

Abstract
WHAT IS KNOWN ABOUT THIS SUBJECT
Clinical pharmacology and clinical therapeutic studies of fingolimod demonstrate that heart rate after the initial dose decreases by about 10–20% while normal circadian rhythm is preserved. With continued daily dosing, heart rate returns to normal over the next 2 weeks.
This negative chronotropic effect is consistent with the binding of fingolimod-phosphate to the sphingosine-1-phosphate receptor on atrial myocytes.
WHAT THIS STUDY ADDS
The present clinical pharmacology study demonstrates that atropine administered at usual therapeutic doses can prevent the decrease in heart rate when given concomitantly with fingolimod and can counteract the decrease in heart rate when give at the time of the typical heart rate nadir, 4 h after the fingolimod dose.
Although therapeutic intervention is rarely needed for reduced heart rate in patients receiving fingolimod, atropine is an option, should this be desired.
AIMS
The authors determined whether intravenous atropine can prevent or counteract the negative chronotropic effect of the immunomodulator fingolimod.
METHODS
In this randomized, placebo-controlled, two-period, crossover study, 12 healthy subjects received 5 mg fingolimod orally concurrently with intravenous atropine (titrated to a heart rate of 110–120 beats min−1) or intravenous placebo. A second group of 12 subjects received atropine/placebo 4 h after the fingolimod dose. Continuous telemetry measurements were made for 24 h after each fingolimod dose.
RESULTS
Fingolimod administration alone yielded a heart rate nadir of 51 ± 5 beats min−1 at a median 4 h postdose with heart rate remaining depressed at 51–64 beats min−1 over the rest of the day. Concurrent administration of fingolimod and atropine yielded a nadir of 66 ± 6 beats min−1 resulting in an atropine : placebo ratio (90% confidence interval) of 1.30 (1.22, 1.36). When atropine was administered at the time of the nadir, it was able to reverse the negative chronotropic effect of fingolimod from a heart rate of 56 ± 9 beats min−1 (placebo) to 64 ± 8 beats min−1 (atropine) resulting in an atropine : placebo ratio of 1.15 (1.04, 1.26). Atropine had no influence on the pharmacokinetics of fingolimod.
CONCLUSIONS
Atropine administered concurrently with fingolimod prevented the heart rate nadir that typically occurs 4 h postdose. Atropine administered at the time of the heart rate nadir was able to reverse the negative chronotropic effect of fingolimod.
Keywords: atropine, bradycardia, fingolimod, heart rate
Introduction
The immunomodulator fingolimod (development code, FTY720) is an orally administered sphingosine-1-phosphate receptor agonist. In a clinical proof-of-concept study, it exerted a favorable effect on the course of relapsing multiple sclerosis compared with placebo [1]. Fingolimod is activated via phosphorylation to fingolimod-phosphate which binds to the sphingosine-1-phosphate receptor on lymphocytes. In the treatment of multiple sclerosis, it is thought that this leads to trapping of encephalitogenic T-lymphocytes in lymph nodes preventing them from migrating to the central nervous system [2].
In addition to its presence on lymphocytes, the sphingosine-phosphate receptor is also found in many tissues including atrial myocytes. Binding of the natural ligand sphingosine-1-phosphate at this site regulates heart rate [3]. Specifically, sphingosine-1-phosphate activates the cardiac G protein-gated potassium channel (IKACh) eliciting a negative chronotropic effect. Fingolimod-phosphate is believed also to activate these channels resulting in lower heart rate. This was observed in the multiple sclerosis proof-of-concept study in which fingolimod-treated patients had a reduction in mean heart rate after the first dose with recovery back to pretreatment baseline during continued daily dosing of fingolimod [1]. It is speculated that heart rate reduction is transient due to subsequent sphingosine-1-phosphate receptor desensitization [4]. This is thought to account for the attenuation of the effect of fingolimod on heart rate over time observed in patients.
Given the above, the clinical effects of fingolimod on the heart would be expected to be similar to vagally-mediated cardiac effects, since both fingolimod-phosphate and vagal stimulation signal the same G protein-gated potassium channel, albeit via different pathways. Vagally-mediated effects would include mild to moderate heart rate slowing and benign atrioventricular blocks (Type I or Wenckebach blocks). These indeed have been reported in clinical pharmacology profiling studies in healthy subjects receiving single and multiple-dose fingolimod [5–7]. Atropine is one option to treat acute symptomatic bradycardia and vagally-mediated bradycardia. We therefore designed the present study to determine if pharmacologic intervention with atropine could either prevent or reverse the negative chronotropic effect of fingolimod.
Methods
Study design
The study consisted of two separate parts, each of which had a single-blind, randomized, two-period, crossover design intended for 12 evaluable subjects. Both periods consisted of a baseline (day –1) and a 48 h treatment phase (days 1–3). Periods were separated by a 33 day washout to eliminate fingolimod and its effects on heart rate.
Study part 1 assessed the ability of atropine to prevent fingolimod negative chronotropy by administering atropine/placebo concurrently with fingolimod. All subjects received placebo orally on day 1 and fingolimod 5 mg orally on day 2 of both periods. They were randomized to receive either atropine or placebo intravenously on days 1 and 2 of period 1 and the alternate treatment on days 1 and 2 of period 2. Hence, all subjects received four oral/intravenous combinations: oral placebo with intravenous placebo to assess normal circadian heart rate, oral placebo with atropine to assess atropine alone, fingolimod with intravenous placebo to assess fingolimod alone, and fingolimod with atropine.
Study part 2 assessed the ability of atropine to reverse fingolimod negative chronotropy by administering atropine at the time when fingolimod exerted its maximal reduction in heart rate. The design was identical to part 1 except that the intravenous atropine/placebo was administered at 4 h after the oral fingolimod/placebo dose rather than simultaneously as in part 1.
The study protocol was reviewed and approved by Research Consultants Review Committee (Austin, TX, USA) and subjects gave written informed consent to participate. The sample size estimate was based on a one-sided, one-sample t-test at the 5% significance level and a coefficient of variation of ≤15% for heart rate nadir observed in previous fingolimod clinical pharmacology studies. Enrolling 12 evaluable subjects in each study part would give 80% power to detect a 17% difference in heart rate nadir between treatments.
Study population and disposition
We enrolled 30 subjects into the study: 16 subjects in part 1 and 14 subjects in part 2. There were 11 men and 19 women, 35.8 ± 9.2 years of age (range 20–48), weighing 71.0 ± 11.4 kg (range, 50.8–91.0), with a body mass index of 24.7 ± 3.2 kg m−2 (range, 18.4–29.0). There were 16 whites, one black, and 13 of other ethnicities. Six subjects did not complete the study: two subjects for protocol violations, one subject withdrew consent to participate, and three subjects were lost to follow-up. Hence, 24 subjects completed the study, 12 subjects in each study part.
Domiciling and drug administration
Subjects were present at the clinical site from the evening of day –1 until the morning of day 3 in both periods. Fingolimod 5 mg (2.5 mg capsules, Novartis Pharmaceuticals) or oral placebo was administered with 240 ml of water after a light breakfast. Atropine was administered intravenously via a peripheral forearm vein. The infusion began 5 min before intake of the oral study medication in part 1 and 4 h after intake of the oral study medication in part 2. Atropine was titrated 0.25 mg every 5 min to achieve a heart rate of 110–120 beats min−1 or a maximum total dose of 2 mg. The placebo infusion of normal saline was an initial 0.25 ml bolus followed by two 0.25 ml titrations every 5 min. Standardized lunch and dinner were served 4 and 9 h after the oral study medications.
Clinical and cardiovascular assessments
Standard biochemistry, haematology and urinalysis laboratory parameters and 12-lead electrocardiography were performed at baseline and day 3 of each period. In both study periods, continuous heart rate data were collected by telemetry for 48 h over days 1–3. Supine systolic and diastolic blood pressure and pulse were measured at baseline before each period, for 12 h after oral study medication on days 1 and 2, and in the morning of day 3.
Cardiovascular data evaluation
From the telemetry recordings of each subject, mean heart rate was calculated over hourly-intervals from the 24 h monitoring on days 1 and 2 of both periods. From these data, heart rate nadirs and the area under the heart rate-time effect curve (AUEC) were calculated over time intervals of interest: 0–4 h postdose (when the absolute nadir is expected), 0–12 h postdose (to capture delayed nadirs due to the atropine intervention), and 0–24 h (to assess the full period of telemetry). From the start of each atropine/placebo infusion to 60 min thereafter, each subject's mean heart rate was derived more frequently, namely, over 5 min intervals. The heart rate maximum was determined over this 60 min interval.
The primary response for concurrent administration was the heart rate nadir from 0–4 h postdose and for delayed administration was the heart rate at the end of the atropine/placebo infusion phase. These responses were log-transformed and compared between fingolimod with intravenous placebo (reference) and fingolimod with intravenous atropine (test) in a linear mixed-effects model with sequence, treatment, and period as fixed effects and subject-within-sequence as a random effect. The geometric mean treatment ratios and 90% confidence intervals were derived. These were interpreted against the conventional bioequivalence framework in that a confidence interval extending outside the interval 0.80–1.25 signals the potential for a clinically relevant treatment effect.
Pharmacokinetic assessments
Blood samples for the determination of fingolimod and fingolimod-phosphate were collected predose and then 2, 4, 6, 12, and 24 h postdose. Samples were inverted several times, the contents transferred to polypropylene vials, and frozen at −18°C.
Bioanalytics
Blood concentrations of fingolimod were determined by a validated liquid chromatography tandem mass spectrometry method (LC-MS) as previously described [7]. As applied in this study for fingolimod, there were seven calibration concentrations (range, 0.08 to 30 ng ml−1) and three quality control concentrations (0.24, 5, 25 ng ml−1). Quality control accuracy ranged from 87.1% to 98.0% and precision from 4.6% to 16.1%. The lower quantification limit was 0.08 ng ml−1 using 0.5 ml blood.
Fingolimod-phosphate was analyzed separately by a validated method using LC-MS in multiple reaction monitoring mode with an API4000 and turboion spray (TIS). Briefly, 0.1 ml blood was mixed with 0.1 ml internal standard in methanol and 1 ml methanol. After centrifugation for 7 min with 20 817 rcf at 5°C the supernatant was transferred into a glass tube and evaporated under nitrogen at 37°C. The dry sample was reconstituted in 0.2 ml of 1 : 1 (v : v) water and methanol. The sample was vortexed, transferred to glass injection vials, and centrifuged for 20 min with 2880 rcf at 5°C before being placed in the cooled autosampler. An aliquot of 0.05 ml sample was injected onto a Asahipak ODP-50 5 µm (125 x 2.0 mm) HPLC column. The mobile phase consisted of acetonitrile and dimethylhexylamin (4.7 mM) in ammonium formiate buffer (1.5 mm). The gradient started at 30% acetonitrile, was kept isocratic for 0.5 min, increased to 95% at 2 min, held at 95% between 2.0 and 4.0 min, stepped down to 30% at 4.1 min and the system was conditioned for the next injection with 30% acetonitrile until 9.5 min. The flow rate was 0.4 ml min−1. The TIS conditions were: 50 psi nebulizer gas, 60 psi turbo gas, 10 psi curtain gas, ionspray voltage 5500 V, and temperature 650°C. There were six calibration concentrations (1.5 to 500 ng ml−1) and the assay performance was judged on the basis of three quality control concentrations (3.5, 35, 350 ng ml−1). Quality control accuracy ranged from 101.9% to 105.9% and precision from 2.7% to 6.3%. The lower quantification limit was 1.5 ng ml−1 using 0.1 ml of blood.
Pharmacokinetic data evaluation
Standard noncompartmental pharmacokinetic parameters included the peak concentration (Cmax), the time of its occurrence (tmax), and the area under the concentration-time curve to 24 h postdose [AUC(0,24 h)] by trapezoidal summation. We did not sample blood for fingolimod beyond 24 h in order to reduce the participation burden on the subjects for repeat clinical visits. We reasoned that any clinically relevant pharmacokinetic influence of atropine on fingolimod would manifest over the 24 h blood sampling interval which included the intravenous treatment. Fingolimod and fingolimod-phosphate Cmax and AUC(0,24 h) were log-transformed and compared between treatments in the model described above for heart rate response parameters.
Results
Safety and tolerability
The study medications were generally well tolerated. There were a total of 48 adverse events reported by 18 subjects. All were mild and resolved spontaneously. The most common were dry mouth, blurred vision, dizziness, and headache. There were no clinically relevant changes in laboratory parameter with the exception of decreased lymphocyte counts, consistent with the mode of action of fingolimod. No clinically relevant changes occurred in blood pressure or electrocardiograms over the study course.
Fingolimod/placebo with concurrent intravenous placebo
On day 1 when oral and intravenous placebo were administered at approximately 08:00, mean heart rate declined slightly from 76 beats min−1 pre-infusion to 68 beats min−1 1 h later due to normal circadian rhythm. The mean heart rate trajectory was slightly steeper in subjects receiving oral fingolimod and intravenous placebo with a preinfusion heart rate of 79 beats min−1 to 61 beats min−1 1 h later indicating the start of the negative chronotropic effect of fingolimod. Figure 1 shows the corresponding full 24-h heart rate trajectories. Subjects receiving oral and intravenous placebo had a normal circadian rhythm. The heart rate trajectory in subjects receiving oral fingolimod showed a clear decline from the placebo group after the first hour but maintained the same circadian pattern over time. The response parameters summarized in Table 1 indicate that fingolimod elicited a mean morning nadir heart rate 18% lower than oral placebo (51 vs. 61 beats min−1) and this occurred a median 4 h postdose.
Figure 1.
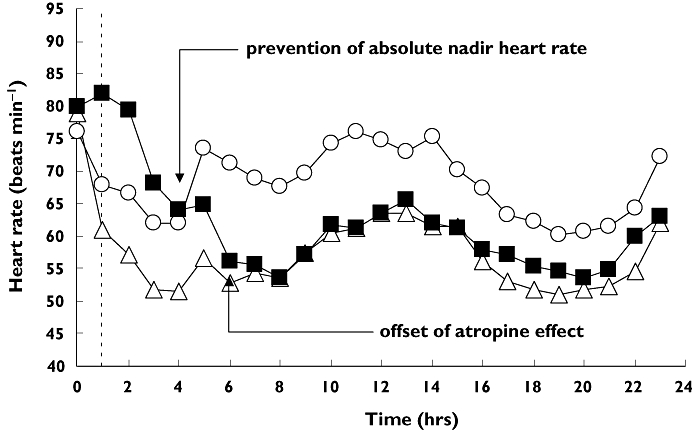
Mean heart rate trajectories after oral placebo (○), fingolimod (▵), and fingolimod with concurrent atropine (▪). Concurrent atropine is able to prevent the absolute nadir heart rate observed for fingolimod alone. The vertical dashed line identifies the end of the intravenous titration phase
Table 1.
Heart rate responses in study part 1: concurrent administration
| Intravenous placebo | Intravenous atropine | |||
|---|---|---|---|---|
| Response | Oral placebo | Oral fingolimod | Oral placebo | Oral fingolimod |
| Pretitration rate (bpm) | 76 ± 8 | 79 ± 8 | 73 ± 6 | 80 ± 11 |
| During titration (1 h): | ||||
| Atropine total dose (mg) | – | – | 1.5 ± 0.6 | 1.5 ± 0.6 |
| Subjects dosed maximally (2 mg) | – | – | 6 | 6 |
| Subjects reaching target rate | – | – | 7 | 1 |
| Maximum rate (beats min−1) | 82 ± 11 | 77 ± 8 | 108 ± 11 | 95 ± 12 |
| Time of maximum rate (min) | 25 (0–60) | 0 (0–40) | 25 (10–60) | 25 (10–60) |
| Full day (24 h): | ||||
| Nadir rate (beats min−1) (0–4 h) | 61 ± 6 | 51 ± 5 | 65 ± 5 | 66 ± 6 |
| AUEC(0–12) (beats min−1 h) | 835 ± 70 | 694 ± 88 | 856 ± 80 | 788 ± 102 |
| AUEC(0–24) (beats min−1 h) | 1566 ± 135 | 1314 ± 173 | 1562 ± 170 | 1434 ± 199 |
Values are arithmetic mean ± SD except for temporal parameters which are median (range). AUEC is area under the heart rate effect–time curve. Target heart rate during atropine titration was 110–120 beats min−1.
Fingolimod/placebo with concurrent atropine
When intravenous atropine was administered with oral placebo in the morning at 08.00 h, five subjects received the maximal allowed atropine dose of 2 mg and did not reach the target heart rate of 110–120 beats min−1 (maximal rates, 87–107 beats min−1). The remaining seven subjects reached the target heart rate with one requiring 2 mg and the others requiring 0.75 to 1.25 mg. The overall average atropine dose was 1.5 mg. The average maximal heart rate was 108 beats min−1 occurring at a median 25 min. By the end of the first hour postdose, average heart rate was 90 beats min−1. These data are summarized in Table 1.
The total dose and titration of atropine when given with oral fingolimod was identical for each subject as when given with oral placebo. Under these conditions, however, only a single subject reached the lower end of the target heart rate range (110 beats min−1). The remaining 11 subjects had maximal heart rates between 68 and 109 beats min−1. The average maximal heart rate was 95 beats min−1 (12% lower than atropine and oral placebo) occurring at a median 25 min. By the end of the first hour postdose, average heart rate was 82 beats min−1.
Figure 1 shows that fingolimod with a concurrent atropine titration raised mean heart rate to normal or above-normal values for a median 5.5 h (individual range, 3–24 h) after fingolimod administration. Thereby the typical heart rate nadir that occurs 3–4 h after the first dose of fingolimod was prevented. This is reflected in the test/reference heart rate nadir (90% confidence interval) in the 0–4 h time interval of 1.30 (1.22, 1.36). By 6 h and thereafter, the effect of an early atropine titration was no longer present and mean heart rate conformed to the typical fingolimod-associated negative chronotropic response pattern fluctuating between 54 and 66 beats min−1.
Fingolimod/placebo with delayed intravenous placebo
When intravenous placebo was given 4 h after oral placebo (approximately 12.00 h), mean heart rate rose slightly from 65 to 68 bats min−1 due to normal circadian rhythm in subjects receiving oral and intravenous placebo. Since the heart rate was at or near its daytime nadir in subjects receiving intravenous placebo 4 h after fingolimod, mean heart rate showed only a slight change over the 60 min observation period (59 to 56 beats min−1). Figure 2 shows the corresponding full 24 h heart rate trajectories. Subjects receiving oral and intravenous placebo had a normal circadian rhythm. The heart rate trajectory in subjects receiving oral fingolimod showed a downward divergence from the placebo group about 2 h postdose but maintained the same circadian pattern. The response parameters summarized in Table 2 indicate that fingolimod elicited a mean morning nadir heart rate 14% lower than oral placebo (54 vs. 63 beats min−1). These responses are consistent with those in the concurrent administration cohort described above. This is reasonable since administration of intravenous placebo at 08.00 h or 12.00 h should not perturb the heart rate trajectories differently in these two treatment groups.
Figure 2.
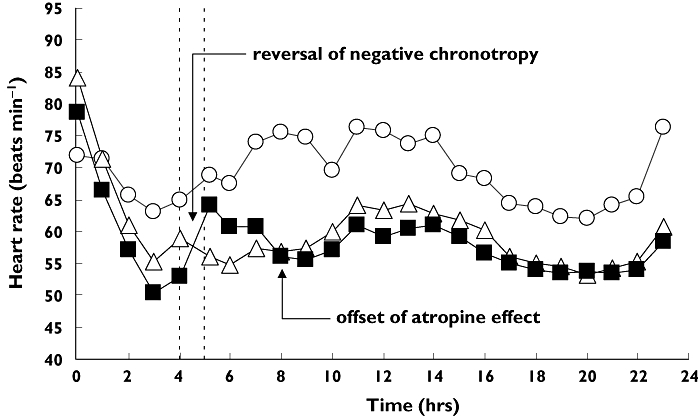
Mean heart rate trajectories after oral placebo (○), fingolimod (▵), and fingolimod with delayed atropine (▪). Atropine is able to reverse the heart rate nadir observed for fingolimod alone. The vertical dashed lines identifies the intravenous titration phase
Table 2.
Heart rate responses in study part 2: delayed administration
| Intravenous placebo | Intravenous atropine | |||
|---|---|---|---|---|
| Response | Oral placebo | Oral fingolimod | Oral placebo | Oral fingolimod |
| Pretitration rate (beats min−1) | 65 ± 10 | 59 ± 11 | 65 ± 10 | 53 ± 8 |
| During titration (1 h): | ||||
| Atropine total dose (mg) | – | – | 1.6 ± 0.5 | 1.9 ± 0.4 |
| Subjects dosed maximally (2 mg) | – | – | 7 | 11 |
| Subjects reaching target rate | – | – | 7 | 0 |
| Maximum rate (beats min−1) | 79 ± 9 | 64 ± 11 | 107 ± 9 | 71 ± 10 |
| Time of maximum rate (min) | 48 (15–60) | 35 (0–60) | 35 (15–55) | 40 (10–60) |
| Rate at 60 min (beats min−1) | 68 ± 13 | 56 ± 9 | 91 ± 8 | 64 ± 8 |
| Full day (24 h): | ||||
| Nadir rate (beats min−1) (0–4 h) | 63 ± 6 | 54 ± 9 | 62 ± 8 | 50 ± 6 |
| AUEC(0–12) (beats min−1 h) | 845 ± 86 | 723 ± 108 | 895 ± 85 | 708 ± 77 |
| AUEC(0,24 h) (beats min−1 h) | 1590 ± 170 | 1363 ± 189 | 1606 ± 161 | 1328 ± 142 |
Values are arithmetic mean ± SD except for tmax which is median (range). AUEC is area under the heart rate effect–time curve. Target heart rate during atropine titration was 110–120 beats min−1.
Fingolimod/placebo with delayed atropine
When intravenous atropine was administered with oral placebo at 12.00 h, five subjects received the maximum allowed atropine dose of 2 mg and did not reach the target heart rate (maximal rates, 89–106 beats min−1). The remaining seven subjects reached the target heart rate of which two required 2 mg and the others required 0.75 to 1.25 mg. The overall average atropine dose was 1.6 mg. The average maximal heart rate was 107 beats min−1 occurring at a median 35 min. By the end of the hour observation period, average heart rate was 91 beats min−1.
When atropine was titrated 4 h after oral fingolimod, 11 subjects received the maximum allowed atropine dose of 2 mg; the remaining subject received 0.75 mg atropine. None of the subjects reached the target heart rate window: maximum heart rates ranged from 51 to 91 beats min−1. The average maximal heart rate was 71 beats min−1 (34% lower than atropine with oral placebo) occurring at a median 40 min. By the end of the hour observation period, average heart rate was 64 beats min−1. Based on the heart rate at the end of the infusion, the response to delayed atropine with oral fingolimod was 30% lower than with oral placebo (64 vs. 91 beats min−1).
Figure 2 shows that when the atropine titration was delayed to 4 h after fingolimod, the typical first-dose nadir had already occurred between 3–4 h postdose. Hence, the test : reference heart rate nadir ratio (90% confidence interval) in the time interval 0–4 h was near unity: 0.98 (0.91, 1.05). Titrating atropine at this time was able to reverse the maximal negative chronotropic effect of fingolimod. Although none of the subjects reached the target 110–120 beats min−1, nonetheless, all subjects reached or exceeded their corresponding normal heart rate at that time of the day (based on the values from the oral and intravenous placebo treatment). The total atropine dose needed to return heart rate to normal averaged 1.4 ± 0.8 mg (individual range, 0.25 to 2 mg). Heart rates remained elevated relative to those after fingolimod alone for a median 3.5 h (individual range, 1–17 h). By 8 h and thereafter, mean heart rate resumed the typical pattern due to the negative chronotropic effect of fingolimod fluctuating between 53 and 61 beats min−1. The test : reference ratio (90% confidence interval) for the heart rate at the end of the infusion was 1.15 (1.04, 1.26). To maintain the higher heart rate would require a longer administration of atropine than used in this study.
Pharmacokinetics
As summarized in Table 3, atropine did not alter any of the fingolimod or fingolimod-phosphate parameters with the single exception of fingolimod-phosphate Cmax with concurrent atropine which was 11% lower than with out atropine and whose lower 90% confidence interval fell slightly outside the equivalence bound. The mean concentration–time profiles from the concurrent administration group are shown in Figure 3. Profiles were similar for the delayed administration group (not shown).
Table 3.
Pharmacokinetics
| Fingolimod | Fingolimod-phosphate | |||||
|---|---|---|---|---|---|---|
| Parameter | Placebo | Atropine | Ratio (90%CI) | Placebo | Atropine | Ratio (90%CI) |
| Concurrent administration: | ||||||
| tmax (h) | 12 (12–12) | 12 (12–12) | – | 6 (6–12) | 12 (6–12) | – |
| Cmax (ng ml−1) | 3.9 ± 0.6 | 3.8 ± 0.5 | 0.97 (0.90, 1.05) | 5.2 ± 1.4 | 4.5 ± 1.1 | 0.89 (0.77, 1.01) |
| AUC(0,24 h) (ng ml−1 h) | 74 ± 11 | 65 ± 9 | 0.90 (0.83, 0.97) | 77 ± 21 | 69 ± 15 | 0.93 (0.83, 1.05) |
| Delayed administration: | ||||||
| tmax (h) | 12 (12–12) | 12 (12–24) | – | 12 (12–12) | 12 (12–12) | – |
| Cmax (ng ml−1) | 4.2 ± 0.7 | 4.0 ± 0.8 | 0.95 (0.86, 1.05) | 5.3 ± 0.9 | 5.3 ± 1.1 | 0.99 (0.88, 1.11) |
| AUC(0,24 h) (ng ml−1 h) | 76 ± 12 | 73 ± 10 | 0.96 (0.89, 1.03) | 81 ± 12 | 81 ± 13 | 1.00 (0.93, 1.07) |
Values are arithmetic mean ± SD except for tmax which is median (range). Ratio (90%CI) is the ratio of geometric means and 90% confidence interval. tmax is time to reach peak concentration, Cmax is peak concentration, AUC is area under the curve.
Figure 3.
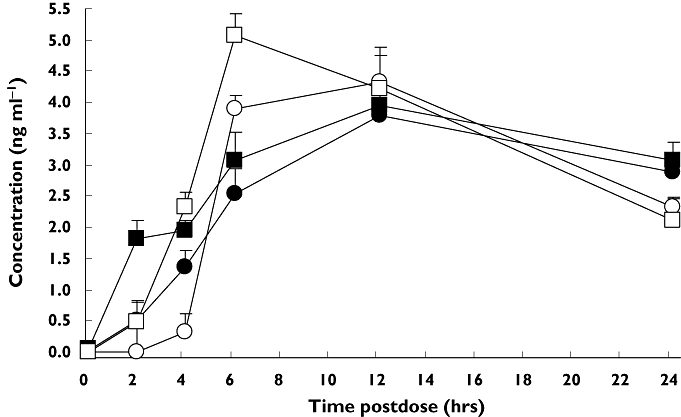
Mean fingolimod and fingolimod-phosphate concentration profiles after concurrent administration of fingolimod with intravenous atropine or placebo. Bars are from the 95% confidence intervals. Fingolimod + IV atropine, (•); fingolimod + IV placebo, (▪); fingolimod-phosphate + IV atropine, (○); fingolimod-phosphate + IV placebo, (□)
Discussion
This study sought to determine whether the antimuscarinic agent atropine can prevent or reverse the negative chronotropic effect of fingolimod in healthy subjects. The combination of fingolimod and atropine was generally well tolerated by healthy subjects. The safety data were remarkable only for decreased lymphocyte counts and decreased heart rate, both anticipated pharmacological effects of fingolimod.
An intravenous titration of atropine at the time of fingolimod administration was able to prevent the typical nadir heart rate that occurs at around 4 h postdose due to the negative chronotropic effect of fingolimod. As applied in this study, the mean atropine total dose of 1.5 mg maintained mean heart rate in the above-normal or normal range for a median 5.5 h relative to the mean heart rate after fingolimod administration alone. Conversely, allowing the nadir heart rate to occur after fingolimod administration and then titrating atropine was able to reverse the maximal negative chronotropic effect of fingolimod at a mean total dose of 1.4 mg. Continuing the atropine titration to a mean total dose of 1.9 mg, as applied in this study, raised and maintained heart rate in the low normal range lasting a median 3.5 h. Both the prophylactic and counteracting effects of atropine on fingolimod-associated negative chronotropy were achievable at atropine doses within the range used clinically to treat sinus bradycardia and acute symptomatic bradycardia [8].
Certain aspects of our study design may limit the generalizability of the results. Our study subjects were young and healthy with normal vagotonus and likely had a robust response to atropine. Elderly patients, on the other hand, often have attenuated vagotonus which may lessen their response to atropine. Patients with multiple sclerosis, however, are generally young and otherwise relatively healthy; hence, our results should serve as a guide to clinicians wishing to use atropine in this setting. Our study focused on acute atropine use after the first dose of fingolimod as this is the likely clinical scenario should pharmacological intervention be needed to increase a patient's heart rate. Consequently, we cannot answer the question whether atropine has any effect on the lessening of the negative chronotropic response that occurs during repeated administration of fingolimod. However, as Figures 2 and 3 show, the heart rate vs. time curve returns within a few hours after the atropine titration to the pattern observed for fingolimod without atropine,suggesting no longterm effect on heart rate from the acute use of atropine.
Assessing the influence of atropine on fingolimod pharmacokinetics was a secondary objective of this study. There is a reversible interconversion of fingolimod and fingolimod-phosphate by sphingosine kinase and irreversible metabolism of fingolimod via CYP4F to metabolites excreted in the urine and faeces [6]. We have not found any published data on whether atropine interferes with sphingosine kinase or CYP4F activity, but we reasoned that any acute influence of atropine would manifest in the first 24 h after the fingolimod dose. We therefore did not require the subjects to return to the clinic for blood sampling to characterize the long elimination phase of fingolimod (half-life 8.8 days). The concentration–time profiles in Figure 3 show the typical slow rise to peak blood levels of both analytes in the presence of intravenous placebo. While delayed atropine did not alter the pharmacokinetics of either analyte, concurrent atropine was associated with a minor 11% lower Cmax for fingolimod-phosphate but no change for its AUC(0,24 h).
We conclude from this study that atropine, over its duration of action, can prevent the negative chronotropic effect of fingolimod when titrated intravenously concurrently with the fingolimod dose. Once the nadir daytime heart rate has occurred after fingolimod administration, atropine is able to reverse the negative chronotropic effect and return heart rate to normal values. Although therapeutic intervention for decreased heart rate was not needed in the multiple sclerosis clinical study [1], the present results indicate that atropine could be used if desired.
Acknowledgments
Conflict of interest: Dr Hunt holds shares in Novartis Pharmaceuticals.
REFERENCES
- 1.Kappos L, Antel J, Comi G. Oral fingolimod (fty720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–40. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 2.Massberg S, von Andrian UH. Fingolimod and sphingosine-1-phosphate: modifiers of lymphocyte migration. N Engl J Med. 2006;355:1088–91. doi: 10.1056/NEJMp068159. [DOI] [PubMed] [Google Scholar]
- 3.Hla T. Signaling and biological actions of sphingosine-1-phosphate. Pharmacol Res. 2003;47:401–7. doi: 10.1016/s1043-6618(03)00046-x. [DOI] [PubMed] [Google Scholar]
- 4.Koyrakh L, Lujan R, Colon J. Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J Neurosci. 2005;25:11468–78. doi: 10.1523/JNEUROSCI.3484-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmouder RL, Barilla D, Wang Y. FTY720: placebo-controlled study of the effect on cardiac rate and rhythm in healthy subjects. J Clin Pharmacol. 2006;46:895–904. doi: 10.1177/0091270006289853. [DOI] [PubMed] [Google Scholar]
- 6.Kovarik JM, Schmouder RL, Slade AJ. Overview of FTY720 clinical pharmacokinetics and pharmacology. Ther Drug Monit. 2004;26:585–7. doi: 10.1097/00007691-200412000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Kovarik JM, Schmouder RL, Barilla D. Single-dose FTY720 pharmacokinetics, food effect, and pharmacological responses in healthy subjects. Br J Clin Pharmacol. 2004;57:586–91. doi: 10.1111/j.1365-2125.2003.02065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Committee ECC, Subcommittees and Task Forces of the American Heart Association. 2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Advanced Cardiac Life Support Part 10.2: toxicology in ECC. Circulation. 2005;112(24):IV126–32. Suppl. [Google Scholar]