

Abstract
Abstract: Pulmonary lymphangitic carcinomatosis is a metastatic lung disease characterized by diffuse spread of the tumour to the pulmonary lymphatic system. We describe the case of a 31-year-old woman who initially received a diagnosis of sarcoidosis based on the results of imaging studies. However, results of a transbronchial biopsy led to the diagnosis of pulmonary lymphangitic carcinomatosis from metastatic colon cancer.
The case: A 31-year-old woman presented to her primary care provider with a nonproductive cough and shortness of breath, which she had experienced intermittently for 1 month. She had no fever, chills or night sweats. She had type 1 diabetes mellitus and hypertension. The patient had experienced multiple miscarriages and was 1 month post partum. She had a 10-pack-year history of smoking. Her father and maternal grandfather both had colon cancer, which, in her father, had been diagnosed after his death at the age of 45. The patient received a diagnosis of exacerbated chronic obstructive pulmonary disease, which was treated with a short course of corticosteroids and broad-spectrum antibiotics. Her condition did not improve.
Two months later, the patient presented to the emergency department with dyspnea. A radiograph of her chest showed interstitial lung disease, thickening of the right paratracheal region and hilar prominence. A computed tomography (CT) scan of her chest revealed extensive paratracheal, subcarinal and para-aortic lymphadenopathy. It also showed diffuse consolidation with a nodular pattern throughout the lungs, particularly in the upper lobes and the lower left lobe. She received a diagnosis of sarcoidosis, which her primary care physician treated with corticosteroids. Her symptoms abated, but less than 1 week later, her condition worsened. Oxygen therapy was started at home.
A respirologist saw the patient 6 months after the onset of her symptoms. She still had exertional dyspnea and a nonproductive cough without hemoptysis. She had lost 20 pounds in the 5 months preceding the visit. She had tachypnea and tachycardia, and was normotensive and afebrile. She did not have enlarged lymph nodes, skin changes or peripheral edema.
Examinations of the patient's abdomen and heart sounds were unremarkable. We heard rhonchi and crackles in the base of both lungs. A radiograph of her chest showed diffuse interstitial and septal thickening in both lungs (Figure 1). A CT scan showed large nodular areas, ground-glass opacities and thickened interlobular septa with interstitial lung disease (Figure 2).
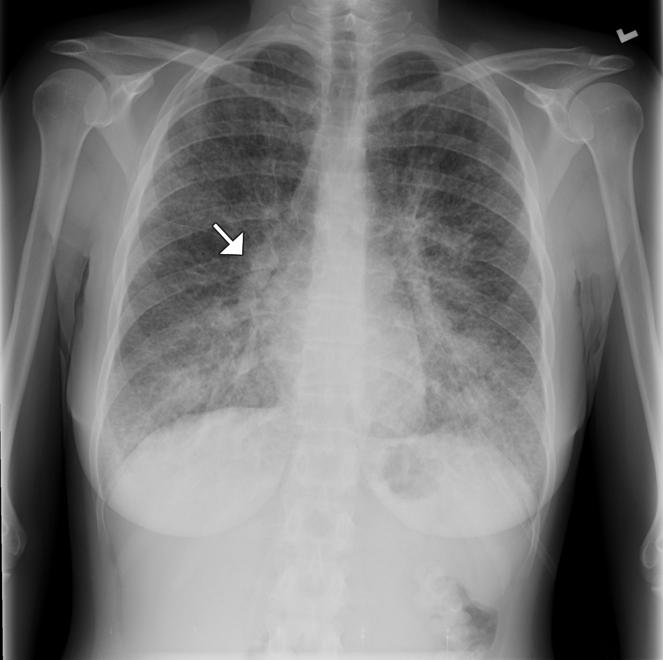
Figure 1: Chest radiograph of a 31-year-old woman showing diffuse interstitial and septal thickening (arrow) in both lungs.
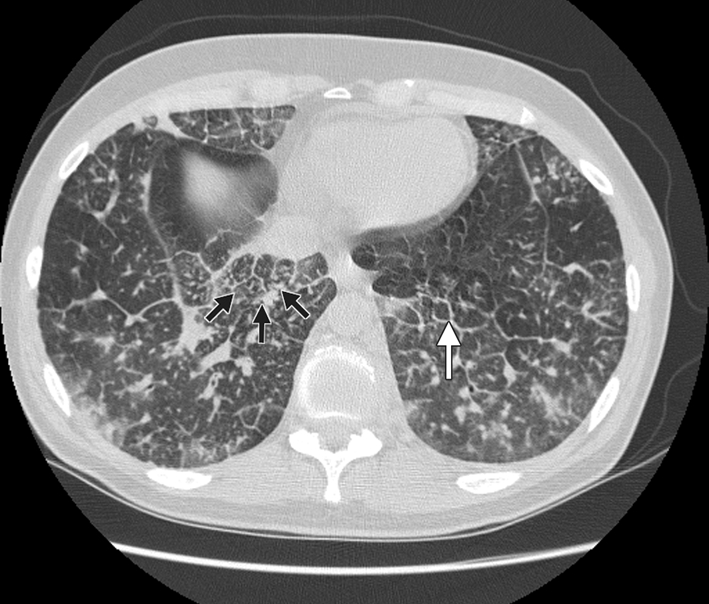
Figure 2: Computed tomography scan showing nodular thickening of interlobular septa (white arrow), seen as polygonal arcades with thickened and nodular limbs, and ground-glass opacities (black arrows).
Results of a transbronchial biopsy showed a moderate to poorly differentiated adenocarcinoma. Immunohistochemical studies of the biopsy specimen showed a pattern of staining consistent with metastatic adenocarcinoma most suggestive of a primary origin in the colon. A colonoscopy showed a mass in her proximal sigmoid colon, and biopsy results confirmed the mass to be a moderately differentiated adenocarcinoma. A bone scan showed multiple tomours in her thorax and lumbar spine.
The patient was admitted to hospital for chemotherapy (FOLFOX4, a regimen of oxaliplatin, leucovorin and 5-fluorouracil). She was transferred the next day to the intensive care unit because of worsening respiratory distress, necessitating mechanical ventilation. The chemotherapy was continued; however, the patient died of respiratory failure 11 days after admission to hospital.
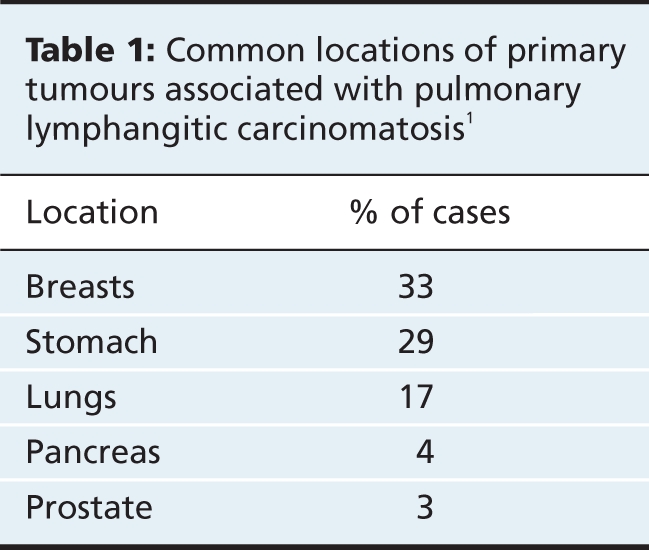
Pulmonary lymphangitic carcinomatosis occurs in 6%–8% of patients with pulmonary metastases.1 The spread of tumour cells to the pulmonary lymphatic system or the adjacent interstitial tissue results in thickening of the bronchovascular bundles and septa. Desmoplastic reaction due to proliferation of neoplastic cells, and lymphatic dilation by edema fluid or tumour secretions contribute to this interstitial thickening. Spread of the neoplasm outside the interstitium and lymphatic spaces into the adjacent parenchyma can result in a nodular pattern.2 Although virtually any metastatic neoplasm can cause pulmonary lymphangitic carcinomatosis, the common locations of the primary tumour are the breasts, stomach, lungs, pancreas and prostate1,3 (Table 1).
Table 1
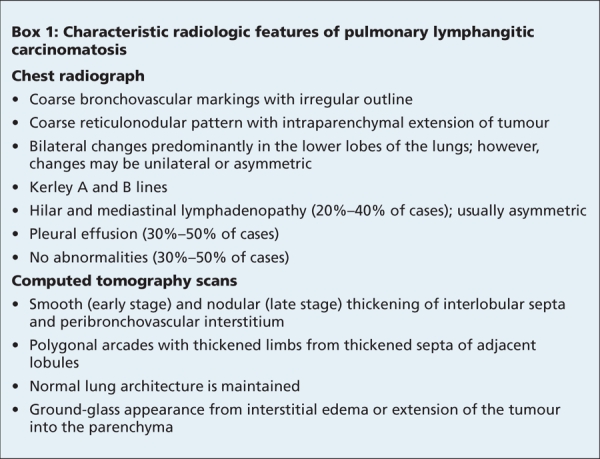
Patients with pulmonary lymphangitic carcinomatosis often present with breathlessness and a nonproductive cough. As with our patient, the onset of pulmonary symptoms may precede diagnosis of the primary tumour; however, the frequency of this presentation is unknown. Although chest radiographs appear normal for 30%–50% of patients with histologically proven disease,2 pulmonary lymphangitic carcinomatosis has several characteristic changes that can be observed on radiographs (Box 1). Transbronchial biopsy is required for a definitive diagnosis.
Box 1.
Pulmonary lymphangitic carcinomatosis can mimic sarcoidosis radiologically. Nodular thickening and ground-glass attenuation are seen in 30%–60% of patients with sarcoidosis. The nodules in sarcoidosis mainly involve central regions of the middle and upper lobes of the lungs. In contrast, changes usually occur in the lower lobes in pulmonary lymphangitic carcinomatosis. Although imaging studies may suggest sarcoidosis, the diagnosis should be confirmed by biopsy results indicating noncaseating granulomas and by the exclusion of other causes of granulomatous disease. Rapid onset and progression of symptoms, asymmetrically enlarged lymph nodes, predominant disease in the lower lobes of the lungs and lack of response to steroids within 2–4 weeks also should alert clinicians to a diagnosis other than sarcoidosis. Thickening of the interlobular septa and peribronchovascular interstitium without a nodular pattern may be seen in other conditions, such as pulmonary edema and idiopathic pulmonary fibrosis.2
Although the diagnosis was delayed for our patient, an earlier diagnosis may not have altered the outcome because of the condition's extremely poor prognosis in most cases. Less than half of patients with pulmonary lymphangitic carcinomatosis who present with respiratory symptoms survive for 3 months.1 However, platinum-based chemotherapy has led to transient remissions in some cases.4
Although our patient did not undergo genetic testing, her young age at presentation and strong family history of colon cancer led to a diagnosis of hereditary nonpolyposis colorectal cancer. This condition accounts for 2%–3% of colorectal cancers and is the most common form of hereditary colorectal cancer.5 It is characterized by an early onset, with an average age at diagnosis of about 45 years and the presence of tumours predominantly on the right side of the colon. Patients with this condition are also at increased risk of having more than 1 primary colorectal tumour at the time of diagnosis (synchronous neoplasms) and of having another primary tumour after successful treatment of the index tumour (metachronous carcinoma). Patients with this condition are more likely than others of the same age and sex to experience tumours of the endometrium, small bowel, stomach, renal pelvis ureter and ovaries. Skin lesions, including carcinomas and nonmalignant lesions such as sebaceous adenomas and keratoacanthomas, are also more common among these patients.6
Our patient had unusual features of pulmonary lymphangitic carcinomatosis, including involvement of the sigmoid colon and metastatis to the lungs and spine without liver involvement. This presentation is seen in only about 9% of rectal cancers.7 If hereditary nonpolyposis colorectal cancer is suspected, a biopsy of the tumour and genetic testing should be carried out to identify mutations. Once identified, relatives at high risk for colorectal cancer should be counselled and screened for these mutations. Those who carry the mutations should have a colonoscopy every 2 years starting at age 20–25.6
Anish Thomas MD Robert Lenox MD Department of Medicine State University of New York Syracuse, New York
Footnotes
Teaching cases are brief case reports that convey clear, practical lessons. Preference is given to common presentations of rare conditions and unusual presentations of common problems. Articles start with the case presentation (500 words maximum) followed by a discussion of the underlying condition (1000 words maximum). We allow about 5 references and encourage visual elements (e.g., tables of the differential diagnosis, clinical features or diagnostic approach). Authors must obtain written consent from patients for publication of their story (form available at www.cmaj.ca/authors/checklist.html). Submit manuscripts online at http://mc.manuscriptcentral.com/cmaj.
This article has been peer reviewed.
Competing interests: None declared.
REFERENCES
- 1.Bruce DM, Heys SD, Eremin O. Lymphangitis carcinomatosa: a literature review. J R Coll Surg Edinb 1996;41:7-13. [PubMed]
- 2.Fraser RS, Muller NL, Colman N, et al. Section VI, Pulmonary neoplasms. In: Fraser and Pare's diagnosis of diseases of the chest. 4th ed. Philadelphia (PA): Elsevier Health Sciences; 1999. p. 1390–7.
- 3.Dennstedt FE, Greenberg SD, Kim HS, et al. Pulmonary lymphangitic carcinomatosis from occult stomach carcinoma in young adults: an unusual cause of dyspnea. Chest 1983;84:787-8. [DOI] [PubMed]
- 4.Kikuchi N, Shiozawa T, Ishii Y, et al. A patient with pulmonary lymphangitic carcinomatosis successfully treated with TS-1 and cisplatin. Intern Med 2007;46:491-4. [DOI] [PubMed]
- 5.Jemal A, Thomas A, Murray T, et al. Cancer statistics, 2002. CA Cancer J Clin 2002;52:23-47. [DOI] [PubMed]
- 6.Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 1998;338:1481-7. [DOI] [PubMed]
- 7.DeVita VT Jr, Hellman. S, Rosenberg SA. Cancers of the gastrointestinal tract. In: Cancer: principles and practice of oncology. 6th ed. Philadelphia (PA): Lippincott Williams & Wilkins; 2001. p. 1229-30.