

Abstract
Bacterial populations produce a small number of persister cells that exhibit multidrug tolerance. Persister cells are largely responsible for the antibiotic recalcitrance of biofilm infections. The mechanism of persister cell formation largely remains unknown due to the challenges in identifying persister genes. We screened an ordered comprehensive library of 3,985 Escherichia coli knockout strains to identify mutants with altered antibiotic tolerance. Stationary-state cultures in 96-well plates were exposed to ofloxacin at a concentration which allows only tolerant persister cells to survive. The persister cell level of each culture was determined. A total of 150 mutants with decreased persistence were identified in the initial screen, and subsequent validation confirmed that neither the growth rate nor the ofloxacin MIC was affected for 10 of them. The genes affected in these strains were dnaJ and dnaK (chaperones), apaH (diadenosine tetraphosphatase), surA (peptidyl-prolyl cis-trans isomerase), fis and hns (global regulators), hnr (response regulator of RpoS), dksA (transcriptional regulator of rRNA transcription), ygfA (5-formyl-tetrahydrofolate cyclo-ligase), and yigB (flavin mononucleotide [FMN] phosphatase). The prominent presence of global regulators among these strains pointed to the likely redundancy of persister cell formation mechanisms: the elimination of a regulator controlling several redundant persister genes would be expected to produce a phenotype. This observation is consistent with previous findings for a possible role of redundant genes such as toxin/antitoxin modules in persister cell formation. ygfA and yigB were of special interest. The mammalian homolog of YgfA (methenyltetrahydrofolate synthetase) catalyzes the conversion of 5-formyl-tetrahydrofolate (THF) into the rapidly degraded 5,10-methenyl-THF, depleting the folate pool. The YigB protein is a phosphatase of FMN which would deplete the pool of this cofactor. Stochastic overexpression of these genes could lead to dormancy and, hence, tolerance by depleting the folate and FMN pools, respectively. Consistent with this scenario, the overexpression of both genes produced increased tolerance to ofloxacin.
Persister cells are multidrug-tolerant cells that contribute to the antibiotic recalcitrance of biofilm infections (12, 27, 53). Biofilms are implicated in many bacterial infections, including those associated with indwelling devices, dental disease, endocarditis, and cystic fibrosis (16, 18, 23, 38, 46). Understanding the the mechanism of persister cell formation/maintenance is likely to lead to new effective therapies for the treatment of biofilm infections. However, research in this area has been hindered by the difficulties in identifying persister genes.
Persisters are phenotypic variants of the wild type that are tolerant to killing by antibiotics (8, 27). They are survivor cells that make up a small part of the population: 10−6 to 10−4 in exponentially growing cultures and ∼10−2 in stationary phase (33). A targeted search for mutants of Escherichia coli with high levels of persistence led to the finding of a hipA7 allele, but a mutant with a knockout of the gene appeared to have no phenotype (9, 10, 39, 40, 49), and the work of the early pioneers in this field remained largely unknown. Persisters are tolerant to all currently available antibiotics and are found in all species examined (33). Recent progress in this area can be summarized as follows: persisters are nongrowing (6), dormant (50) cells; persistence is not heritable (27); persisters probably form a single subpopulation tolerant to unrelated antibiotics (65); a transcriptome of isolated persisters points to toxin/antitoxin modules as possible persister genes (28, 50); and expression cloning points to enzymes of phospholipid synthesis being involved in persister maintenance (54).
Analysis of a transposon-mutagenized library seemed to be a straightforward way to identify persister genes. However, initial efforts to find specific persister genes by analyzing the levels of persisters in clones from a transposon insertion library have been unsuccessful (25, 54).
We decided to revisit this approach using an ordered knockout library of all open reading frame (ORF)-coding nonessential E. coli gene disruptions (Keio collection) (4) and a sensitive method for detecting tolerance. We found that several global regulators (DnaK, DnaJ, DksA, histone-like protein, integration host factor [IHF]), as well as the phosphatase YigB and the 5-formyl-tetrahydrofolate (5-formyl-THF) cyclo-ligase YgfA, are involved in persister cell formation.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The Keio collection of E. coli knockout strains (4) was generously provided by Hirotada Mori. E. coli K-12 BW25113 is the parental strain of the ordered knockout library. Precise deletion-replacement of ssrS was created by the method of Datsenko and Wanner (17). To create double deletions, the kanamycin resistance cassette, which replaces one gene, was removed by FLP recombinase, leaving a FLP recognition target site. A second deletion allele was then moved into that strain by P1 transduction, which selected for kanamycin resistance. Bacteria were cultured with aeration at 37°C in Luria-Bertani (LB) medium or morpholinepropanesulfonic acid (MOPS) minimal medium (42) supplemented with 0.1% succinate and 0.05% Casamino Acids. When supplementation was required, the medium was supplemented with MgSO4 at a final concentration of 20 mM and with the following antibiotics at the indicated final concentrations: amdinocillin (mecillinam), 4 μg/ml; ofloxacin, 5 or 10 μg/ml; chloramphenicol; 30 μg/ml, kanamycin, 50 μg/ml; streptomycin 10 μg/ml; benzalkonium chloride, 25 μg/ml; ampicillin, 100 μg/ml, ciprofloxacin, 1 μg/ml. Amdinocillin was a gift from Leo Pharmaceutical Products.
Frequency of amdinocillin-resistant mutants.
The fraction of spontaneous amdinocillin-resistant mutants was determined by plating a serially diluted stationary-state culture on LB agar supplemented with 4 μg/ml amdinocillin. A sample of the culture was diluted and spotted onto LB agar to determine the initial cell count.
Persistence assay.
Persistence was measured by determining survival upon exposure to antibiotics.
To determine the number of persister cells in stationary phase, cells were cultured in 1-ml or 200-μl volumes for one overnight (ON) in LB medium; diluted 1,000- and 200-fold, respectively, in MOPS minimal medium; and cultured for two additional ONs. The cultures were exposed to 5 μg/ml or 10 μg/ml ofloxacin for the indicated times. Ten-microliter samples were removed before and after ofloxacin challenge, diluted, and spot plated onto LB agar. To determine the number of persister cells in a concentration-dependent assay, the concentration of ofloxacin varied as indicated, and the time of challenge was 6 h. Strains harboring expression vector pALS21 and derivatives thereof were assayed in the presence of 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG).
To determine the number of persister cells in exponential phase, cells were cultured in 1-ml volumes for one ON in LB medium and diluted 1:100 into MOPS minimal medium for 5 h (final concentration, ∼2 × 108 CFU/ml). The cultures were exposed to various antibiotics, as indicated, for 3 h. Ten-microliter samples were removed before and after ofloxacin challenge, diluted, and spot plated onto LB agar.
Screen for persistence mutants.
The mutant library was grown in 200 μl LB medium in 96-well plates ON at 37°C, diluted in 200 μl MOPS minimal medium, and incubated for two additional ONs. The library was challenged with 5 μg/ml ofloxacin for 6 h. The contents of the wells were then plated by transferring 10-μl volumes with a 96-pin replicator on LB agar containing 20 mM MgSO4 and 4 μg/ml amdinocillin and were incubated ON. The number of amdinocillin-resistant mutants was established from the colony counts. The growth rate of the mutant strains was determined by measuring the optical density at 600 nm MOPS minimal medium. The MIC of ofloxacin was determined by the standard NCCLS broth microdilution method (1). Strains which produced no amdinocillin-resistant mutants or fewer mutants than the wild type were retested in persistence assays, as described above.
Construction of YigB and YgfA overexpression vectors.
pALS21 was constructed by inserting the lacIq fragment from pCA24N (29) into the ClaI and BspHI sites of pACYC184 with primers 5′-ATGCATCGATTTCGCGGTATGGCATGATAG-3′ and 5′-ATCGTCATGATACGAGCCGGAAGCATAAAG-3′ and the multiple-cloning site from pAH153 (22) into the ScaI and EagI sites with primers 5′-ACGTAGTACTAAGCAGAAGGCCATCCTGAC-3′ and 5′-TGACCGGCCGACCCAGCCTCGCTTTGTAAC-3′. Primers pORFup (5′GAGCTCGCCCTTTCGTCTTCAC-3′) and pORFdown (5′-CCCGGGTCAGTCACGATGAATTCC-3′) were used to insert the yigB and ygfA ORFs, including the PT5-lac promoter region from pCA24N (29), into the SmaI and SacI sites of pALS21, creating pALS22 and pALS23, respectively. The vectors were transformed into strain BW25113 by standard procedures.
RESULTS
Screening of an ordered library for persister genes.
A screen of an ordered library (Keio collection) was developed to identify all single-knockout mutants exhibiting low-level persistence. The Keio collection contains 3,985 strains created by replacing single genes with a kanamycin resistance cassette (17) in E. coli K-12 BW25113 (4, 5). The library was designed to create in-frame single-gene-deletion mutants after excision of the kanamycin resistance cassette with FLP recombinase to avoid polar effects. Persister cell levels can vary strongly in parallel samples within a single experiment (65), which made our initial screening attempts unmanageable due to large numbers of false-positive results. Apparently, each culture creates slightly different conditions in the process of growth and cannot be controlled for. We therefore reasoned that simplifying the growth medium may decrease culture-to-culture variation. Several conditions were tested, and we found that variation in the level of persister cells was decreased in a defined MOPS minimal medium (42) with 0.1% succinate and 0.05% Casamino Acids compared to the variation in LB medium (data not shown). The level of persister cells strongly increases with cell density, reaching a maximum in stationary state (27). Variations are therefore likely to be less prominent once all cultures reach the same density at stationary state, and cells in cultures in this state were chosen for testing. Another problem with screening is that multiple serial dilutions of a culture must be made and plated to count the surviving colonies. Given the large number of strains and the need for replication of the results, this becomes a very laborious task. We therefore developed a screening approach which allowed direct plating without dilutions. The rationale is to plate cells on a medium containing an antibiotic and count the surviving colonies of spontaneous antibiotic-resistant mutants. Since the number of resistant mutants is a fraction of the number of cells of the parent strain, this sharply reduces the number of colonies and obviates the need for dilution. We chose amdinocillin, a beta-lactam antibiotic that targets penicillin-binding protein 2 (PBP 2), as a selecting agent. Spontaneous resistance to amdinocillin occurred at a frequency of 5 × 10−5 in the wild type, as determined by serially diluting the wild type in twofold steps and plating 10-μl drops on LB agar supplemented with 4 μg/ml amdinocillin. A good correlation between cell density and the occurrence of amdinocillin-resistant mutants was observed (Fig. 1A), which allowed us to use this indirect enumeration method in the screen.
FIG. 1.

Screen for mutants with low levels of persistence. (A) Frequency of amdinocillin-resistant mutants in E. coli K-12 BW25113. Stationary-phase culture in LB medium was serially diluted in twofold steps and plated onto LB agar supplemented with 4 μg/ml amdinocillin. Samples from each dilution were also removed for determination of the colony counts. The frequency was 5 × 10−5 on the basis of the initial cell counts and the number of amdinocillin-resistant mutants arising on LB agar supplemented with amdinocillin. (B) Survival of E. coli BW25113 stationary-phase cells challenged with ofloxacin. Stationary-phase cultures grown in MOPS minimal medium were treated with ofloxacin at the indicated concentrations for 6 h and plated for determination of the colony counts. The experiment was performed in triplicate, and error bars represent standard deviations. (C) Survival of strains from the Keio collection. Mutant strains cultured in MOPS minimal medium were treated with ofloxacin for 6 h, and samples were plated on LB agar supplemented with 4 μg/ml amdinocillin and 20 mM MgSO4. Two hits are circled.
To screen the Keio collection for strains with low-level persistence, the mutant library was incubated in MOPS minimal medium. After growth for two ONs, 5 μg/ml ofloxacin (50× MIC) was added to the stationary-phase cultures for 6 h, killing the bulk of the population. At this concentration and within this time period, only persister cells survive (Fig. 1B) (27). The contents of the wells were plated by transferring 10-μl drops to LB agar containing 20 mM MgSO4 and 4 μg/ml amdinocillin, and the plates were incubated at 37°C. Mg2+ inhibits the penetration of ofloxacin and minimizes carryover effects (32). The number of spontaneous amdinocillin-resistant mutants from the wild-type and deletion strains was then counted. In a pilot experiment, the wild-type control produced 5 × 108 CFU/ml after growth for 48 h in MOPS minimal medium. After ofloxacin challenge, the persister cell level of the wild type was 3 × 107 CFU/ml, which allowed 15 spontaneously amdinocillin-resistant colonies to grow on LB medium supplemented with amdinocillin and MgSO4 from the 10-μl aliquot. Thus, under these conditions a mutant with low-level persistence either does not produce any amdinocillin-resistant colonies or at least produces fewer amdinocillin-resistant colonies than the wild type (Fig. 1C). Since slight day-to-day variations in the numbers of amdinocillin-resistant colonies of the wild type were observed, the relative numbers rather than the absolute numbers of resistant mutants from the wild type and knockout strains were compared.
Mutants with low-level persistence.
A total of 150 mutants with reduced or no colony counts were identified in the screen (hit rate, 3.7%). To distinguish between mutants that were impaired in persistence and those that produced fewer amdinocillin-resistant colonies due to lower starting cell numbers or increased sensitivity to ofloxacin or amdinocillin, the growth rates and the MICs of these hits were examined. Strains that grew poorly in the minimal medium were identified (atpE, atpF, carB, cyaA, purA, purD, purF, purK, purL, pyrD, pyrF, pyrI, sdhA, sdhB, sdhC, sdhD). The expected increased sensitivity to ofloxacin was found in strains from which genes involved in recombination and repair (recA, recB, recG, recJ, recN, recQ, ruvA, ruvB, xseA, xseB) were deleted (47, 14, 26, 35, 36, 62) and in a strain from which tolC, an outer membrane component of several multidrug resistance pumps that extrude fluoroquinolones (43), was deleted. These mutants were not considered further. The expected increased sensitivity to amdinocillin was found in a strain from which ponB, which codes for PBP 1B (20), was deleted. That strain was retested in subsequent persistence assays. The wild-type phenotype of three mutants impaired in cysteine metabolism (ΔcysHIJ) was restored when 0.8 mM cysteine was added to the growth medium, showing that impaired survival was due to cysteine limitation (data not shown).
Identification of these mutants, which were expected to fail to produce colonies on amdinocillin-supplemented medium, helped validate the screen. The strain with the glpD deletion, which was previously linked to persistence by our group (54), was among the remaining mutants with low-level persistence and served as a further internal control.
All remaining hits were retested by performing persistence assays in stationary phase with 10 μg/ml ofloxacin in 96-well plates. A considerable number of hits did not show significant changes in their persister cell levels when they were retested.
The genes affected in strains that were validated and that had the greatest reduction in persister cell levels compared to the levels for the wild type were dnaJ and dnaK (chaperones), apaH (diadenosine tetraphosphatase), surA (peptidyl-prolyl cis-trans isomerase), fis and hns (global regulators), hnr (response regulator of RpoS), dksA (transcriptional regulator of rRNA transcription), ygfA (5-formyl-THF cyclo-ligase), and yigB (flavin mononucleotide [FMN] phosphatase) (Fig. 2).
FIG. 2.
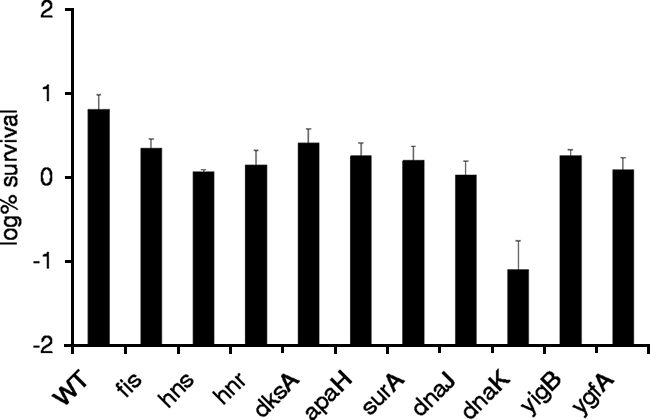
Survival of mutants affected in putative persister genes. Cells were grown to stationary phase in MOPS minimal medium in 96-well plates (final concentration, ∼1× 109 CFU/ml) and treated with 5 μg/ml ofloxacin for 6 h. Samples were diluted and spot plated on LB agar. The experiment was performed in triplicate, and error bars represent standard deviations. WT, wild type.
Time-dependent killing studies of mutants with low-level persistence.
The low-level-persistence phenotype of the mutant strains was further characterized in time-dependent killing studies, which were performed in MOPS minimal medium in 1-ml cultures in 24-well plates. Stationary-phase cultures were challenged with ofloxacin and sampled for determination CFU counts after 1, 3, and 6 h of incubation. The absolute survival rate observed in the time-dependent killing studies was different from that observed in the original selection. In our experience, the level of persistence is highly dependent on the culture conditions, such as the volume, level of aeration, carbon source, and pH. Increasing the culture volume from 200 μl in 96-well plates to 1 ml in 24-well plates mostly affects aeration. In 1-ml cultures, the low-level-persistence phenotype was lost in strains with deletions of dnaJ, fis, hns, and hnr (data not shown). The persistence of strains with deletions of apaH and surA were moderately affected after 6 h of exposure to ofloxacin, with survival rates of 1.3% and 1.7%, respectively, whereas the survival rate for the wild type was 3.3%. Four strains whose persister genes were affected exhibited a prominent phenotype of an at least fourfold decrease in the level of persistence compared to that of the wild type. The persister genes affected were yigB (30-fold), dnaK (22-fold), dksA (6-fold), and ygfA (4-fold) (Fig. 3).
FIG. 3.
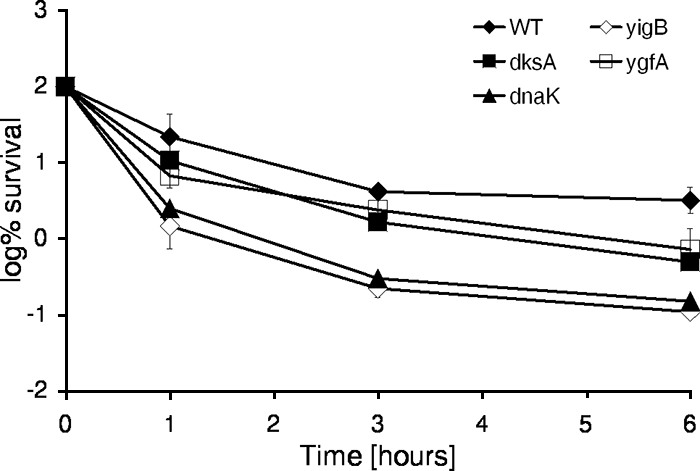
Time-dependent killing studies of mutants whose persister genes were affected. One-milliliter cultures were grown to stationary phase in MOPS minimal medium (final concentration, ∼1 × 109 CFU/ml) and treated with 10 μg/ml ofloxacin for the indicated times. Samples were diluted and spot plated on LB agar. The experiment was performed in triplicate, and error bars represent standard deviations. WT, wild type.
The frequency of persister cells for the strain with the dnaK deletion was highly variable in parallel replicates. Replicates either had a reduced growth rate at 37°C and had lower levels of persistence than the wild type or picked up suppressor mutations which restored normal growth at 37°C and the level of persistence of the wild type. dnaK-null strains have been reported to frequently acquire suppressor mutations, for example, in rpoH and rpoD (13, 51).
ygfA is cotranscribed with ssrS, which codes for a small RNA, 6S RNA, which acts as a repressor of σ70-dependent transcription in stationary phase (63). The location of ygfA directly downstream of ssrS is conserved in members of the classes Alpha- and Gammaproteobacteria as well as certain members of the class Betaproteobacteria, which suggests that the operon structure has functional relevance (7). Indeed, YgfA has been shown to stabilize 6S RNA in an overexpression system (63). Thus, the deletion of ygfA apparently diminishes the level of the ssrS transcript. In order to test the phenotype of an ssrS mutant (which is absent from the Keio library, which only contains strains with deletions in protein-coding ORFs), strains with an ssrS deletion (strain KLE910) as well as an ssrS ygfA double deletion (strain KLE911) were created. Persistence was tested in 200 μl MOPS minimal medium, where ΔygfA had a stronger effect on persistence. We did not detect a phenotype for ΔssrS. The growth as well as the persistence of the ΔssrS ΔygfA strain appeared to be affected (Fig. 4).
FIG. 4.

Effects of ΔygfA and ΔssrS mutations on persister cell formation. E. coli BW25113 wild type (WT), a ΔygfA mutant (ygfA), strain KLE910 (ssrS), and strain KLE911 (ssrS ygfA) were grown to stationary phase in MOPS minimal medium and treated with 10 μg/ml ofloxacin for 6 h. Samples were diluted and spot plated on LB agar. The experiment was performed in triplicate, and error bars represent standard deviations.
To test for the robustness of the low-level-persistence phenotype of the mutants identified, we investigated whether the number of persister cells produced by the mutant strains was also lower in growing cultures. We diluted the strains with deletions 1:100 in MOPS minimal medium and exposed them to 5 μg/ml ofloxacin when they were in exponential phase, after 5 h of growth. We found that the persistence of all mutants was also impaired (at least a fourfold decrease) during exponential phase (Fig. 5A). Similar to what we observed in stationary phase, after 3 h of exposure to ofloxacin, strains with deletions of dnaK (27-fold), ygfA (17-fold), dksA (11-fold), and yigB (11-fold) showed the most prominent reduction in persistence compared with that of the wild type. Additionally, the persistence of a strain with a deletion of dnaJ was also strongly affected (41-fold decrease). We validated the reduction in persistence in exponential phase in these strains in a time-dependent killing assay and observed the typical persister cell plateau for all strains (Fig. 5B).
FIG. 5.
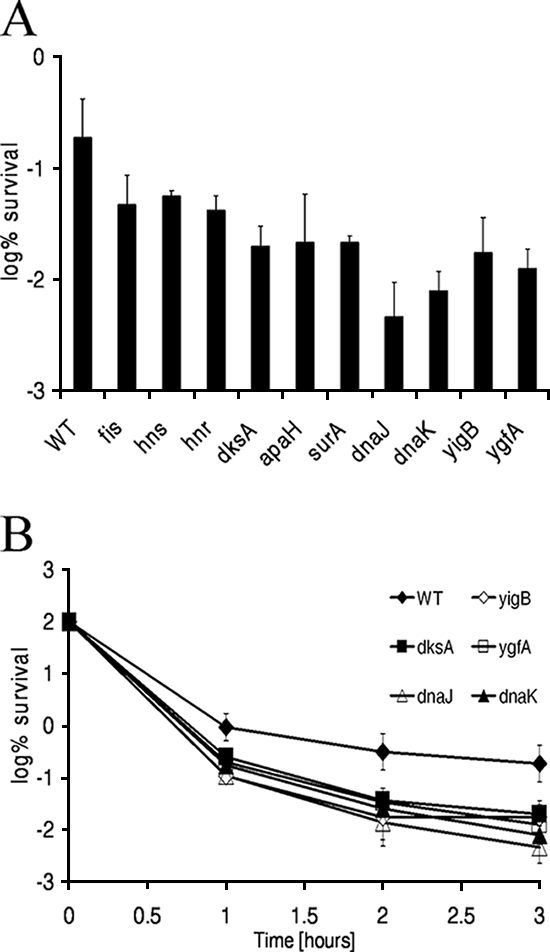
Survival of mutants whose putative persister genes were affected in exponential phase. (A) One-milliliter cultures were grown to exponential phase in MOPS minimal medium and treated with 5 μg/ml ofloxacin for 3 h. (B) Time-dependent killing studies of mutants whose persister genes were affected. Mutants with a prominent low-level-persistence phenotype in log phase were exposed to 5 μg/ml ofloxacin for the indicated times. Samples were diluted and spot plated on LB agar. The experiment was performed in triplicate, and error bars represent standard deviations. WT, wild type.
Survival of mutants exposed to different classes of antibiotics.
Persister cells are characterized by multidrug tolerance. The screen for mutants with low levels of persistence was performed in stationary phase, where the choice of antibiotic limited us to fluoroquinolones, which are able to kill nongrowing cells. In growing cultures, however, we were able to test whether the tolerance of the mutants identified was affected when they were subjected to different antibiotics. We cultured the wild type and the five mutants with the strongest decrease in persistence in exponential phase as described above and exposed them to different classes of antibiotics for 3 h (Fig. 6): another fluoroquinolone, ciprofloxacin; the aminoglycoside streptomycin; the beta-lactam antibiotic ampicillin; and the quaternary ammonium compound benzalkonium chloride. When they were exposed to ciprofloxacin, all mutants showed a prominent decrease in persistence compared to that for the wild type, ranging from 4-fold (ΔygfA) to a 111-fold (ΔdnaK) (Fig. 6A). After exposure to streptomycin, the ΔygfA mutant again showed the weakest decrease (3-fold), whereas the survival of mutants ΔdksA (6-fold), ΔyigB (10-fold), ΔdnaK (66-fold), and ΔdnaJ (87-fold) was strongly affected (Fig. 6B). The ΔygfA mutant, however, showed a prominent effect after ampicillin challenge (82-fold). The persistence of the ΔyigB mutant was highly variable in this set of experiments, and the decrease was an average of twofold. The persistence levels of mutants ΔdksA (40-fold), ΔdnaJ (13-fold), and ΔdnaK (46-fold) were strongly affected (Fig. 6C). The difference in the levels of persisters between the wild type and the mutants was less pronounced when they were exposed to benzalkonium chloride. ΔdnaK had the strongest effect, with an 11-fold decrease (Fig. 6D). In summary, these results show that not only ofloxacin tolerance but also multidrug tolerance were affected in all mutants identified.
FIG. 6.

Survival of mutants with low levels of persistence exposed to different classes of antibiotics. One-milliliter cultures were grown to exponential phase in MOPS minimal medium and challenged for 3 h with 1 μg/ml ciprofloxacin (A), 10 μg/ml streptomycin (B), 100 μg/ml ampicillin (C), and 25 μg/ml benzalkonium chloride (D). Samples were diluted and spot plated on LB agar. The experiment was performed in triplicate, and error bars represent standard deviations. WT, wild type.
Effects of YgfA and YigB overexpression on persistence.
ygfA and yigB were overexpressed in order to further characterize their effects on persistence. The overexpression of persister genes has been shown to be challenging since overproduction of some proteins causes nonspecific toxicity, shutting down cell growth and leading to tolerance (60). Thus, a low-copy-number vector (pACYC184) was used to clone these genes under their native promoters in order to achieve moderate overexpression. Both constructs conferred high-level persistence in stationary phase after exposure to ofloxacin (Fig. 7). Strains overexpressing global regulators involved in persistence were not tested due to the multitude of phenotypes associated with proteins of global regulatory function and the genes that these regulators control.
FIG. 7.

Effects of ygfA and yigB overexpression on persister cell formation. E. coli BW25113 cells transformed with the pALS21 control (empty vector), pALS22 (yigB), and pALS23 (ygfA) were grown in MOPS minimal medium in the presence of 0.1 mM IPTG to stationary phase. The cultures were treated with ofloxacin for the indicated times. Samples were diluted and spot plated on LB agar. Closed squares, wild type; open squares, KLE921(pALS21); open diamonds, KLE922 (pALS22); closed diamonds, KLE923 (pALS23). The experiment was performed in triplicate, and error bars represent standard deviations.
Roles of HU and IHF in persistence.
Two of the mutants with low levels of persistence had deletions of hns and fis, which encode nucleoid binding proteins with global regulatory functions. In stationary phase, the low-level-persistence phenotype was apparent only when the mutants were tested in 200 μl medium in 96-well plates. When they were tested in 1-ml cultures, the persistence of the strains with the hns and fis deletions was almost indistinguishable from that of the wild type. In exponentially growing cultures, both Δhns and Δfis strains had a fourfold reduction in the levels of persister cells. Interestingly, deletions of the HupA subunit of HU and the IhfB subunit of IHF, two other nucleoid binding proteins which are global regulators as well, also showed up as hits in the screen. However, when it was retested, the strain with the hupA deletion was not significantly impaired in persister formation, and the strain with the ihfB deletion even showed an increase in persistence. We decided to revisit the persistence of strains with the hupA and ihfB deletions and test the persistence of strains with deletions of their respective second subunits, hupB and ihfA, and of strains without HU (strain KLE912) and IHF (strain KLE913). It is known that HupB and HupA and that IhfA and IhfB have partial functionality when they are expressed individually (15, 64, 67). We observed a decrease in the level of persistence of the ΔhupB strain, and this decrease was even more pronounced in the mutant with the double mutation (Fig. 8A). We confirmed the increase in the level of persistence of the ΔihfB strain and the IHF-null strain, whereas the ihfA mutant showed no phenotype (Fig. 8B). We then tested the IHF- and HU-null strains in a time-kill study in 1-ml volumes and found that both null strains exhibited robust phenotypes (Fig. 8C). In exponentially growing cultures, the HU null strain had strongly reduced persistence levels, 40-fold compared to those of the wild type, when it was exposed to ofloxacin for 3 h, whereas the persistence of the IHF-null strain fluctuated highly in parallel replicates of the same experiment and from day to day in exponential phase (data not shown).
FIG. 8.

Effects of mutations in global regulators IHF and HU on persister formation. (A and B) Cultures of E. coli BW25113 wild type and mutant strains were grown to stationary phase and treated with ofloxacin for 6 h. Samples were diluted and spot plated on LB agar. (A) HU mutants ΔhupA (hupA) ΔhupB (hupB), and KLE912 hupA hupB (hupAB); (B) IHF mutants ΔihfA (ihfA) ΔihfB (ihfB), and KLE913 ihfA ihfB (ihfAB). (C) Time-dependent killing studies of HU and IHF mutants. The cultures were grown to stationary phase in MOPS minimal medium and treated with 10 μg/ml for the indicated times. Samples were diluted and spot plated on LB agar. Closed squares, wild type; closed triangles, KLE912 hupA hupB; open squares, KLE913 ihfA ihfB. All experiments were performed in triplicate, and error bars represent standard deviations.
DISCUSSION
Persister cells are dormant, multidrug-tolerant cells and play a major role in biofilm recalcitrance to antimicrobials (12, 27, 53, 50, 33). In this study, we examined a comprehensive knockout library of E. coli ORFs with the aim of identifying persister genes. The detection of genes coding for a complex biological function is usually straightforward, and the screening of transposon insertion libraries has been very successful in identifying components of sporulation, the SOS response, biofilm formation, and many others. Indeed, most spo genes, for example, were discovered by examining mutants defective in sporulation (37). However, the screening of libraries of knockout strains for changes in persister levels has largely been unsuccessful (25, 34). The detection of mutants with diminished persister cell levels proved to be highly challenging. The possible reason for this is the high level of redundancy in the mechanism coding for persistence. Previous research suggests that this may indeed be the case. Thus, transcription profiling of isolated persisters pointed to the overexpression of toxin/antitoxin modules; and the ectopic expression of several different toxins, such as RelA, MazF, HipA, and YgiU, causes reversible stasis and multidrug tolerance, emulating persistence (28, 50, 60). Another problem with screening for persister genes is the high level of variability in persister cell levels among parallel samples, making the background false-positive and -negative results unmanageable for a large experiment (65).
We decided to revisit the screening approach by taking advantage of the recently created complete, ordered knockout library of E. coli ORFs. We introduced several changes that considerably improved the quality of screening: (i) we simplified the growth medium, which decreased variability among parallel samples; (ii) we screened strains in stationary state, where persisters are most abundant and all cultures reach a similar growth end point; and (iii) we introduced a method that allows for sensitive screening without multiple dilutions. This involved the plating of persister cells that survived antibiotic challenge on a medium containing a different antibiotic, amdinocillin. In this way, only spontaneous mutants resistant to amdinocillin survived, which reduced the number of surviving cells, obviating the need for multiple dilutions.
It is important to stress that none of the knockout strains had a complete lack of persisters; in most cases, the drop was about 10-fold. The preponderance of global regulators and chaperones among the genes affecting persistence strongly suggests that the function is encoded by redundant genes/mechanisms. Persister genes are probably among the many that are controlled by these regulators. This strongly points to redundancy in persister genes. Note that there have been reports of strong phenotypes of single-gene deletions. We reported previously that a hipA deleted strain had a significantly decreased persister level in stationary state; however, that deletion extended into the noncoding dif region, which is important for chromosome partitioning. The strain with a precise deletion of hipA did not show a persister phenotype. A recent report described phoU as a gene whose disruption caused the complete loss of persister cells in the presence of an antibiotic (34). However, the strain with the phoU disruption grew slowly and had decreased MICs toward a number of antibiotics tested. Strains that showed considerably slower growth or changes in MICs may be pleiotropic mutants with changes in resistance rather than strains whose persister genes controlling dormancy and antibiotic tolerance are affected.
The most prominent finding from the screening was the identification of genes that act as global regulators. The regulators identified were DksA, SsrS-YgfA, DnaKJ, HupAB, and IhfAB.
DksA is a ppGpp-dependent modulator of RNA polymerase. ppGpp is formed in response to carbon and amino acid starvation, which leads to the inhibition of transcription from rRNA and tRNA promoters and which directs transcription toward the genes needed for the synthesis and transport of amino acids (44, 45). Considering that persisters are dormant cells, this makes sense, since a stochastic increase in ppGpp or DksA, or both, would contribute to dormancy, and then knocking out dksA would decrease the level of persister cells. The strain whose ppGpp synthetase RelA was deleted was identified as a hit in the primary screen. However, a ΔrelA strain grows very poorly in minimal medium (unless suppressor mutations in RNA polymerase subunits are acquired) (24); moreover, resistance to amdinocillin is conferred by an increase in the ppGpp concentration (61). Thus, the ΔrelA strain was removed from our set of hits of strains with low levels of persistence. Note that relA and relA spoT deletions have previously been linked to persistence in a hipA7 mutant and in the wild type (30).
The knockout of ygfA produced a robust phenotype of decreased persister cell formation. YgfA apparently codes for a 5-formyl-THF cyclo-ligase involved in folate biosynthesis. The mammalian homolog of YgfA, methenyltetrahydrofolate synthetase, catalyzes the conversion of the stable folate storage form 5-formyl-THF into the rapidly degraded 5,10-methenyl-THF, thereby depleting cellular folate pools (2). If the E. coli enzyme has the same function, then in a simple scenario, the stochastic overexpression of YgfA will lead to folate deficiency, which will impair the biosynthesis of purines, thymidilate, and methionine (7) and will contribute to persister cell formation. The artificial overexpression of YgfA indeed produced a phenotype of increased tolerance to ofloxacin. The knocking out of ygfA would be expected to decrease the level of persister cells, which we have observed. Interestingly, ygfA is a member of a two-gene ssrS ygfA operon. ssrS codes for the small regulatory 6S RNA which interacts with σ70-RNA polymerase and thereby inhibits transcription of most σ70-dependent promoters in stationary phase. Common among most inhibited promoters is an extended −10 element, which is present in more than 100 σ70-dependent promoters (59, 63). The 6S RNA is present throughout all growth phases but is most abundant during stationary phase, when >75% of the σ70-RNA polymerase complexes are associated with 6S RNA (63). In addition, 6S RNA also indirectly activates the transcription of σS-dependent promoters (59). It appears that these two elements potentially control two different aspects of RNA synthesis: 6S RNA acts as an inhibitor of RNA polymerase, while YgfA can inhibit the synthesis of nucleotides. This two-gene module seems like an attractive candidate for a persister operon. The knocking out of ssrS produced no phenotype. However, it is known that ygfA stabilizes ssrS (63) and that a ygfA knockout is therefore functionally closer to an ssrS ygfA double knockout. It may be that in vivo, changes in both of these functionally similar redundant components are necessary to observe a change in persister cell levels.
Another interesting gene that can potentially affect dormancy is yigB. The YigB protein was shown to act as a phosphatase of FMN (31), which would deplete the pool of this important cofactor in the cell upon overproduction, potentially contributing to dormancy. The overproduction of YigB produced high-level persistence, consistent with a possible role in persister cell production. Three other phosphatases of the same group, YnjI, Cof, and YrfG, were also shown to have specificity for FMN. To investigate whether involvement in persister cell formation is specific to YigB or is shared among FMN phosphatases, we retested strains with deletions of ynjI, cof, and yrfG. None of the strains had a phenotype in regards to low-level persistence (data not shown).
The DnaK chaperone assists in protein folding of nascent polypeptides and requires ATP and its cochaperones, DnaJ and GrpE (55, 56). DnaK also acts as a regulator of a large set of genes induced by the heat shock and general stress response by regulating the activity and stability of the alternative sigma factors σ32 and σS, respectively (41, 57, 58, 66). A decreased level of persister cells in a dnaK deleted strain suggests that it may be required for the maintenance of persister cells and is thus a persister maintenance gene (similar to the previously described persister maintenance gene plsB [54]). It is also possible that DnaK controls several redundant persister genes, and the phenotype is observed only when their regulators are absent. A decrease in persistence was also found in a Staphylococcus aureus strain with a dnaK deletion when it was challenged with oxacillin (52).
Other global regulators which showed a phenotype when they were deleted were HU and IHF. A ΔhupB strain showed a decrease in persistence, and this decrease was even more pronounced in the mutant with the hupAB double mutation. The opposite effect was observed in an IHF-null strain and a strain with a ihfB single deletion, which were both high-level-persistence mutants. IHF binds to specific DNA sequences of 30 to 35 bp (21) and directly or indirectly regulates the transcription of more than 100 genes (3). Thus, a fluctuation in the level of persistence in an exponentially growing culture of an IHF-null strain might be explained by the deregulation of numerous genes. In contrast, HU binding to DNA is nonspecific (11). HU acts as a regulator by promoting and inhibiting the DNA binding of transcriptional regulators, such as the cyclic AMP repressor protein, the Lac repressor, the Gal repressor, the Trp repressor, and LexA (19, 48).
The genes identified by this screen provide good clues to the mechanism of persister cell formation and maintenance and serve as a starting point for a more detailed investigation of the molecular mechanism of multidrug tolerance. What this screen missed are possible highly redundant persister genes, such as the toxin/antitoxin modules, the essential genes, and most regulatory RNAs. It appears that a combination of several independent approaches will be required to obtain a comprehensive set of genes that participate in persistence.
Acknowledgments
We thank Hirotada Mori for providing the Keio and the ASKA collections and Amy L. Spoering for providing pALS21. We thank Sevda Kantarci for performing some of the screening experiments.
We thank Leo Pharmaceutical Products for providing amdinocillin. This work was supported by NIH grant R01 GM061162.
Footnotes
Published ahead of print on 2 June 2008.
REFERENCES
- 1.Andrews, J. M. 2001. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 48(Suppl. 1):5-16. [DOI] [PubMed] [Google Scholar]
- 2.Anguera, M. C., J. R. Suh, H. Ghandour, I. M. Nasrallah, J. Selhub, and P. J. Stover. 2003. Methenyltetrahydrofolate synthetase regulates folate turnover and accumulation. J. Biol. Chem. 278:29856-29862. [DOI] [PubMed] [Google Scholar]
- 3.Arfin, S. M., A. D. Long, E. T. Ito, L. Tolleri, M. M. Riehle, E. S. Paegle, and G. W. Hatfield. 2000. Global gene expression profiling in Escherichia coli K12. The effects of integration host factor. J. Biol. Chem. 275:29672-29684. [DOI] [PubMed] [Google Scholar]
- 4.Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knock-out mutants the Keio collection. Mol. Systems Biol. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed]
- 5.Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Systems Biol 2:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balaban, N. Q., J. Merrin, R. Chait, L. Kowalik, and S. Leibler. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622-1625. [DOI] [PubMed] [Google Scholar]
- 7.Barrick, J. E., N. Sudarsan, Z. Weinberg, W. L. Ruzzo, and R. R. Breaker. 2005. 6S RNA is a widespread regulator of eubacterial RNA polymerase that resembles an open promoter. RNA 11:774-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bigger, J. W. 1944. Treatment of staphylococcal infections with penicillin. Lancet ii:497-500. [Google Scholar]
- 9.Black, D. S., B. Irwin, and H. S. Moyed. 1994. Autoregulation of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J. Bacteriol. 176:4081-4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Black, D. S., A. J. Kelly, M. J. Mardis, and H. S. Moyed. 1991. Structure and organization of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA synthesis. J. Bacteriol. 173:5732-5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonnefoy, E., and J. Rouviere-Yaniv. 1991. HU and IHF, two homologous histone-like proteins of Escherichia coli, form different protein-DNA complexes with short DNA fragments. EMBO J. 10:687-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brooun, A., S. Liu, and K. Lewis. 2000. A dose-response study of antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 44:640-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bukau, B., and G. C. Walker. 1989. Cellular defects caused by deletion of the Escherichia coli dnaK gene indicate roles for heat shock protein in normal metabolism. J. Bacteriol. 171:2337-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chase, J. W., and C. C. Richardson. 1977. Escherichia coli mutants deficient in exonuclease VII. J. Bacteriol. 129:934-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claret, L., and J. Rouviere-Yaniv. 1997. Variation in HU composition during growth of Escherichia coli: the heterodimer is required for long term survival. J. Mol. Biol. 273:93-104. [DOI] [PubMed] [Google Scholar]
- 16.Costerton, J., and D. Keller. 2007. Oral periopathogens and systemic effects. Gen. Dent. 55:210-215. [PubMed] [Google Scholar]
- 17.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donlan, R. M., and J. W. Costerton. 2002. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15:167-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flashner, Y., and J. D. Gralla. 1988. DNA dynamic flexibility and protein recognition: differential stimulation by bacterial histone-like protein HU. Cell 54:713-721. [DOI] [PubMed] [Google Scholar]
- 20.Garcia del Portillo, F., and M. A. de Pedro. 1991. Penicillin-binding protein 2 is essential for the integrity of growing cells of Escherichia coli ponB strains. J. Bacteriol. 173:4530-4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodrich, J. A., M. L. Schwartz, and W. R. McClure. 1990. Searching for and predicting the activity of sites for DNA binding proteins: compilation and analysis of the binding sites for Escherichia coli integration host factor (IHF). Nucleic Acids Res. 18:4993-5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haldimann, A., and B. L. Wanner. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J. Bacteriol. 183:6384-6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hall-Stoodley, L., J. W. Costerton, and P. Stoodley. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2:95-108. [DOI] [PubMed] [Google Scholar]
- 24.Hernandez, V. J., and M. Cashel. 1995. Changes in conserved region 3 of Escherichia coli sigma 70 mediate ppGpp-dependent functions in vivo. J. Mol. Biol. 252:536-549. [DOI] [PubMed] [Google Scholar]
- 25.Hu, Y., and A. R. Coates. 2005. Transposon mutagenesis identifies genes which control antimicrobial drug tolerance in stationary-phase Escherichia coli. FEMS Microbiol. Lett. 243:117-124. [DOI] [PubMed] [Google Scholar]
- 26.Iwasaki, H., T. Shiba, A. Nakata, and H. Shinagawa. 1989. Involvement in DNA repair of the ruvA gene of Escherichia coli. Mol. Gen. Genet. 219:328-331. [DOI] [PubMed] [Google Scholar]
- 27.Keren, I., N. Kaldalu, A. Spoering, Y. Wang, and K. Lewis. 2004. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230:13-18. [DOI] [PubMed] [Google Scholar]
- 28.Keren, I., D. Shah, A. Spoering, N. Kaldalu, and K. Lewis. 2004. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186:8172-8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitagawa, M., T. Ara, M. Arifuzzaman, T. Ioka-Nakamichi, E. Inamoto, H. Toyonaga, and H. Mori. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12:291-299. [DOI] [PubMed] [Google Scholar]
- 30.Korch, S. B., T. A. Henderson, and T. M. Hill. 2003. Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol. Microbiol. 50:1199-1213. [DOI] [PubMed] [Google Scholar]
- 31.Kuznetsova, E., M. Proudfoot, C. F. Gonzalez, G. Brown, M. V. Omelchenko, I. Borozan, L. Carmel, Y. I. Wolf, H. Mori, A. V. Savchenko, C. H. Arrowsmith, E. V. Koonin, A. M. Edwards, and A. F. Yakunin. 2006. Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J. Biol. Chem. 281:36149-36161. [DOI] [PubMed] [Google Scholar]
- 32.Lecomte, S., M. H. Baron, M. T. Chenon, C. Coupry, and N. J. Moreau. 1994. Effect of magnesium complexation by fluoroquinolones on their antibacterial properties. Antimicrob. Agents Chemother. 38:2810-2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis, K. 2007. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5:48-56. [DOI] [PubMed] [Google Scholar]
- 34.Li, Y., and Y. Zhang. 2007. PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob. Agents Chemother. 51:2092-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lloyd, R. G., and C. Buckman. 1991. Genetic analysis of the recG locus of Escherichia coli K-12 and of its role in recombination and DNA repair. J. Bacteriol. 173:1004-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lloyd, R. G., M. C. Porton, and C. Buckman. 1988. Effect of recF, recJ, recN, recO and ruv mutations on ultraviolet survival and genetic recombination in a recD strain of Escherichia coli K12. Mol. Gen. Genet. 212:317-324. [DOI] [PubMed] [Google Scholar]
- 37.Losick, R., P. Youngman, and P. J. Piggot. 1986. Genetics of endospore formation in Bacillus subtilis. Annu. Rev. Genet. 20:625-669. [DOI] [PubMed] [Google Scholar]
- 38.Lyczak, J. B., C. L. Cannon, and G. B. Pier. 2002. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 15:194-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moyed, H. S., and K. P. Bertrand. 1983. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 155:768-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moyed, H. S., and S. H. Broderick. 1986. Molecular cloning and expression of hipA, a gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 166:399-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muffler, A., D. D. Traulsen, R. Lange, and R. Hengge-Aronis. 1996. Posttranscriptional osmotic regulation of the sigma (σ) subunit of RNA polymerase in Escherichia coli. J. Bacteriol. 178:1607-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neidhardt, F. C., P. L. Bloch, and D. F. Smith. 1974. Culture medium for enterobacteria. J. Bacteriol. 119:736-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okusu, H., D. Ma, and H. Nikaido. 1996. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 178:306-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paul, B. J., M. M. Barker, W. Ross, D. A. Schneider, C. Webb, J. W. Foster, and R. L. Gourse. 2004. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 118:311-322. [DOI] [PubMed] [Google Scholar]
- 45.Paul, B. J., M. B. Berkmen, and R. L. Gourse. 2005. DksA potentiates direct activation of amino acid promoters by ppGpp. Proc. Natl. Acad. Sci. USA 102:7823-7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peters, G., R. Locci, and G. Pulverer. 1981. Microbial colonization of prosthetic devices. II. Scanning electron microscopy of naturally infected intravenous catheters. Zentralbl. Bakteriol. Mikrobiol. Hyg. B 173:293-299. [PubMed] [Google Scholar]
- 47.Piddock, L. J., and R. N. Walters. 1992. Bactericidal activities of five quinolones for Escherichia coli strains with mutations in genes encoding the SOS response or cell division. Antimicrob. Agents Chemother. 36:819-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Preobrajenskaya, O., A. Boullard, F. Boubrik, M. Schnarr, and J. Rouviere-Yaniv. 1994. The protein HU can displace the LexA repressor from its DNA-binding sites. Mol. Microbiol. 13:459-467. [DOI] [PubMed] [Google Scholar]
- 49.Scherrer, R., and H. S. Moyed. 1988. Conditional impairment of cell division and altered lethality in hipA mutants of Escherichia coli K-12. J. Bacteriol. 170:3321-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah, D. V., Z. Zhang, K. Kurg, N. Kaldalu, A. Khodursky, and K. Lewis. 2006. Persisters: a distinct physiological state of E. coli. BMC Microbiol. 6:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shiozawa, T., C. Ueguchi, and T. Mizuno. 1996. The rpoD gene functions as a multicopy suppressor for mutations in the chaperones, CbpA, DnaJ and DnaK, in Escherichia coli. FEMS Microbiol. Lett. 138:245-250. [DOI] [PubMed] [Google Scholar]
- 52.Singh, V. K., S. Utaida, L. S. Jackson, R. K. Jayaswal, B. J. Wilkinson, and N. R. Chamberlain. 2007. Role for dnaK locus in tolerance of multiple stresses in Staphylococcus aureus. Microbiology 153:3162-3173. [DOI] [PubMed] [Google Scholar]
- 53.Spoering, A. L., and K. Lewis. 2001. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 183:6746-6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spoering, A. L., M. Vulic, and K. Lewis. 2006. GlpD and PlsB participate in persister cell formation in Escherichia coli. J. Bacteriol. 188:5136-5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Straus, D., W. Walter, and C. A. Gross. 1990. DnaK, DnaJ, and GrpE heat shock proteins negatively regulate heat shock gene expression by controlling the synthesis and stability of sigma 32. Genes Dev. 4:2202-2209. [DOI] [PubMed] [Google Scholar]
- 56.Szabo, A., T. Langer, H. Schroder, J. Flanagan, B. Bukau, and F. U. Hartl. 1994. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc. Natl. Acad. Sci. USA 91:10345-10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tilly, K., N. McKittrick, M. Zylicz, and C. Georgopoulos. 1983. The dnaK protein modulates the heat-shock response of Escherichia coli. Cell 34:641-646. [DOI] [PubMed] [Google Scholar]
- 58.Tilly, K., J. Spence, and C. Georgopoulos. 1989. Modulation of stability of the Escherichia coli heat shock regulatory factor sigma. J. Bacteriol. 171:1585-1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trotochaud, A. E., and K. M. Wassarman. 2004. 6S RNA function enhances long-term cell survival. J. Bacteriol. 186:4978-4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vazquez-Laslop, N., H. Lee, and A. A. Neyfakh. 2006. Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. J. Bacteriol. 188:3494-3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vinella, D., R. D'Ari, A. Jaffe, and P. Bouloc. 1992. Penicillin binding protein 2 is dispensable in Escherichia coli when ppGpp synthesis is induced. EMBO J. 11:1493-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Viswanathan, M., and S. T. Lovett. 1998. Single-strand DNA-specific exonucleases in Escherichia coli. Roles in repair and mutation avoidance. Genetics 149:7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wassarman, K. M., and G. Storz. 2000. 6S RNA regulates E. coli RNA polymerase activity. Cell 101:613-623. [DOI] [PubMed] [Google Scholar]
- 64.Werner, M. H., G. M. Clore, A. M. Gronenborn, and H. A. Nash. 1994. Symmetry and asymmetry in the function of Escherichia coli integration host factor: implications for target identification by DNA-binding proteins. Curr. Biol. 4:477-487. [DOI] [PubMed] [Google Scholar]
- 65.Wiuff, C., R. M. Zappala, R. R. Regoes, K. N. Garner, F. Baquero, and B. R. Levin. 2005. Phenotypic tolerance: antibiotic enrichment of noninherited resistance in bacterial populations. Antimicrob. Agents Chemother. 49:1483-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yura, T., H. Nagai, and H. Mori. 1993. Regulation of the heat-shock response in bacteria. Annu. Rev. Microbiol. 47:321-350. [DOI] [PubMed] [Google Scholar]
- 67.Zulianello, L., E. de la Gorgue de Rosny, P. van Ulsen, P. van de Putte, and N. Goosen. 1994. The HimA and HimD subunits of integration host factor can specifically bind to DNA as homodimers. EMBO J. 13:1534-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]