicits only weak antibody responses compared with those of adult animals. In addition the quality of neonatal and adult antibody responses are distinct, with an apparent age-dependent limitation of IgG2/IgG2a responses to conventional vaccines in humans and mice respectively. Despite these poor primary responses, it has been reported previously that mice immunized at 7 days of age showed enhanced antibody responses after further vaccine challenge, indicating the generation of immunological memory [22].
Here we show that infection of neonatal mice with RSV induced a considerably weaker IgM and IgG response than in adult animals. This may be due to the well-documented small numbers of B lymphocytes and other immune cells present in peripheral lymphoid tissue of neonatal mice or to the immaturity of the follicular dendritic cell network [23]. The neonatal spleen is a haematopoietic organ, and during the first weeks of life production of B cells from fetal precursors crosses over with B cell generation from adult bone marrow progenitors [24]. In addition, in the first 2 weeks of life a high proportion of splenic B cells in the neonate are CD5+, and these cells are believed to derive mainly from the fetal liver [25]. Neonatal spleen thus contains a high proportion of fetal precursor-derived B cells and for this reason antibody responses of neonatal mice might be expected to differ from the responses of adult mice. However, splenic B cells may not be the major contributors to neonatal antibody responses, as splenectomy has little affect on IgG1-dominant secondary responses [26]. B cells in neonatal lymph nodes are more mature phenotypically and functionally than those in the spleen and may be responsible for antibody responses to some antigens in early life. This suggests that the route of immunization may also be important for vigorous neonatal antibody responses. Indeed, it has been shown that intranasal immunization of neonatal mice with a bacterial conjugate vaccine induced stronger, adult-like responses than immunization via the subcutaneous route [27]. We observed that neonatal IgG responses showed delayed kinetics compared with those of adults. Similar findings have been reported for responses to vaccine antigens [4]. These differences in kinetics may reflect the late development of germinal centres in neonatal lymphoid tissue, as these are vital for isotype switching.
Murine neonatal vaccine studies have shown a Th2 bias in immune responses characterized by preferential production of IgG1 antibodies [8]. Our data, however, demonstrate that following intranasal RSV infection the primary neonatal IgG response to RSV, although weak, is dominated by IgG2a antibodies as in the adult. This bias towards a Th1 type of response may possibly reflect the effects of innate antiviral responses on adaptive responses of T and B lymphocytes. When we undertook experiments to investigate responses to a second challenge with RSV we found that, despite the weak primary IgG response of neonatal mice, neonatally primed mice made swifter and greater IgG responses than unprimed adults, demonstrating the generation of memory B cells in early life. Like the primary response, this enhanced secondary response was also dominated by IgG2a antibodies. Thus, despite the widely reported preferential induction of Th2 responses on exposure of neonatal mice to a variety of antigens and vaccines [8], we show that neonatal mice make a predominantly Th1-like IgG2a antibody response to infection with live RSV and this bias is maintained when they are rechallenged with RSV in adulthood. Investigation of cytokine production in these animals revealed a strong IFN-γ response of restimulated cells, from spleen and lymph node, equivalent to the response of cells from RSV-infected unprimed mice, but this was significantly weaker than the response of cells from adult-primed mice. Moreover, IL-5 secretion and IL-4-secreting cells were detected only in restimulation cultures of cells from neonatally primed mice. We interpret these findings as induction of RSV-specific Th2 memory responses after neonatal RSV infection. Other investigators have shown, in a model of neonatal priming with murine leukaemia virus, a transient production of IL-4 after secondary viral challenge [28]. However, this IL-4 production was not virus-specific and subsided 3 weeks after challenge. In contrast, we showed that the IL-4- and IL-5-secreting cells in both spleen and lymph node to be RSV-specific and that they required antigen for their induction. The fact that we observed both Th1 and Th2 cytokine secretion in both spleen and in the draining lymph node of the lung is significant and interesting. Primary immunization of neonates with keyhole limpet haemocyanin induces the generation of both Th1 and Th2 effectors in lymph node but only Th2 effectors in the spleen, while the same immunization protocol induces mixed responses in both organs of adult mice [29]. In a further report both splenic and lymph node T cell memory responses of mice immunized as neonates were Th2-biased [30]. The Th2-biased primary responses of the spleen were not required for the Th2-biased memory in the lymph node and it was shown that neonates are impaired selectively in the development of Th1 memory in both organs, suggesting an intrinsic property of lymph node T cells or the neonatal environment which leads to a Th2-dominated secondary response. One possible explanation is the recent finding that Th1 cells generated during neonatal life express IL-13Rα1 selectively, through which IL-4 induces Th1 cell apoptosis, leading thereby to a Th2-biased response if re-encounter of the antigen occurs in an IL-4-containing environment [31]. CD5+ B cells, which are more abundant in early life, may also play a role in the inefficient induction of Th1 responses in neonatal mice [32]. The authors report that CD5+ B cells produce IL-10 in response to TLR-9 signalling, which limits the priming ability of neonatal dendritic cells by reducing their capacity to produce IL-12, thereby providing a mechanism for less efficient Th1 priming. Our data, which show a pronounced boosting of Th2 cytokine production in spleen and lymph node following secondary challenge, are in agreement with the wide body of data describing the Th2 skewing of neonatal immune responses. In the present study, despite the presence of Th2 cytokine-producing Th cells after rechallenge, recall antibody responses in neonatally primed mice as well as primary responses are biased strongly towards the IgG2a isotype, probably because of the significant IFN-γ production.
Studies by others have shown that neonatal mice are able to mount type 1 CD8 T cell responses against viral, bacterial or parasite antigens in the presence of strong Th1-promoting agents [9,33,34] and also in response to more natural challenges such as virus [12] and against alloantigens [11]. CD8+ CTL are the main effector cells involved in the resolution of most viral infections, but studies have shown that in mice the CD8 T cell response to RSV infection during primary infection is also responsible for a significant amount of immunopathology [35]. Here we have demonstrated clearly that neonatal mice infected with RSV can generate RSV-specific CD8+ T cells. These cells are maintained in the weeks following infection allowing a boosted response after secondary challenge in adulthood. The kinetics of both the appearance of RSV-specific CD8 T cells in lung and spleen and the presence of RSV-specific IFN-γ secreting cells after primary infection of neonatal animals are similar to those observed following primary adult infection. This suggests that priming of CD8 T cells is taking place within the neonatal period and is not due to the presence of virus remaining later in life.
We show here that neonatal mice infected with RSV make weak B and T cell responses; however, this is sufficient to generate a protective memory response on secondary challenge during adult life. This is in contrast to the findings of a human study which demonstrated the lack of boosting of RSV-specific T cell responses upon natural reinfection [36]. In a mouse RSV infection model, age of first infection was shown to be important in the pattern of cell-mediated responses generated during reinfection later in life [17]: a Th2-biased response in the lung and increased inflammatory cell recruitment, including eosinophilia, was observed after neonatal priming. More recently it has been demonstrated that neonatal priming results in enhanced airway hyperresponsiveness (AHR), airway eosinophilia, increased mucus production and increased IL-13 production after reinfection with RSV [21]. Our data extend these findings and we show here that reinfection during adulthood of neonatally primed mice is associated with a profound increase in the concentration of inflammatory cytokines and chemokines in the lung at an early time-point after the secondary challenge. We found that concentrations of MCP-1, TNF, MIP-1α and RANTES were significantly greater than concentrations of the same inflammatory mediators in the lungs of adult-primed mice undergoing secondary challenge. Each of these mediators has been implicated in the development of RSV-mediated lung pathology. Inhibition of TNF has been shown to reduce the severity of RSV-specific lung immunopathology during a secondary RSV infection [37]; however, absence of TNF was not sufficient to reduce CTL-mediated immunopathology during primary RSV infection [38]. Mediators identified so far in playing a role in lung immunopathology during primary infection of adult mice include TNF-α[39] and MIP-1α[18] and these mediators were shown to be involved in inflammation; RANTES was also implicated as its inhibition leads to reduced AHR [40]. In most cases the cellular source of inflammatory mediators has not been identified, but in vitro infection of human airway epithelial cells has shown that a number of chemokines can be induced, including RANTES, MIP-1α and MCP-1 [19,41]. Analysis of chemokines in bronchoalveolar lavage samples from infants with RSV bronchiolitis identified a number of chemokines, including MIP-1α and MCP-1 [20]. Apart from one study [37], none have addressed the roles played by inflammatory mediators in lungs of neonatally primed mice undergoing secondary challenge. Chemokines are involved in leucocyte trafficking, and have emerged as important regulatory molecules in the immune processes of the lung [42]. Here we show that, following rechallenge, the concentrations of the mononuclear leucocyte-attracting chemokines MCP-1, MIP-1α and RANTES were increased significantly in the lungs of neonatally primed mice. In this study we did not analyse leucocyte recruitment, but our findings provide a possible explanation for the previously reported enhanced cellular recruitment that occurs in neonatally primed, rechallenged mice and is associated with enhanced pathology and illness [17]. The role played by individual chemokines in cellular recruitment and enhanced pathology seen following secondary challenge are currently the subject of further investigation.
In conclusion, the data presented here provide further confirmation that neonates are able to mount efficient immune responses against infection and that these can lead to the development of immunological memory. However, neonatal priming leads to a differential enhancement of Th1 and Th2 responses following secondary challenge. Secondary challenge with RSV is associated with a profound increase in chemokines and inflammatory cytokines in the lungs of neonatally primed mice and these may be involved in the histological and inflammatory features of RSV-induced airway disease. Future studies are required to investigate the mechanisms responsible for the particularly marked enhancement of Th2 cytokine production in response to secondary RSV infection and the associated enhanced lung inflammation.
Acknowledgments
This work was supported by the Edward Jenner Institute for Vaccine Research.
References
- 1.Billingham RE, Brent L, Medawar PB. Activity acquired tolerance of foreign cells. Nature. 1953;172:603–6. doi: 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- 2.Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. 2004;4:553–64. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- 3.Marshall-Clarke S, Reen D, Tasker L, Hassan J. Neonatal immunity: how well has it grown up? Immunol Today. 2000;21:35–41. doi: 10.1016/s0167-5699(99)01548-0. [DOI] [PubMed] [Google Scholar]
- 4.Siegrist CA. Neonatal and early life vaccinology. Vaccine. 2001;19:3331–46. doi: 10.1016/s0264-410x(01)00028-7. [DOI] [PubMed] [Google Scholar]
- 5.Garcia AM, Fadel SA, Cao S, Sarzotti M. T cell immunity in neonates. Immunol Res. 2000;22:177–90. doi: 10.1385/IR:22:2-3:177. [DOI] [PubMed] [Google Scholar]
- 6.Adkins B. Development of neonatal Th1/Th2 function. Int Rev Immunol. 2000;19:157–71. doi: 10.3109/08830180009088503. [DOI] [PubMed] [Google Scholar]
- 7.Powell TJ, Jr, Streilein JW. Neonatal tolerance induction by class II alloantigens activates IL-4-secreting, tolerogen-responsive T cells. J Immunol. 1990;144:854–9. [PubMed] [Google Scholar]
- 8.Barrios C, Brawand P, Berney M, Brandt C, Lambert PH, Siegrist CA. Neonatal and early life immune responses to various forms of vaccine antigens qualitatively differ from adult responses: predominance of a Th2-biased pattern which persists after adult boosting. Eur J Immunol. 1996;26:1489–96. doi: 10.1002/eji.1830260713. [DOI] [PubMed] [Google Scholar]
- 9.Martinez X, Brandt C, Saddallah F, et al. DNA immunization circumvents deficient induction of T helper type 1 and cytotoxic T lymphocyte responses in neonates and during early life. Proc Natl Acad Sci USA. 1997;94:8726–31. doi: 10.1073/pnas.94.16.8726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ridge JP, Fuchs EJ, Matzinger P. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science. 1996;271:1723–6. doi: 10.1126/science.271.5256.1723. [DOI] [PubMed] [Google Scholar]
- 11.Adkins B, Jones M, Bu Y, Levy RB. Neonatal tolerance revisited again: specific CTL priming in mouse neonates exposed to small numbers of semi- or fully allogeneic spleen cells. Eur J Immunol. 2004;34:1901–9. doi: 10.1002/eji.200324271. [DOI] [PubMed] [Google Scholar]
- 12.Sarzotti M, Robbins DS, Hoffman PM. Induction of protective CTL responses in newborn mice by a murine retrovirus. Science. 1996;271:1726–8. doi: 10.1126/science.271.5256.1726. [DOI] [PubMed] [Google Scholar]
- 13.Forsthuber T, Yip HC, Lehmann PV. Induction of TH1 and TH2 immunity in neonatal mice. Science. 1996;271:1728–30. doi: 10.1126/science.271.5256.1728. [DOI] [PubMed] [Google Scholar]
- 14.Openshaw PJ, Culley FJ, Olszewska W. Immunopathogenesis of vaccine-enhanced RSV disease. Vaccine. 2001;20(Suppl. 1):S27–31. doi: 10.1016/s0264-410x(01)00301-2. [DOI] [PubMed] [Google Scholar]
- 15.Sigurs N. Epidemiologic and clinical evidence of a respiratory syncytial virus-reactive airway disease link. Am J Respir Crit Care Med. 2001;163:S2–6. doi: 10.1164/ajrccm.163.supplement_1.2011109. [DOI] [PubMed] [Google Scholar]
- 16.Holt PG, Sly PD. Interactions between respiratory tract infections and atopy in the aetiology of asthma. Eur Respir J. 2002;19:538–45. doi: 10.1183/09031936.02.00229302. [DOI] [PubMed] [Google Scholar]
- 17.Culley FJ, Pollott J, Openshaw PJ. Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J Exp Med. 2002;196:1381–6. doi: 10.1084/jem.20020943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haeberle HA, Kuziel WA, Dieterich HJ, Casola A, Gatalica Z, Garofalo RP. Inducible expression of inflammatory chemokines in respiratory syncytial virus-infected mice: role of MIP-1alpha in lung pathology. J Virol. 2001;75:878–90. doi: 10.1128/JVI.75.2.878-890.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller AL, Bowlin TL, Lukacs NW. Respiratory syncytial virus-induced chemokine production: linking viral replication to chemokine production in vitro and in vivo. J Infect Dis. 2004;189:1419–30. doi: 10.1086/382958. [DOI] [PubMed] [Google Scholar]
- 20.McNamara PS, Flanagan BF, Hart CA, Smyth RL. Production of chemokines in the lungs of infants with severe respiratory syncytial virus bronchiolitis. J Infect Dis. 2005;191:1225–32. doi: 10.1086/428855. [DOI] [PubMed] [Google Scholar]
- 21.Dakhama A, Park JW, Taube C, et al. The enhancement or prevention of airway hyperresponsiveness during reinfection with respiratory syncytial virus is critically dependent on the age at first infection and IL-13 production. J Immunol. 2005;175:1876–83. doi: 10.4049/jimmunol.175.3.1876. [DOI] [PubMed] [Google Scholar]
- 22.Barrios C, Brandt C, Berney M, Lambert PH, Siegrist CA. Partial correction of the TH2/TH1 imbalance in neonatal murine responses to vaccine antigens through selective adjuvant effects. Eur J Immunol. 1996;26:2666–70. doi: 10.1002/eji.1830261118. [DOI] [PubMed] [Google Scholar]
- 23.Pihlgren M, Tougne C, Bozzotti P, et al. Unresponsiveness to lymphoid-mediated signals at the neonatal follicular dendritic cell precursor level contributes to delayed germinal center induction and limitations of neonatal antibody responses to T-dependent antigens. J Immunol. 2003;170:2824–32. doi: 10.4049/jimmunol.170.6.2824. [DOI] [PubMed] [Google Scholar]
- 24.Kincade PW, Owen JJ, Igarashi H, Kouro T, Yokota T, Rossi MI. Nature or nurture? Steady-state lymphocyte formation in adults does not recapitulate ontogeny. Immunol Rev. 2002;187:116–25. doi: 10.1034/j.1600-065x.2002.18710.x. [DOI] [PubMed] [Google Scholar]
- 25.Herzenberg LA. B-1 cells: the lineage question revisited. Immunol Rev. 2000;175:9–22. [PubMed] [Google Scholar]
- 26.Adkins B, Bu Y, Vincek V, Guevara P. The primary responses of murine neonatal lymph node CD4+ cells are Th2-skewed and are sufficient for the development of Th2-biased memory. Clin Dev Immunol. 2003;10:43–51. doi: 10.1080/10446670310001598474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakobsen H, Bjarnarson S, Del Giudice G, Moreau M, Siegrist CA, Jonsdottir I. Intranasal immunization with pneumococcal conjugate vaccines with LT-K63, a nontoxic mutant of heat-labile enterotoxin, as adjuvant rapidly induces protective immunity against lethal pneumococcal infections in neonatal mice. Infect Immun. 2002;70:1443–52. doi: 10.1128/IAI.70.3.1443-1452.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fadel SA, Ozaki DA, Sarzotti M. Enhanced type 1 immunity after secondary viral challenge in mice primed as neonates. J Immunol. 2002;169:3293–300. doi: 10.4049/jimmunol.169.6.3293. [DOI] [PubMed] [Google Scholar]
- 29.Adkins B, Bu Y, Cepero E, Perez R. Exclusive Th2 primary effector function in spleens but mixed Th1/Th2 function in lymph nodes of murine neonates. J Immunol. 2000;164:2347–53. doi: 10.4049/jimmunol.164.5.2347. [DOI] [PubMed] [Google Scholar]
- 30.Adkins B, Bu Y, Guevara P. The generation of Th memory in neonates versus adults: prolonged primary Th2 effector function and impaired development of Th1 memory effector function in murine neonates. J Immunol. 2001;166:918–25. doi: 10.4049/jimmunol.166.2.918. [DOI] [PubMed] [Google Scholar]
- 31.Li L, Lee HH, Bell JJ, et al. IL-4 utilizes an alternative receptor to drive apoptosis of Th1 cells and skews neonatal immunity toward Th2. Immunity. 2004;20:429–40. doi: 10.1016/s1074-7613(04)00072-x. [DOI] [PubMed] [Google Scholar]
- 32.Sun CM, Deriaud E, Leclerc C, Lo-Man R. Upon TLR9 signaling, CD5+ B cells control the IL-12-dependent Th1-priming capacity of neonatal DCs. Immunity. 2005;22:467–77. doi: 10.1016/j.immuni.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 33.Bot A, Bot S, Garcia-Sastre A, Bona C. Protective cellular immunity against influenza virus induced by plasmid inoculation of newborn mice. Dev Immunol. 1998;5:197–210. doi: 10.1155/1998/50472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brazolot Millan CL, Weeratna R, Krieg AM, Siegrist CA, Davis HL. CpG DNA can induce strong Th1 humoral and cell-mediated immune responses against hepatitis B surface antigen in young mice. Proc Natl Acad Sci USA. 1998;95:15553–8. doi: 10.1073/pnas.95.26.15553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graham BS, Rutigliano JA, Johnson TR. Respiratory syncytial virus immunobiology and pathogenesis. Virology. 2002;297:1–7. doi: 10.1006/viro.2002.1431. [DOI] [PubMed] [Google Scholar]
- 36.Bont L, Versteegh J, Swelsen WT, et al. Natural reinfection with respiratory syncytial virus does not boost virus-specific T-cell immunity. Pediatr Res. 2002;52:363–7. doi: 10.1203/00006450-200209000-00009. [DOI] [PubMed] [Google Scholar]
- 37.Hussell T, Pennycook A, Openshaw PJ. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur J Immunol. 2001;31:2566–73. doi: 10.1002/1521-4141(200109)31:9<2566::aid-immu2566>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 38.Ostler T, Davidson W, Ehl S. Virus clearance and immunopathology by CD8(+) T cells during infection with respiratory syncytial virus are mediated by IFN-gamma. Eur J Immunol. 2002;32:2117–23. doi: 10.1002/1521-4141(200208)32:8<2117::AID-IMMU2117>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 39.Rutigliano JA, Graham BS. Prolonged production of TNF-alpha exacerbates illness during respiratory syncytial virus infection. J Immunol. 2004;173:3408–17. doi: 10.4049/jimmunol.173.5.3408. [DOI] [PubMed] [Google Scholar]
- 40.Tekkanat KK, Maassab H, Miller A, Berlin AA, Kunkel SL, Lukacs NW. RANTES (CCL5) production during primary respiratory syncytial virus infection exacerbates airway disease. Eur J Immunol. 2002;32:3276–84. doi: 10.1002/1521-4141(200211)32:11<3276::AID-IMMU3276>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 41.Olszewska-Pazdrak B, Casola A, Saito T, et al. Cell-specific expression of RANTES, MCP-1, and MIP-1alpha by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J Virol. 1998;72:4756–64. doi: 10.1128/jvi.72.6.4756-4764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oppenheim JJ, Zachariae CO, Mukaida N, Matsushima K. Properties of the novel proinflammatory supergene ‘intercrine’ cytokine family. Annu Rev Immunol. 1991;9:617–48. doi: 10.1146/annurev.iy.09.040191.003153. [DOI] [PubMed] [Google Scholar]