

Abstract
Oncolytic adenoviral mutants have considerable activity in ovarian cancer. However, the mechanisms by which they induce cell death remain uncertain. dl922-947, which contains a 24 bp deletion in E1A CR2, is more potent than both E1A wild-type adenoviruses and the E1B-55K deletion mutant dl1520 (Onyx-015). We investigated the mode of death induced by three E1A CR2-deleted replicating adenoviruses in models of ovarian cancer and also the importance of E3 11.6 (adenovirus death protein) in determining this mode of death. Ovarian cancer cells were infected with dl922-947 (E3 11.6+) and dlCR2 (E3 11.6−). We also generated dlCR2 tSmac, which also encodes the gene for processed Smac/DIABLO. Classical apoptosis does not occur in adenoviral cell death and there is no role for mitochondria. Expression of Smac/DIABLO does not enhance cytotoxicity nor increase apoptotic features. A role for cathepsins and lysosomal membrane permeability was excluded. Autophagy is induced, but is not the mode of death and may act as a cell survival mechanism. There is no evidence of pure necrosis, while the presence of E3 11.6 does not modulate the mode or extent of cell death. Thus, E1A CR2-deleted oncolytic adenoviral cytotoxicity in ovarian cancer may define a novel mode of programmed cell death.
Keywords: adenovirus, ovarian cancer, apoptosis, autophagy, necrosis
Introduction
Oncolytic viruses can infect cancer cells, multiply selectively within them and cause cell death, with released mature viral particles that infect neighbouring cells. We have previously shown that the E1A CR2 deletion adenoviral mutant dl922-947 has considerable activity in ovarian cancer, and is more potent than both E1A wild-type adenoviruses and the E1B-55K mutant dl1520 (Onyx-015) (Leyton et al., 2006; Lockley et al., 2006). dl922-947 replicates selectively in cells with abnormalities of the Rb pathway and the consequent G1−S checkpoint, findings seen in over 80% of human cancers (Sherr and McCormick, 2002), including ovarian (Yaginuma et al., 1997). However, the exact mechanisms by which adenoviruses cause cell death remain uncertain. A phase I trial of dl922-947 in women with relapsed ovarian cancer is imminent and understanding these mechanisms will be pivotal for further clinical development.
Adenovirus-induced cell death was long presumed to be classically apoptotic (Hall et al., 1998). E1A induces potent apoptosis in many cell systems (Rao et al., 1992), while E4orf4 can induce caspase-dependent cell death (Robert et al., 2002). Adenoviruses also express several proteins that inhibit cell death early after infection; for example, E1B-19K is the viral homologue of Bcl-2 (Chiou et al., 1994) and E1B-55K, in concert with E4orf6, targets p53 for proteasomal destruction (Steegenga et al., 1998). However, many of these studies investigated the expression of viral proteins in isolation, rather than in the context of productive infection, which may have led to oversimplified conclusions. Recently, two groups have made more systematic attempts to monitor selectively replicating viral mutants in human cancer cells. One used Δ24 and Δ24RGD in H460 non-small cell lung carcinoma cells (Abou El Hassan et al., 2004). Both these vectors contain the same 24 bp E1A CR2 deletion as dl922-947, but Δ24 is entirely E3 deleted (Fueyo et al., 2000), whereas Δ24RGD has an intact E3 region and an RGD motif in the fibre knob to redirect binding to αv integrins (Fueyo et al., 2003). The second group used a mutant with a wild-type E1A region under the control of the human telomerase reverse transcriptase (hTERT) promoter in telomerase-positive cancer cells (Ito et al., 2006). Their conclusions were dramatically different; Abou El Hassan used the term ‘necrosis-like programmed cell death’ to describe the actions of Δ24 viruses, whereas Ito concluded that autophagy contributed significantly to the death of glioma cells.
Here we show clearly that classical apoptosis does not occur in ovarian cancer cells treated with E1A CR2 mutant adenoviruses, nor do mitochondria play any determining role. We exclude a role for proteases such as cathepsins, and our data suggest that pure necrosis is not evident. We also show that autophagy is not the mechanism of cell death. Rather, autophagy appears to act as a cell survival response to adenovirus infection in ovarian cancer. Our overall conclusion is that adenovirus-induced cytotoxicity in ovarian cancer cells may constitute a novel mode of programmed cell death.
Results
Biochemical features of classical apoptosis in adenoviral cytotoxicity
We have previously shown that dl922-947 produces greater cytotoxicity in ovarian cancer cells than wild-type adenovirus type 5 and dl1520 (Lockley et al., 2006). Infection of IGROV1 and OVCAR4 cells with dl922-947 and dlCR2 induced similar dose-dependent reductions in cell survival (Figure 1a). To investigate whether co-expression of the potent pro-apoptotic protein Smac/DIABLO could enhance viral activity, we constructed dlCR2 tSmac; cytotoxicity induced by this virus, however, never exceeded that induced by dlCR2 or dl922-947 (Figure 1a), despite a dose- and time-dependent increase in processed Smac/DIABLO expression (not shown).
Figure 1.
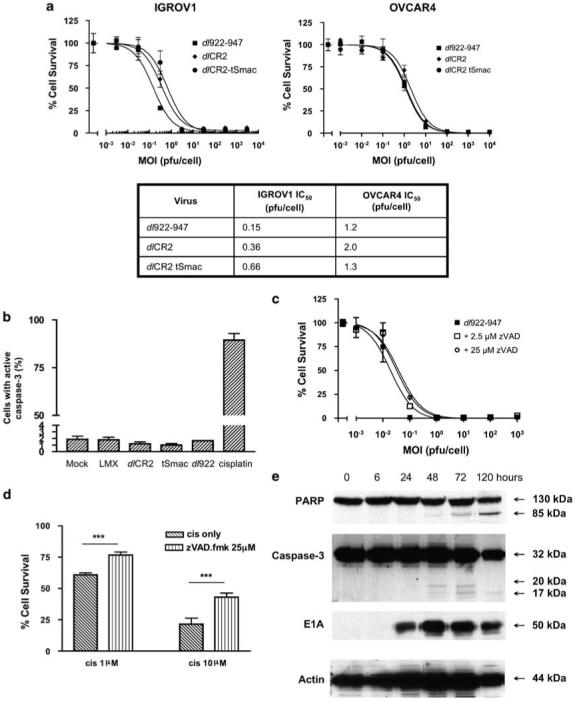
Activity of E1A CR2-deleted adenoviral vectors in ovarian cancer. (a) IGROV1 and OVCAR4 cells were infected with dl922-947, dlCR2 and dlCR2 tSmac (MOI 0.001-10000 plaque-forming unit (PFU) per cell). Cell viability was assessed 120 h later. Results are presented as percentage cell survival compared to mock-infected cells (mean±s.d. n=3). IC50 results are presented in tabular form. (b) IGROV1 cells were infected with dl922-947, dlCR2, dlCR2 tSmac and Ad LM-X (all MOI 10), mock-infected or incubated with cisplatin (10 μm) in triplicate. After 72 h, caspase activation was measured using PhiPhi Lux-G1D2. (c) Following infection with dl922-947 (MOI 0.001-1000), IGROV1 cells were treated with zVAD-fmk (0-25 μm). Cell viability was assessed 144 h later. (d) IGROV1 cells were treated with cisplatin (1 and 10 μm) with and without 25 μm zVAD-fmk. Cell survival was assessed after 72 h by MTT assay. ***P<0.001. (e) IGROV1 cells were harvested up to 120 h following infection with dlCR2 (MOI 10). About 20 μg of protein was separated on sodium dodecyl sulphate (SDS)-PAGE gels and analysed for expression of caspase-3, E1A and PARP by immunoblot.
Since classical apoptosis had been the presumed mechanism of adenovirus-induced cell death and expression of Smac had no effect on viral efficacy, we investigated the role of apoptosis in adenoviral cytotoxicity. Using PhiPhi lux-G1D2, we showed that there was no detectable activation of caspase-3 72 h after infection of IGROV1 or OVCAR4 cells with any of the E1A CR2-deleted viruses, including dlCR2 tSmac. By contrast, 10 μm cisplatin, a classical apoptosis inducer, caused 90% of cells to activate caspase-3 (Figure 1b). The pan-caspase inhibitor zVAD-fmk had no effect on viral cytotoxicity (Figure 1c), but was able to reduce cisplatin-induced death highly significantly (Figure 1d). Western blotting showed only very minor activation of procaspase-3, but with some evidence of poly-ADP ribose polymerase (PARP) cleavage at 72 h (Figure 1e).
IGROV1 and OVCAR4 cells were analysed for exposure of phosphatidylserine (PS) with Annexin V and change in mitochondrial membrane potential by tetramethylrhodamine ethyl ester perchlorate (TMRE) staining. Following viral infection, PS exposure was trivial and only found at late time points once cell membrane permeability had increased (Figure 2a), a process that permits Annexin V entry into the cell. By contrast, 75% of cells treated with cisplatin for 72 h were Annexin V-positive (Figure 2c). Similarly, the number of cells that had lost mitochondrial membrane potential, but not cell membrane potential, never exceeded 12% (Figure 2a), whereas 19% of intact cells treated with cisplatin lost mitochondrial membrane potential (Figure 2c). There was minimal hypodiploid DNA up to 96 h post infection, which coincided with the number of cells that had lost cell membrane integrity (Figure 2b). By contrast, 90% of cisplatin-treated cells had hypodiploid DNA (Figure 2c). These data suggest that classical apoptosis is not induced in ovarian carcinoma cells following infection with replicating adenovirus. To investigate whether adenovirus-infected cells were capable of undergoing apoptosis, IGROV1 cells co-treated with dl922-947 multiplicity of infection of 10 plaque-forming units per cell (MOI 10) and 3 μm cisplatin were assayed for markers of apoptosis. Results indicated that dual treated cells displayed enhanced features of apoptosis, suggesting that cisplatin was able to act in a ‘dominant’ manner (Figure 2d).
Figure 2.

Markers of classical apoptosis. (a) IGROV1 cells were infected with dlCR2 (MOI 10). Cell surface exposure of PS and loss of mitochondrial membrane permeability were analysed using Annexin V-FITC and TMRE over 120 h. DAPI-positive cells were gated out. (b) The presence of hypodiploid DNA in IGROV1 cells infected with dlCR2 (MOI 10) was assessed following incubation with PI. (c) Markers of classical apoptosis in IGROV1 cells treated with cisplatin (10 μm) for 72 h are shown. PS, phosphatidylserine exposure, MMP, mitochondrial membrane potential loss and sub-G1, sub-G1 DNA levels. (d) IGROV1 cells were treated with dl922-947 (MOI 10), cisplatin (3 μm) alone and in combination. After 72 h, cells were harvested and analysed for markers of apoptosis as above. (e) OVCAR4 and OVCAR4-Bcl-2 cells were infected with viruses (all MOI 10) or treated with cisplatin (10 μm). Cell viability was assessed 72 h later. All results represent mean±s.d. n=3.
Mitochondria have no defining role in cell death caused by replicating adenovirus
We further tested the role of mitochondria in viral cytotoxicity. Overexpression of Bcl-2 had no effect on viral cytotoxicity, but significantly reduced cisplatin sensitivity (Figure 2e). This is particularly significant, as the anti-apoptotic effects of Bcl-2 are mediated through mitochondria (Gross et al., 1999) and Bcl-2 is an important mediator of chemotherapy resistance in ovarian cancer (Opipari et al., 2004). Second, cytochrome c release did not increase in virus-treated IGROV1 cells compared to untreated controls (Supplementary Figure B). Finally, cyclosporin A, the cyclophilin-D inhibitor, was found to have no effect on dl922-947 efficacy (Supplementary Figure C). These data do not support a role for mitochondria or Bcl-2 in adenoviral cytotoxicity, as does the failure of Smac overexpression to augment cell death levels.
Lysosomes and autophagy act to promote cell survival
Lysosomal proteases, such as cathepsins, can mediate non-classical apoptosis (Castino et al., 2002). The cysteine protease inhibitor E64 (Figure 3a) and the aspartic acid protease inhibitor pepstatin A (data not shown) had minimal effects dlCR2- or dl922-947-mediated cell death in IGROV1 and OVCAR4 cells, suggesting that the main lysosomal cathepsins, B and D, have little role in virus-mediated cell death. Examination of lysosomal stability using acridine orange (AO) staining indicated that infection with virus, in contrast to cisplatin treatment, caused lysosomes to stabilize (Figure 3b), suggesting that lysosomal number or size increased in response to virus, despite an increase in oxidative stress.
Figure 3.

Assessment of non-classical apoptosis. (a) Following infection with dlCR2 (MOI 10), IGROV1 cells were treated with E64 cysteine protease inhibitor (0-100 μm). Cell viability was assessed up to 144 h later by MTT assay (72 h results shown). Each bar represents normalized survival compared to mock-transfected cells at the same dose of E64; mean±s.d. n=3. (b) IGROV1 cells were infected with dlCR2 (MOI 10) or treated with cisplatin (10 μm). After 72 h, lysosomal stability was assessed following AO staining.
Autophagy is a mechanism by which cells recycle organelles and can also be induced to combat infection (Terman et al., 2003). It has been suggested as the main pathway of cell death with replicating adenoviral vectors in glioma cells (Ito et al., 2006). During autophagy, a double-membraned autophagosome forms, engulfing cytoplasmic constituents, which later fuses with a lysosome to degrade the contents (Levine and Yuan, 2005). To examine the role of autophagy in viral cytotoxicity, we first examined cells with transmission electron microscopy (TEM) and stained autophagosomes with monodansylcadaverine (MDC). Both techniques showed an increase in the number of autophagosomes with replicating virus infection. TEM showed a general increase in the number of vacuoles and lysosomes in the cytoplasm of virus-infected cells compared to controls (Figure 4a). Many of these were double-membraned and had contents that included other membranes, which forms the definition of an autophagosome, the early stepof autophagy (Kroemer et al., 2005). MDC staining confirmed early autophagosome formation (Figure 4b), while co-infection of IGROV1 cells with dl922-947 and Ad GFP-LC3 showed punctate LC3 staining as it associates with autophagosome membranes (Figure 4c). We then used 3-methyladenine (3MA), the class III phosphoinositide-3-kinase inhibitor, to block early autophagocytic signalling (Stroikin et al., 2004), and chloroquine (CQ), a lysosomotropic drug that raises lysosomal pH and impairs autophagic protein degradation, the last step in the autophagic process (Amaravadi et al., 2007). Both drugs augmented virus-induced death in a dose dependent manner (Figure 4d), confirming that autophagy occurs in ovarian cancer cells following viral infection, but that it may have a protective role. Furthermore, inhibition of autophagy with 3MA and CQ did not increase the features of apoptosis, suggesting no alteration in the mode of cell death (Supplementary Figure D).
Figure 4.
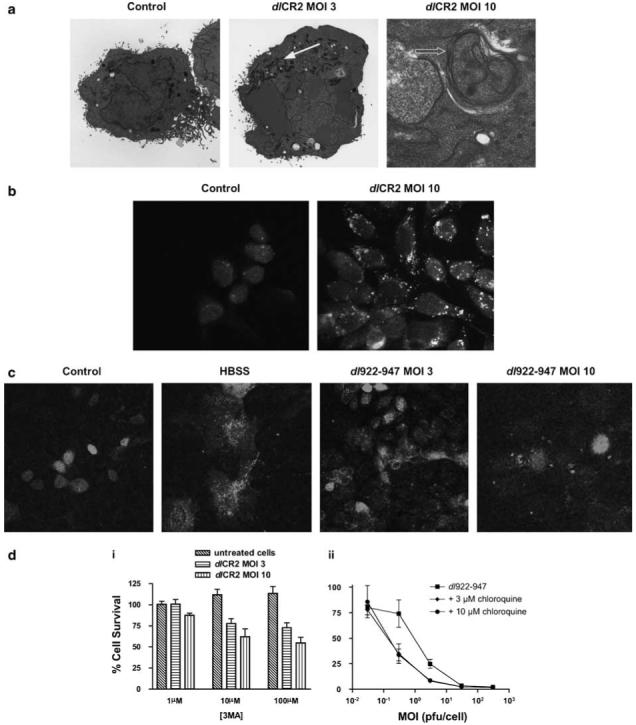
Autophagy. (a) IGROV1 cells were prepared for TEM 72 h following infection with dlCR2 and Ad LM-X (both MOIs 3 and 10). Features of autophagy include increased numbers of lysosomes (closed arrow), the presence of autophagosomes with double membranes (open arrow). (b) IGROV1 cells were infected with dl922-947 (MOI 10) and imaged 48 h later following staining with monodansylcadaverine. (c) IGROV1 cells were co-infected with dl922-947 (MOI 10) and Ad GFP-LC3 (MOI 0.2) and assayed 72 h later for the formation of autophagosomes by confocal microscopy. As positive control, cells infected with Ad GFP-LC3 only were incubated in HBSS for 16 h prior to confocal imaging. (d) (i) IGROV1 cells were infected with dlCR2 (MOI 3 and 10) in the presence of 3-methyladenine (3MA; 0-100 μm). Cell viability was assessed 72 h later. (ii) IGROV1 cells were infected with dl922-947 (MOI 0.03-300) and re-fed with 0-10 μm chloroquine 3 h later. Cell survival was assayed 120 h later and results represent survival normalized for mock-transfected cells at each dose of chloroquine.
Cell morphology and necrosis
Necrosis is often regarded as the default mode of cell death in the absence of apoptosis, and there are few well-validated assays compared to apoptosis. We examined gross cell morphology using phase-contrast microscopy. Virus-infected cells were rounded with some membrane blebbing. Cell rounding became more apparent with time, but there was no obvious rupture of membranes (Figure 5a). We further examined cellular morphology using TEM. dlCR2 produced marked changes: viral particles were seen throughout the cell, with virions assembling in the nucleus. Other features included chromatin condensation, dilation of mitochondrial cristae, Golgi and endoplasmic reticulum, and disorganization of the cytoplasm (Figure 5b). These features have all previously been described in association with apoptosis (Cereghetti and Scorrano, 2006). By contrast, the non-replicating control virus Ad LM-X had a very limited effect. Hoechst 33342 staining did demonstrate some nuclear swelling in virus-infected cells (Supplementary Figure E). This change is a potential marker of necrosis, but the TEM images suggest that this results from viral particle assembly. Hoechst 33342 staining following virus infection also showed some apoptotic features in occasional cells, but these were dramatically less than seen with cisplatin treatment and were absent in the vast majority of cells.
Figure 5.
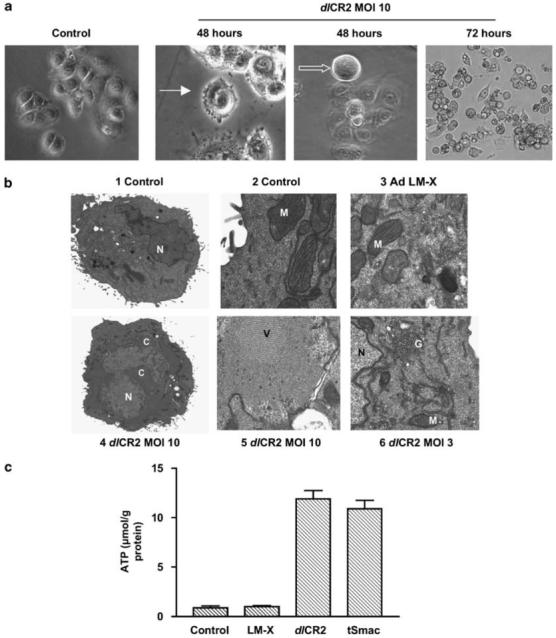
Cell architecture and necrosis. (a) Cells were infected with dlCR2 and imaged up to 72 h later under phase contrast microscopy. After 48 h, individual cells show membrane blebbing (solid arrow) and appear rounded (open arrow). By 72 h, widespread cell rounding and detachment is seen, but without disruption to cell membranes. (b) IGROV1 cells were infected with Ad LM-X (MOI 10) or dlCR2 (MOI 3 and 10) and prepared for TEM 72 h later. (i and ii) Mock-infected control cells; (iii) Ad LM-X (MOI 10); (iv and v) dlCR2 (MOI 10); (vi) dlCR2 (MOI 3). C, condensed chromatin; G, disrupted Golgi; M, mitochondria; N, nucleus; V, intra-nuclear virus assembly. (c) IGROV1 cells were infected with Ad LM-X, dlCR2 and dlCR2 tSmac (all MOI 10). Intracellular adenosine triphosphate (ATP) levels were measured 72 h later using a luciferase-based assay as detailed in the ‘Materials and methods’ section. Results represent mean±s.d., n=3.
There are few biochemical markers of necrosis. However, a fall in intracellular adenosine triphosphate (ATP) levels is frequently seen (Lelli et al., 1998). ATP levels increased following infection with all E1A CR2-deleted viruses and never fell below the level of the untreated control (Figure 5c).
Discussion
These results provide evidence that classical apoptosis is not the mechanism by which replicating adenoviruses cause cell death. Some apoptotic morphological features are found, but there are dramatically fewer than with cisplatin treatment. The morphology is clearly strongly influenced by virus, as shown by the swollen nuclei with intra-nuclear virus assembly, as well as the large number of virions in cytoplasm and the increase in lysosomes and autophagosomes.
The complete lack of a defining role for mitochondria is especially striking, since they are central both to classical apoptosis and also some forms of necrosis (Baines et al., 2005). Mitochondrial changes were induced by the virus, with some mitochondrial membrane potential loss and ballooning of cristae on TEM. However, these events did not drive cell death, as their modulation had no effect on overall cytotoxicity. IGROV1 and OVCAR4 cells can undergo mitochondria-dependent classical apoptosis in the presence of virus, as was evident in dominant response to cisplatin following dual treatment. However, this is selective, as co-expression of Smac had no effect, implying that viruses can inhibit some, but not all, forms of mitochondrial-dependent death. The lysosomal studies showed that cathepsins did not play a major role.
We also showed that autophagy may be a cell survival mechanism in the presence of virus, rather than a cell death pathway, as suggested recently (Ito et al., 2006; Jiang et al., 2007). Treatment with 3MA and chloroquine caused a significant increase in cytotoxicity. Others have previously shown autophagy to be a survival mechanism in varied tumour models (Bauvy et al., 2001; Amaravadi et al., 2007). Cells are also known to instigate autophagy as a defence mechanism in response to bacterial or viral infections (Talloczy et al., 2002). Ito et al. suggested that autophagy had the defining role in cell death: an increase in acidic and autophagic vacuoles was demonstrated, but showing that this was the main cause of death rather than simply a cellular response to virus proved difficult. A single high concentration of 3MA (1 mm) was used, which caused only 18.3% change in cell viability; 1 mm 3MA is likely to have many non-specific effects (Caro et al., 1988). By contrast, we were able to show that the 3MA and chloroquine effects depended upon both dose of virus and drug and that these drugs caused no change in the mode of cell death.
Proving the existence of necrosis is difficult in the absence of classical markers such as exist for apoptosis. We examined cellular morphology using three techniques and changes in ATP concentration. Although the eventual result of viral infection is lysis, adenoviral cytotoxicity does not fit a purely necrotic profile, as there is no wholesale disruption of cellular membranes and organelles nor a reduction in ATP concentration, typical features of necrosis. It was recently suggested that the E1A CR2-deleted virus Δ24 caused a type of cell death described as necrosis-like programmed cell death (Abou El Hassan et al., 2004). Infection with viruses did not result in the appearance of apoptotic features, with the exception of PS exposure. However, the authors failed to examine any of the key features that would constitute pure necrosis and used the term necrosis to signify absence of (classical) apoptosis rather than necrosis itself. They also did not investigate other types of cell death or potential mechanisms of ‘programming’. They themselves say the use of the term necrosis is arbitrary.
Others have previously suggested that viruses lacking E3 11.6, the adenovirus death protein (ADP), are less potent than those with an intact E3 region (Tollefson et al., 1996). Our results also indicate that ADP does not affect either the mode or extent of viral cytotoxicity in ovarian cancer. All parameters investigated were evident in cells infected with both dl922-947 and dlCR2: indeed, dlCR2 induced many effects more rapidly than dl922-947, the cause of which is uncertain. Abou El Hassan also found that the nature of cell death was the same in glioma cells infected with Δ24 and Δ24RGD (Abou El Hassan et al., 2004) and suggested that the presence of E3 in Δ24RGD increased potency. However, this increased potency could equally have resulted from increased infectivity afforded by the RGD motif in glioma cells rather than the presence of E3. The mechanism of ADP activity remains elusive, although it is believed to bind to MAD2B (Ying and Wold, 2003). Recent results suggest that deletions in the E1 region could compensate for absence of ADP in A549 cells (Subramanian et al., 2006) and, although E1A CR2 mutations were not specifically investigated, it is possible that deletions in this region could also enhance viral spread.
Replicating adenovirus-induced cell death may therefore be described as a strongly virus-controlled version of non-classical cell death, one that has not been fully characterized before. It is programmed, but with the ultimate control coming from virus rather than cell. This may explain the early cleavage of PARP, found despite the significant caspase activation. If PARP were not inactivated, necrosis could have occurred, as PARP activity can result in depletion of NAD+ and ATP levels (Zong et al., 2004). The cells make a valiant attempt to undergo apoptosis, as witnessed by some of the morphological features found, but the control by the virus is sufficiently thorough that biochemical characteristics are effectively abolished. The virus uses the cell processes for its own ends, to replicate and assemble itself, and then causes cell lysis, although the final pathway of this lysis remains to be fully determined.
Materials and methods
Cell culture, adenoviral construction and cell viability assays
IGROV1 and OVCAR4 were incubated at 37 °C with 5% CO2 in air, in Dulbecco’s modified Eagle’s medium with 10% foetal bovine serum (FBS). Bcl2-overexpressing OVCAR4 cells were produced previously (McNeish et al., 2003). dl922-947 is an Ad5 vector deleted in the region encoding amino acids 122-129 of the E1A CR2 domain as well as in E3B (Heise et al., 2000). dlCR2 contains the same E1A CR2 deletion as dl922-947 and its construction is described elsewhere (Leyton et al., 2006). To generate dlCR2 tSmac, an expression cassette consisting of the cytomegalovirus immediate-early promoter, the gene for processed Smac and a polyadenylation signal were ligated as a SalI fragment into pShuttle dlCR2 immediately downstream of E1, to create pShuttle dlCR2 tSmac. Viruses dlCR2 and dlCR2 tSmac were created by linearizing pShuttle dlCR2/pShuttle dlCR2 tSmac with PmeI and recombining with pAd Easy1 in electrocompetent Escherichia coli. Virus was rescued following transfection of the recombinant plasmid into 293 cells, followed by amplification and purification using double density CsCl ultracentrifugation. Both dlCR2 and dlCR2 tSmac are entirely E3 deleted, corresponding to nucleotides 28130-30820 of the Ad5 genome (He et al., 1998). PCR evaluation of E3 genes in dl922-947 and dlCR2 is presented in Supplementary Figure A. The E1-deleted control vector Ad LM-X is described elsewhere (McNeish et al., 2001). Ad GFP-LC3 is described elsewhere (Bampton et al., 2005) and was a kind gift of Dr Aviva Tolkovsky (University of Cambridge, UK).
For cell viability assays, 104 cells were infected with adenovirus in serum-free medium and re-fed 2 h later with 5% FBS-containing medium. Cell viability was assayed by MTT and Trypan blue exclusion assays; both assays gave identical results. All viability assays were performed in both IGROV1 and OVCAR4 triplicate at least three times. Results from the two cell lines were always similar.
Western blots
Cells were lysed in 150 mm NaCl, 50 mm Tris (pH 7.5), 0.05% SDS, 1% Triton X-100. Antibody binding was visualized using enhanced chemiluminescence (Amersham Pharmacia, Bucks, UK). Caspase-3 antibody was obtained from Alexis Biochemicals, Nottingham, UK (catalogue no. ALX-804-305) and PARP and Bcl-2 antibodies from Santa Cruz Biotechnology, CA, USA (catalogue nos. sc-8007 and sc-509, respectively).
Flow cytometry
For AO analysis, cells were incubated for 15 min in the dark at 37 °C with 15 μgml-1 AO, then analysed on a FacsCalibur (Becton Dickinson, Oxford, UK). For other analyses, cells were incubated with 40 nm TMRE (Invitrogen, Oregon, USA) for 10 min, washed in phosphate-buffered saline (PBS) and resuspended with Annexin V-Alexa 647 conjugate (2.5 μgml-1) for 15 min. 4-6-Diamidino-2-phenylindole (DAPI; 1 μgml-1) was then added. Cells were also incubated with 20 μm dihydroethidium for 30 min before washing and Annexin V and DAPI addition. For caspase activation analysis, cells were incubated for 60 min with 10 μm PhiPhiLux-G1D2 substrate solution (OncoImmunin Inc., Gaithersburg, MD, USA) as per the manufacturer’s instructions before DAPI addition, then analysed on a FACS LSRII (Becton Dickinson) and the data processed using FlowJo software (Tree Star, OR, USA). Samples were then fixed in 70% ice-cold ethanol for 30 min, washed twice in sub-G1 buffer (192 μm Na2HPO4, 4 μm citric acid), treated with 100 μgml-1 RNase and analysed for hypodiploid DNA after addition of 50 μgml-1 propidium iodide (PI).
Assays of necrosis
For ATP assays, protein extracts were suspended in standard reaction buffer containing luciferase and luciferin according to the manufacturer’s instructions (ATP Determination Kit; Invitrogen), and the luminescence read on a Thermo Labsystems Luminoskan Ascent luminometer.
Confocal microscopy
Cells were fixed in 4% paraformaldehyde (PFA) or a 6:3:1 ethanol:chloroform:acetic acid mix for 30 min at room temperature, then washed three times with PBS and incubated with 10 μgml-1 Hoechst 33342 or 50 μm MDC for 15 min, washed three times in PBS and mounted onto slides. Slides were examined on a Zeiss LSM 510 Laser Scanning microscope.
Transmission electron microscopy
IGROV1 cells were incubated with virus for 72 h. They were fixed in 4% PFA and 2.5% gluteraldehyde, postfixed in reduced osmium tetroxide followed by 1% tannic acid, washed in 1% sodium sulphate then water and dehydrated in a graded ethanol series. The preparation was then washed in propylene oxide, infiltrated with araldite/propylene oxide overnight and then with araldite alone and embedded in resin. Sections were cut, placed on copper grids and stained with uranyl acetate in methanol followed by Reynold’s lead citrate and viewed in a JEOL 1010 TEM.
Statistics
All dose-response curves, Kaplan-Meier survival curves and statistical analyses were generated using GraphPad Prism version 4 (GraphPad Software, San Diego, CA, USA). All comparisons are two-tailed t-tests, unless otherwise stated.
Supplementary Material
Acknowledgements
This research was funded by Cancer Research UK (grant C9423/A4119). We acknowledge the excellent technical assistance of Ken Blight in the Electron Microscopy Department, Cancer Research UK London Research Institute.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- Abou El Hassan MAI, van der Meulen-Muileman I, Abbas S, Kruyt FAE. Conditionally replicating adenoviruses kill tumor cells via a basic apoptotic machinery-independent mechanism that resembles necrosis-like Programmed Cell Death. J Virol. 2004;78:12243–12251. doi: 10.1128/JVI.78.22.12243-12251.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- Bauvy C, Gane P, Arico S, Codogno P, Ogier-Denis E. Autophagy delays sulindac sulfide-induced apoptosis in the human intestinal colon cancer cell line HT-29. Exp Cell Res. 2001;268:139–149. doi: 10.1006/excr.2001.5285. [DOI] [PubMed] [Google Scholar]
- Caro LH, Plomp PJ, Wolvetang EJ, Kerkhof C, Meijer AJ. 3-Methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. Eur J Biochem. 1988;175:325–329. doi: 10.1111/j.1432-1033.1988.tb14200.x. [DOI] [PubMed] [Google Scholar]
- Castino R, Pace D, Demoz M, Gargiulo M, Ariatta C, Raiteri E, et al. Lysosomal proteases as potential targets for the induction of apoptotic cell death in human neuroblastomas. Int J Cancer. 2002;97:775–779. doi: 10.1002/ijc.10139. [DOI] [PubMed] [Google Scholar]
- Cereghetti GM, Scorrano L. The many shapes of mitochondrial death. Oncogene. 2006;25:4717–4724. doi: 10.1038/sj.onc.1209605. [DOI] [PubMed] [Google Scholar]
- Chiou SK, Tseng CC, Rao L, White E. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J Virol. 1994;68:6553–6566. doi: 10.1128/jvi.68.10.6553-6566.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA, et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J Natl Cancer Inst. 2003;95:652–660. doi: 10.1093/jnci/95.9.652. [DOI] [PubMed] [Google Scholar]
- Fueyo J, Gomez-Manzano C, Alemany R, Lee P, McDonnell T, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Hall A, Dix B, O’Carroll S, Braithwaite A. p53-dependent cell death/apoptosis is required for a productive adenovirus infection. Nat Med. 1998;4:1068–1072. doi: 10.1038/2057. [DOI] [PubMed] [Google Scholar]
- He T-C, Zhou S, Da Costa L, Yu J, Kinzler K, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A, et al. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6:1134–1139. doi: 10.1038/80474. [DOI] [PubMed] [Google Scholar]
- Ito H, Aoki H, Kuhnel F, Kondo Y, Kubicka S, Wirth T, et al. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J Natl Cancer Inst. 2006;98:625–636. doi: 10.1093/jnci/djj161. [DOI] [PubMed] [Google Scholar]
- Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007;99:1410–1414. doi: 10.1093/jnci/djm102. [DOI] [PubMed] [Google Scholar]
- Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;12(Suppl 2):1463–1467. doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed] [Google Scholar]
- Lelli JL, Jr, Becks LL, Dabrowska MI, Hinshaw DB. ATP converts necrosis to apoptosis in oxidant-injured endothelial cells. Free Radic Biol Med. 1998;25:694–702. doi: 10.1016/s0891-5849(98)00107-5. [DOI] [PubMed] [Google Scholar]
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyton J, Lockley M, Aerts JL, Baird SK, Aboagye EO, Lemoine NR, et al. Quantifying the activity of adenoviral E1A CR2 deletion mutants using renilla luciferase bioluminescence and 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography imaging. Cancer Res. 2006;66:9178–9185. doi: 10.1158/0008-5472.CAN-06-1539. [DOI] [PubMed] [Google Scholar]
- Lockley M, Fernandez M, Wang Y, Li NF, Conroy SE, Lemoine NR, et al. Activity of the adenoviral E1A deletion mutant dl922-947 in ovarian cancer: comparison with adenovirus wild-type, bioluminescence monitoring and intraperitoneal delivery in icodextrin. Cancer Res. 2006;66:989–998. doi: 10.1158/0008-5472.CAN-05-2691. [DOI] [PubMed] [Google Scholar]
- McNeish IA, Bell S, McKay T, Tenev T, Marani M, Lemoine NR. Expression of Smac/DIABLO in ovarian carcinoma cells induces apoptosis via a caspase-9-mediated pathway. Exp Cell Res. 2003;286:186–198. doi: 10.1016/s0014-4827(03)00073-9. [DOI] [PubMed] [Google Scholar]
- McNeish IA, Tenev T, Bell S, Marani M, Vassaux G, Lemoine N. Herpes simplex virus thymidine kinase/ganciclovir-induced cell death is enhanced by co-expression of caspase-3 in ovarian carcinoma cells. Cancer Gene Ther. 2001;8:308–319. doi: 10.1038/sj.cgt.7700305. [DOI] [PubMed] [Google Scholar]
- Opipari AW, Jr, Tan L, Boitano AE, Sorenson DR, Aurora A, Liu JR. Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res. 2004;64:696–703. doi: 10.1158/0008-5472.can-03-2404. [DOI] [PubMed] [Google Scholar]
- Rao L, Debbas M, Sabbatini P, Hockenbery D, Korsmeyer S, White E. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc Natl Acad Sci USA. 1992;89:7742–7746. doi: 10.1073/pnas.89.16.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Miron M-J, Champagne C, Gingras M-C, Branton PE, Lavoie JN. Distinct cell death pathways triggered by the adenovirus early region 4 ORF 4 protein. J Cell Biol. 2002;158:519–528. doi: 10.1083/jcb.200201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- Steegenga WT, Riteco N, Jochemsen AG, Fallaux FJ, Bos JL. The large E1B protein together with the E4orf6 protein target p53 for active degradation in adenovirus infected cells. Oncogene. 1998;16:349–357. doi: 10.1038/sj.onc.1201540. [DOI] [PubMed] [Google Scholar]
- Stroikin Y, Dalen H, Loof S, Terman A. Inhibition of autophagy with 3-methyladenine results in impaired turnover of lysosomes and accumulation of lipofuscin-like material. Eur J Cell Biol. 2004;83:583–590. doi: 10.1078/0171-9335-00433. [DOI] [PubMed] [Google Scholar]
- Subramanian T, Vijayalingam S, Chinnadurai G. Genetic identification of adenovirus type 5 genes that influence viral spread. J Virol. 2006;80:2000–2012. doi: 10.1128/JVI.80.4.2000-2012.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talloczy Z, Jiang W, Virgin HW, Leib DA, Scheuner D, Kaufman RJ, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman A, Dalen H, Eaton JW, Neuzil J, Brunk UT. Mitochondrial recycling and aging of cardiac myocytes: the role of autophagocytosis. Exp Gerontol. 2003;38:863–876. doi: 10.1016/s0531-5565(03)00114-1. [DOI] [PubMed] [Google Scholar]
- Tollefson AE, Ryerse JS, Scaria A, Hermiston TW, Wold WS. The E3-11.6-kDa adenovirus death protein (ADP) is required for efficient cell death: characterization of cells infected with adp mutants. Virology. 1996;220:152–162. doi: 10.1006/viro.1996.0295. [DOI] [PubMed] [Google Scholar]
- Yaginuma Y, Hayashi H, Kawai K, Kurakane T, Saitoh Y, Kitamura S, et al. Analysis of the Rb gene and cyclin-dependent kinase 4 inhibitor genes (p16INK4 and p15INK4B) in human ovarian carcinoma cell lines. Exp Cell Res. 1997;233:233–239. doi: 10.1006/excr.1997.3560. [DOI] [PubMed] [Google Scholar]
- Ying B, Wold WSM. Adenovirus ADP protein (E3-11.6K), which is required for efficient cell lysis and virus release, interacts with human MAD2B. Virology. 2003;313:224–234. doi: 10.1016/s0042-6822(03)00287-3. [DOI] [PubMed] [Google Scholar]
- Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.