

Abstract
The transcription factor NF-κB is a pivotal regulator of inflammatory responses. While the activation of NF-κB in the arthritic joint has been associated with rheumatoid arthritis (RA), its significance is poorly understood. Here, we examine the role of NF-κB in animal models of RA. We demonstrate that in vitro, NF-κB controlled expression of numerous inflammatory molecules in synoviocytes and protected cells against tumor necrosis factor α (TNFα) and Fas ligand (FasL) cytotoxicity. Similar to that observed in human RA, NF-κB was found to be activated in the synovium of rats with streptococcal cell wall (SCW)-induced arthritis. In vivo suppression of NF-κB by either proteasomal inhibitors or intraarticular adenoviral gene transfer of super-repressor IκBα profoundly enhanced apoptosis in the synovium of rats with SCW- and pristane-induced arthritis. This indicated that the activation of NF-κB protected the cells in the synovium against apoptosis and thus provided the potential link between inflammation and hyperplasia. Intraarticular administration of NF-kB decoys prevented the recurrence of SCW arthritis in treated joints. Unexpectedly, the severity of arthritis also was inhibited significantly in the contralateral, untreated joints, indicating beneficial systemic effects of local suppression of NF-κB. These results establish a mechanism regulating apoptosis in the arthritic joint and indicate the feasibility of therapeutic approaches to RA based on the specific suppression of NF-κB.
Keywords: arthritis/apoptosis/tumor necrosis factor α/Fas/gene therapy
Inflammation and hyperplasia of the synovium are the hallmarks of rheumatoid arthritis (RA) (1). The normal synovium is a delicate tissue lining the joint capsule; however, in RA, the synovium transforms into an aggressive, tumor-like structure called the pannus (1, 2). It is a common belief that inflammation promotes hyperplasia in the synovium, but the underlying mechanisms for this are largely unknown.
Impaired regulation of apoptosis has been associated with RA (3–5). Experimental evidence suggests that tumor necrosis factor α (TNFα)- and Fas-mediated apoptosis is important in the regulation of hyperplasia in the synovium (5, 6). The human RA synovium contains a significant proportion of apoptotic cells (7, 8), and elevated expression of TNFα protein and functional FasL in the synovium is a common feature of RA (1, 9).
Gene expression is transcriptionally controlled by inducible transcription factors. The transcription factor NF-κB (reviewed in ref. 10) is particularly important in the regulation of inflammation. In unstimulated cells, NF-κB is retained in the cytoplasm through an interaction with inhibitory proteins known as IκB. Cell stimulation causes degradation of IκB, leading to nuclear translocation of NF-κB and activation of transcription. Inducers of NF-κB include interleukin (IL)-1, TNFα, platelet-derived growth factor, lipopolysaccharide, oxidative stress, and viral products. In turn, NF-κB can activate transcription of IL-1, TNFα, IL-6, IL-8, the adhesion molecules ICAM-1 (intercellular adhesion molecule-1), VCAM-1 (vascular cell adhesion molecule 1), E-selectin, the growth factor granulocyte/macrophage colony-stimulating factor, and inducible nitric oxide synthase. Since NF-κB is activated in human RA synovium (11, 12), and the list of inducers and targets of NF-κB almost perfectly matches the profile of mediators of inflammation in RA, this suggests an important role of NF-κB in the control of inflammation in the synovium. Furthermore, the recent discovery of a protective function of NF-κB against the cytotoxicity of TNFα (13–16) suggests the involvement of this transcription factor in the regulation of apoptosis in the synovium.
Here, we used animal models of RA to examine the relationship between inflammation, activation of NF-κB, and apoptosis in the synovium. We demonstrate that in primary synovial fibroblasts, NF-κB is required for induction of multiple inflammatory molecules, including IL-1β and TNFα. Specific suppression of NF-κB potentiated TNFα- and FasL-mediated apoptosis. Similar to that in human RA, NF-κB was found activated in the synovium of rats with streptococcal cell wall (SCW) arthritis. Suppression of NF-κB by intraarticular (i.a.) administration of proteasomal inhibitors and i.a. adenoviral gene transfer of super-repressor IκBα (srIκBα) profoundly enhanced apoptosis in the synovium of rats with SCW- and pristane-induced arthritis. This indicated that activation of NF-κB by inflammation inhibited apoptosis in the synovium, thereby potentially contributing to hyperplasia. Administration (i.a.) of NF-κB decoy oligodeoxynucleotides (ODNs) significantly inhibited the severity of recurrent SCW arthritis in treated joints. Unexpectedly, the severity of arthritis also was inhibited in untreated, contralateral joints, indicating systemic therapeutic effects of local treatment.
MATERIALS AND METHODS
Adenoviral (Ad) Vectors.
The Ad vectors were derivatives of Ad5 adenovirus with deleted E1A and E1B regions. Expression of the transgene was driven by the cytomegalovirus (CMV) promoter. The srIκBα cDNA was obtained by a double point mutation of cDNA of IκBα (Ser 32/36 to Ala 32/36) (17, 18). The resulting construct was subcloned into pAdCMV transfer vector (Genvec, Rockville, MD) and cotransfected with the linearized vector IN34051 into the HEK 293 cells. The Ad virus was purified on a CsCl gradient and dialyzed. The recombinant AdLacZ and AdGFP vectors (gift of Doug McCarty, Vector Core facility, University of North Carolina) were derivatives of Ad5 with deleted E1 and E3 regions.
NF-κB Decoys.
The ODNs were synthesized as phosphorothioate (PT) derivatives, which are resistant to degradation by endonucleases. The single-stranded (ss) ODNs sequences were as follows (5′ → 3′): wt κB-PT: CTGGGGACTTTCCAT (underlined is κB-binding site from the HIV long terminal repeat); mut κB-PT: TAACCGACTTTGCAT; fluorescein-labeled wt κB-PT: fluorescein-CTGGGGACTTTCCAT. Desalted ODNs were extracted with phenol/chloroform and purified on the C18 Sephac cartridges (Waters). The double-stranded (ds) ODNs were prepared by annealing the ss ODNs in 150 mM NaCl, and unannealed ss ODNs were removed by using Probind columns (Waters). For in vivo delivery, the ds ODNs were mixed with DOTAP (Boehringer Mannheim) at the ratio of 1:1 (wt/wt) in the buffer containing 140 mM NaCl/7 mM Hepes, pH 7.5, and injected i.a. in a volume of 10 μl.
Synoviocytes.
Fibroblast-like synoviocytes were established by enzymatically dispersing synovial explants from rats with SCW arthritis (19). Cells were maintained in RPMI 1640 medium supplemented with 10% FBS and antibiotics.
Luciferase Assay.
The reporter plasmids 3x wild-type (wt) κBLUC and 3x mutated (mut) κBLUC containing three repeats of the wt or mut κB sites from the class I major histocompatibility complex enhancer were described previously (20). The plasmids were purified from endotoxin by using Detoxigel (Pierce). Cells were transiently transfected with DOTAP. One day later, cells were stimulated and the luciferase activity was determined by using a chemiluminometer and normalized on total protein.
Electrophoretic Mobility-Shift Assay (EMSA).
The nuclear extracts of stimulated cells were incubated with a 32P-labeled ds UV21 probe containing the κB-binding site and resolved on nondenaturing polyacrylamide gel (19). Where indicated, a competitive cold probe was included along with the radiolabeled probe at the molar ratio of 100:1. For the supershift analysis, anti-RelA antibody (Ab) sc109 (Santa Cruz Biotechnology) was included into the reactions.
Northern Blotting.
Synoviocytes were transduced at the third passage with the AdCMV or AdsrIκBα adenovirus at the multiplicity of infection of 100. Two days later, cells were stimulated and total RNA was extracted and resolved on a formaldehyde agarose gel (10 μg/lane). Blots were consecutively hybridized with 32P-labeled DNA probes to rat IL-1β (gift of A. Shaw, Glaxo), rat IL-6 [American Type Cell Collection (ATCC)], rat TNFα (gift of K. Decker, Universitat Freiburg, Germany), rat VCAM-1 (gift of T. Collins, Harvard Medical School, Cambridge, MA), and mouse GAPDH (ATCC).
Cytotoxicity Assay.
Cell viability in vitro was determined by using the MTT assay. Synoviocytes (second to fourth passages) were transduced with Ad vectors and 2 days later stimulated with rat TNFα (rTNFα) (BioSource) or FasL (Alexis) for an additional 24 hr. At the end of treatment, cells were incubated for 4 hr in the growth medium containing MTT (0.5 mg/ml). The precipitate was solubilized, and the number of live cells was determined with a spectrophotometer.
Western Blotting.
The animals were sacrificed at the indicated intervals after i.a. injection of Ad vectors. Excised joints were pulverized in liquid nitrogen and extracted in a buffer (20 mM Tris, pH 7.6/140 mM NaCl/2.5% Triton X-100) supplemented with protease and proteasomal inhibitors [2 mM phenylmethylsulfonyl fluoride/4 μg/ml aprotinin/2 μg/ml leupeptin/10 μM MG132 (Peptides International)]. The extracts were resolved on SDS/PAGE, transferred to nitrocellulose, and immunodetected using primary Ab against human IκBα (Rockland, Gilbertsville, PA) and green fluorescent protein (GFP) (CLONTECH).
In Situ Detection of Apoptosis.
Synovial tissue explants were cut into 5-μm cryostat sections, and apoptosis was evaluated by using a quantitative terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) assay. After a brief fixation in acetone/methanol, samples were stained with a fluorescein In Situ Cell Death Detection kit (Boehringer Mannheim) and examined under a fluorescence microscope. Discrimination and count of bright apoptotic cells were facilitated by using a digital charge-coupled device camera with image 1/metamorph software (Universal Imaging, Media, PA). The total number of cells in the field was counted based on a blue fluorescence of 4′,6-diamidino-2-phenylindole-counterstained nuclei.
Immunodetection of Activated NF-κB and AdsrIκBα Expression.
Cryostat sections of synovial explants were air-dried, fixed in the paraformaldehyde, and immunostained with a primary Ab against human IκBα (Rockland) (final dilution 1:200). The expression of srIκBα was assessed by immunostaining with an mAb against human RelA NLS (12, 21) (Boehringer Mannheim) (final dilution 1:100). Immunostaining was visualized by using the HRP ABC Vectastain kit (Vector). The specificity of NF-κB immunodetection was assessed by immunostaining serial cryostat sections with the primary Ab preabsorbed with the RelA NLS peptide (21) (Quality Control Biochemicals, Hopkington, MA).
Rat Models of Arthritis.
The model of recurrent SCW arthritis was reproduced as described (19, 22). Female Lewis rats (Charles River Breeding Laboratories) weighing ≈150 g were injected into both ankle joints with 5 μg of a rhamnose equivalent of peptidoglycan–polysaccharide complex (PG-APS) (fraction 10s) (19, 21) isolated from group A streptococci. Four weeks later, reactivation of arthritis was induced by i.v. injection of PG-APS at a dose of 200–300 μg. The severity of arthritis was assessed as described (19). The model of pristane polyarthritis was reproduced as described (23). Female DA rats (Harlan Breeders, Indianapolis) weighing ≈180 g received a s.c. injection of 0.15 ml of pristane into the base of the tail (Sigma). At day 12, animals were divided into groups with a similar degree of inflammation in ankle joints and i.a. injected with 10 μl of Ad vectors.
RESULTS
NF-κB Controls Inducible Expression of Inflammatory Molecules in Synoviocytes in Vitro.
To assess the role of NF-κB in controlling inflammatory responses in synoviocytes, NF-κB activation was suppressed by expression of srIκBα. The srIκBα is a mutant form of wt IκBα (17). In contrast to wt IkBa, srIκBα cannot be phosphorylated and degraded and thus affords strong inhibition of NF-κB (18). Stimulation of primary rat synovial fibroblasts with IL-1, TNFα, and lipopolysaccharide induced DNA-binding and transcriptional activity of NF-κB (Fig. 1A and B). The DNA-binding activity was composed of two major complexes, both of which contained the RelA (p65) subunit of NF-κB (Fig. 1B). Expression of srIκBα ablated NF-κB activation (Fig. 1C) and strongly inhibited the induction of IL-1β, TNFα, IL-6, and VCAM-1 messages by TNFα and IL-1 (Fig. 1D). This indicated the requirement of NF-κB activation for induction of IL-1, IL-6, and TNFα in synovial fibroblasts and suggested the involvement of NF-κB in VCAM-1-mediated recruitment of inflammatory cells to the RA joints.
Figure 1.

Suppression of NF-κB inhibits inflammatory responses in primary rat synovial fibroblasts. (A) Induction of NF-κB-mediated transcription in vitro. For luciferase assay, cells were transfected with a 3x wt κBLUC reporter vector followed by an overnight incubation in a low serum (0.5% FBS) medium and stimulated for 3 hr with IL-1β (10 ng/ml), lipopolysaccharide (10 μg/ml), and TNFα (10 ng/ml). The specificity of NF-κB activation was determined by assessing the expression of a 3x mut κBLUC vector (data not shown). (B) Induction of NF-κB DNA-binding activity. For EMSA, quiescent synoviocytes were stimulated for 1 hr with lipopolysaccharide (10 μg/ml) and TNFα (10 ng/ml). The nuclear extracts were preincubated with a radiolabeled κB probe and resolved on PAGE. The specificity of NF-κB binding was assessed by including an excessive amount (100:1) of cold wt and mut NF-κB decoys into the binding reactions. Both major NF-κB bands (arrows) contained RelA subunits, as determined by the supershift by a RelA (p65) Ab (data not shown). (C) AdsrIκBα expression inhibits NF-κB activation in vitro. Cells were transduced with indicated Ad vectors and 2 days later stimulated with TNFα (100 ng/ml) for 30 min. NF-κB DNA-binding activity was determined by EMSA as in B. (D) Suppression of NF-κB inhibits inflammatory responses in vitro. For Northern blotting, cells were transduced as indicated in the legend to C and stimulated with IL-1β (10 ng/ml) and TNFα (10 ng/ml). Total RNA was analyzed by sequentially hybridizing the blot with indicated radiolabeled probes.
Suppression of NF-κB Activation Potentiates the Cytotoxicity of TNFα and FasL in Rat Synoviocytes.
Activation of NF-κB provides protection against the cytotoxicity of TNFα in a variety of cell types (13–16). TNFα and FasL are important mediators of apoptosis in RA (5–9), and, therefore, we examined the potential involvement of NF-κB in TNFα- and FasL-mediated apoptosis in synovial fibroblasts. We found that similar to TNFα (Fig. 1 B and C), recombinant FasL (Fig. 2B) induced NF-κB activation in synovial fibroblasts, although with a slower kinetics (Fig. 2B). Untransduced cells and those transduced with a control AdCMV vector were equally resistant to TNFα and FasL cytotoxicity, but AdsrIkBα expression rendered the cells susceptible to both TNFα- and FasL-induced apoptosis (Fig. 2 A and B). These findings demonstrate that in primary synovial fibroblasts, activation of NF-κB serves as a protective mechanism against the cytotoxicity of TNFα and FasL. Since TNFα is a potent mitogen in synovial fibroblasts (24), NF-κB activation appears to be a master switch determining whether TNFα elicits mitogenic or cytotoxic responses. Similar to that with TNFα, activation of NF-κB in synoviocytes by FasL provided a negative feedback to the cytotoxic action of FasL. Therefore, these results strongly suggested the potential involvement of NF-κB in the protection of synovial cells against apoptosis in the RA synovium, where both FasL and TNFα are overexpressed (1, 5, 9).
Figure 2.

Suppression of NF-κB potentiates the TNFα- and FasL-induced cytotoxicity in rat synovial fibroblasts. (A) The expression of srIκBα potentiates the cytotoxicity of TNFα. Cells were transduced in duplicate with the AdsrIκBα vector and, 2 days later, stimulated with indicated concentrations of TNFα for an additional 24 hr. The cell viability was determined by using the MTT assay. (Bars = SD.) The significance of difference was calculated by using unpaired two-tailed Student’s t test. Data are representative of two experiments. (B) Activation of NF-κB by FasL inhibits the cytotoxicity of FasL. (Upper) Induction of NF-κB DNA-binding activity by FasL. For EMSA, cells were stimulated with FasL (100 ng/ml), and NF-κB DNA-binding activity was determined by EMSA as described in the legend to Fig. 1B. The nuclear extracts of TNFα-stimulated cells (see Fig. 1C) were used as a positive control. (Lower) srIκBα expression potentiates cytotoxicity of FasL. Cells were transduced in duplicate with the AdsrIκBα vector and, 2 days later, stimulated with the indicated concentrations of FasL for an additional 24 hr. Control, untransduced cells. The cell viability was determined by using the MTT assay. The significance of difference was calculated by using unpaired two-tailed Student’s t test. (Bars = SD.) Data are representative of three experiments.
The Onset of SCW Arthritis Coincides with Activation of NF-κB in the Synovium.
Based on our in vitro data, we next assessed the role of NF-κB in the regulation of apoptosis in vivo. For these studies, we employed the model of recurrent SCW arthritis in rats, which truly reproduces many features of human RA (21). In this model, initial i.a. injection of PG-APS produces transient acute joint inflammation. Several weeks later, a recurrence of chronic inflammation in the preinjured joint can be induced by i.v. injection of PG-APS. This model provides a predictable reactivation of chronic inflammation that reaches a peak in 2–4 days after the i.v. injection (21).
The activation of NF-κB in human RA synovium has been well documented (11, 12). To assess NF-κB activation in the SCW rat synovium, we used immunostaining with an mAb specifically recognizing activated NF-κB (12). Activated NF-κB was absent in the normal rat synovium (Fig. 3a) and hardly detectable in rats at 5 weeks after the initial i.a. injection of PG-APS (data not shown). Reactivation of SCW arthritis caused strong activation of NF-κB (Fig. 3c), which persisted for at least 4 days (data not shown). Thus, similar to that in human RA, SCW arthritis is associated with activation of NF-κB in the synovium.
Figure 3.
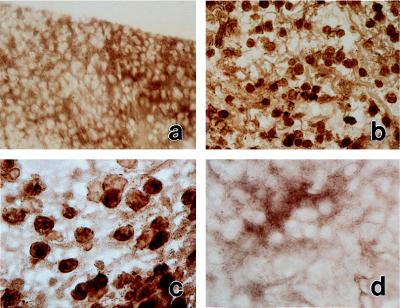
Induction of the recurrence of SCW arthritis is associated with activation of NF-κB in the synovium. Activated NF-κB was detected by HPR immunostaining by using primary mAb against RelA NLS (21). (a) Normal synovium. Original magnification, ×200. (b) Inflamed synovium at day 1 after reactivation of SCW arthritis. Original magnification, ×200. (c) Same as b; original magnification, ×400. (d) Control immunostaining of the inflamed synovium (day 1 after reactivation) by using an mAb preabsorbed with a RelA NLS peptide. Original magnification, ×200. Data are representative of two experiments.
Inhibitors of NF-κB Accelerate Apoptosis in the Inflamed Synovium.
Numerous studies demonstrated the presence of apoptosis in human RA synovium (7, 8). Similar to that in human RA, we found that SCW rat arthritic joints contained a significant proportion of apoptotic cells. As assessed by a quantitative TUNEL assay, 1 day after reactivation of SCW arthritis, the average frequency of apoptosis in the synovium was approximately 6.6%. This was independently confirmed by detecting apoptotic fragmentation in genomic DNA isolated from the whole joint (D.K. and S.M., unpublished data).
To explore the role of NF-κB in the regulation of apoptosis in vivo, we utilized two approaches. In an initial experiment, we assessed proteasome inhibitor peptide aldehyde MG132, which inhibits NF-κB by preventing IκBα degradation (25). Administration (i.a.) of MG132 dramatically increased the frequency of apoptosis in SCW arthritic joints (9.3% in the control, vehicle-treated rats vs. 25.8% in MG132-treated rats, P < 0.01) (Fig. 4A and D). To substantiate these data, we employed a more specific approach based on Ad gene transfer of srIκBα. Ad gene transfer (i.a.) affords efficient transduction in vivo, targeting both fibroblast- and macrophage-like synoviocytes (26, 27). In our study, low titers (≤107 plaque-forming units/joint) of i.a. injected Ad vectors did not cause apparent gross inflammation (data not shown), yet afforded an appreciable expression of srIκBα protein (Fig. 4B). I.a. injected AdsrIκBα did not increase apoptosis in the normal rat joint (data not shown), but significantly accelerated apoptosis in the SCW arthritic joints (8.9% vs. 29.5% in the AdCMV- and AdsrIκBα-transduced joints, P < 0.01) (Fig. 4 C and D). To rule out the possibility that increased apoptosis was a nonspecific effect of overexpressing an exogenous transgene, we additionally examined gene transfer of reporter Ad vectors expressing GFP and β-galactosidase genes. It was found that neither vector significantly increased apoptosis (data not shown). To ascertain that our findings were not restricted to the SCW model, the consequences of suppression of NF-κB were examined in pristane-induced arthritis in rats (22). Similar to that found in SCW model, expression of srIκBα drastically accelerated apoptosis in pristane-induced arthritis (data not shown). We reasoned that if exogenous srIκBα expression “deprotected” transduced cells, srIκBα protein would be rapidly lost. Indeed, while the expression of a reporter GFP protein persisted in the arthritic joints for at least 1 week, srIκBα protein was detectable only during the first 2 days after gene transfer (Fig. 4E). Taken together, these data demonstrate the antiapoptotic function of NF-κB activation in the arthritic synovium.
Figure 4.
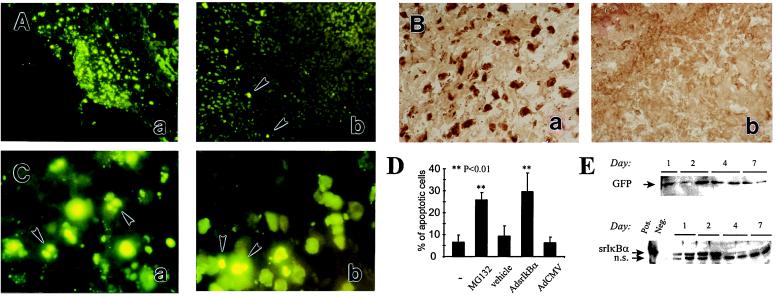
Inhibitors of NF-κB accelerate apoptosis in the inflamed rat synovium. (A) Proteasomal inhibitor MG132 induces apoptosis in the SCW arthritic joint. At day 1 after reactivation of the recurrence of SCW arthritis, ankle joints were injected either with MG132 dissolved in 10% DMSO (5 μg/joint) (a and c) or with the vehicle (b). One day later, synovial tissue was explanted and apoptosis was assessed by using TUNEL assay. Some of apoptotic cells are indicated by the arrowheads. Original magnification, ×100. (B) In vivo adenovirus-mediated gene transfer of srIκBα into the SCW arthritic joints. One day after reactivation of SCW arthritis, ankle joints were injected with the AdsrIκBα vector. Two days later, synovial tissue was explanted and srIκBα expression was assessed by HRP immunostaining for human IκBα (b). The specificity detection was confirmed by immunostaining the AdCMV-transduced joints (a). Data are representative of two experiments. Original magnification, ×200. (C) In vivo gene transfer of srIκBα induces apoptosis in the SCW arthritic joint. At day 1 after reactivation of SCW arthritis, ankle joints were injected either with the AdsrIκBα (a) or with the AdCMV (b) vectors. One day later, synovial tissue was explanted and apoptosis was assessed by using TUNEL assay. Some of apoptotic cells are indicated by the arrowheads. Note nuclear fragmentation in apoptotic cells. Data are representative of two experiments. Original magnification, ×1,000. (D) Quantitative assessment of apoptosis in SCW arthritic joints. A summary of the experiments is described in the legends to A and C. Each bar represents the average percentage of apoptotic cells counted in randomly chosen fields. (Bars = SD.) The frequency of apoptosis was counted at a low magnification in the indicated numbers of randomly chosen fields and compared with that in untransduced SCW arthritic rat synovium (the first bar in the row). MG 132, 12 fields in the explants of four joints; vehicle, 12 fields in the explants of four joints; AdsrIκBa, nine fields in the explants of three joints; AdCMV, nine fields in the explants of three joints. The significance of the difference between treated and untreated SCW joints was calculated by using unpaired two-tailed Student’s t test. (E) Time course of AdGFP (Upper) and AdsrIκBα (Lower) expression in rats with pristane-induced arthritis. Rats with established pristane arthritis received AdsrIκBα or AdGFP vectors into both ankle joints. Expression of the transgene was analyzed by Western blotting. (Upper) Immunodetection of Ad GFP in AdGFP-transduced joints. GFP, the arrow indicates the position of the band of recombinant GFP protein (not shown). (Lower) Immunodetection of srIκBα in AdsrIκBα-transduced joints. Pos., a positive control (in vitro AdsrIκBα-transduced cells); Neg., a negative control (in vitro AdCMV-transduced cells); srIκBα, the arrow indicates the position of the band of the srIκBα protein; n.s., a nonspecific band unrelated to human or rat IκBα, as determined by immunodetection with a peptide-blocked primary Ab (data not shown).
Suppression of Recurrent SCW Arthritis in Rats by NF-κB Decoys.
While Ad gene transfer of srIκBα afforded effective suppression of NF-κB in vitro, inflammatory reaction to the Ad vectors itself represented a serious obstacle for in vivo therapeutic protocols (27). This has been particularly detrimental for delivery of intracellular inhibitors, such as srIkBα, when exogenous protein has to be expressed in the majority of targeted cells. In pilot experiments, we found that high titers of either AdsrIκBα or AdCMV vectors provoked recurrence of SCW arthritis, whereas low titers of AdsrIkBα were not effective. Thus, we attempted an alternative approach that is based on the use of NF-κB decoys, the ds ODNs containing NF-κB-binding sites (28, 29).
Synthesized decoys were found to be functionally active in competing for binding NF-κB in vitro (see Fig. 1B). Liposomal delivery of fluorescein-labeled decoys into the SCW arthritic joint afforded efficient transduction of the synovium (Fig. 5A). The highest intensity of fluorescence was observed at the interface of the pannus and subchondral cartilage in the injected joint (data not shown), while no fluorescence was detected in the contralateral, uninjected ankle joints (data not shown). The ODNs persisted in the synovium for at least 8 days, as determined by hybridization of recovered ODNs with a complementary radiolabeled probe (data not shown). However, consistent with the data by others (27), we observed significant variations in the efficacy of liposomal delivery between experiments.
Figure 5.
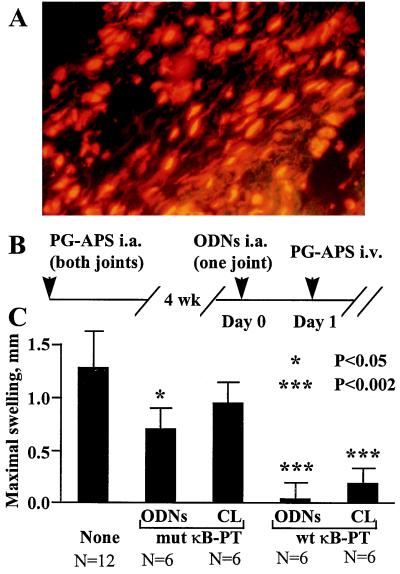
I.a. administration of NF-κB decoys inhibit the recurrence of SCW arthritis. (A) Distribution of fluorescein-labeled ODNs in the synovium. Ankle joints of rats with established SCW arthritis were injected with liposomal complexes of fluorescein-labeled NF-κB decoys. One day later, synovial explants were isolated, sectioned, counterstained with propidium iodide, and analyzed under a fluorescent microscope equipped with a fluorescein filter set. Note the bright-yellow fluorescent nuclei and red fluorescence of cytoplasmic RNA. Original magnification, ×200. (B and C) Therapeutic protocol. (B) Schedule of injections. Rats were injected into both ankle joints with PG-APS. Four weeks later, at Day 0, animals were divided into three groups and monoarticularly injected with liposomal complexes of NF-κB decoys (10 μg ODNs/joint). In the experimental group, animals were injected with wt-κB decoys, and two control groups received mut κB-PT decoys and saline. The contralateral ankle joints were injected with saline. At Day 1, reactivation of arthritis was induced by i.v. injection of PG-APS. (C) The severity of recurrent SCW arthritis in treated rats. The average mean values of joint swelling were calculated in each group as dn = <Dn − D1>, where Dn and D1 are joint diameters at day n and day 1, respectively. After reactivation, the maximal joint swelling in all groups was registered at day 4. ODNs, liposome-treated joints; CL, contralateral joints; N, number of joints per group. The significance of difference with the control, saline-treated group was calculated by using unpaired two-tailed Student’s t test. (Bars = SEM.) Results of a single experiment are shown. The therapeutic effect of wt NF-κB decoys was reproduced in an identical experiment.
The therapeutic efficacy of NF-κB decoys was assessed by using the recurrent SCW arthritis rat model. Rats were injected in both ankle joints with PG-APS, and 4 weeks later, reactivation of arthritis in preinjured joints was induced by the i.v. injection of PG-APS (Fig. 5B). I.a. injection of liposomal complexes containing NF-κB decoys 24 hr before reactivation markedly inhibited development of arthritis in treated joints. Unexpectedly, the severity of inflammation was also inhibited in the contralateral, untreated ankle joints (Fig. 5C). This suppression was NF-κB-specific, since the wt NF-κB decoys were more effective as compared with the mut decoys (Fig. 5C), albeit some suppression of inflammation was evident in the mut NF-κB decoy-treated joints. In a separate experiment, it was found that i.a. injection of “empty” liposomes did not influence the severity of inflammation (data not shown). These data demonstrate therapeutic efficacy of specific inhibitors of NF-κB in SCW arthritis and indicate systemic therapeutic effects of local treatment.
DISCUSSION
In the present study, we examined the role of NF-κB in the arthritic joint in animal models of RA. We demonstrated that reactivation of arthritis caused activation of NF-κB in the synovium and that specific suppression of NF-κB activation induced apoptosis. We thus concluded that the activation of NF-κB in the inflamed synovium protected the cells against apoptosis, thereby providing a putative link between inflammation and hyperplasia. Our in vitro studies in primary synovial fibroblasts demonstrated that one conceivable mechanism whereby NF-κB exerted its protective function was suppression of the cytotoxicity of TNFα and FasL, factors strongly implicated in apoptosis in the RA synovium (5, 6). Our data suggest a dual role of NF-κB in RA pathology (Fig. 6). Activation of NF-κB in synoviocytes is required for the induction of inflammatory cytokines, including IL-1β, IL-6, and TNFα. On the other hand, the activation of NF-κB by inflammation inhibits TNFα- and FasL-mediated apoptosis, thereby promoting hyperplasia. The requirement of NF-κB for the induction of VCAM-1 suggests its potential role in the recruitment of inflammatory cells into the synovium. Our preliminary studies have revealed a requirement of NF-κB for mitogenic activity of growth factors in synovial fibroblasts (J. Romashkova and S.M., unpublished observations), thus implicating NF-κB in the control of proliferation of the inflamed synovium.
Figure 6.

NF-κB in the regulation of inflammation and apoptosis in the arthritic joint.
Importantly, our data indicate that NF-κB activation in synoviocytes is protective against the cytotoxicity of both TNFα and FasL. The protective function of NF-κB in TNFα apoptosis has been recognized (13–16), but the role of NF-κB in Fas-mediated apoptosis remains obscure. As with TNFα, engagement of Fas receptors can activate NF-κB, at least in some cells (30); however, activation of NF-κB by Fas does not correlate with FasL cytotoxicity (30). Our data suggest that this mechanism may be cell type-specific.
The present study focused on the net aspects of activation of NF-κB and apoptosis. Further analysis should determine the role of NF-κB in regulation of apoptosis in distinct cell populations in the inflamed synovium, macrophage- and fibroblast-like synoviocytes, chondrocytes, recruited leukocytes, and APC and T cells. Another important question is the relevance of our findings to human RA. Since suppression of NF-κB accelerates apoptosis in distinctively different models of RA and SCW- and pristane-induced arthritis, we believe that our observations are not limited to a particular model of RA, but are directly relevant to human disease.
In line with the evidence of general involvement of NF-κB in the pathogenic processes in the synovium, it was not surprising that inhibitors of NF-κB alleviated SCW arthritis in treated joints, but systemic therapeutic effects of local treatment were completely unexpected. However, our data are consistent with findings by Ghivizzani et al. (31), who demonstrated that the combined i.a. gene transfer of soluble IL-1 and TNFα in adjuvant-induced arthritis in rabbits exerted beneficial effects in distal joints. Although the mechanisms underlying the systemic therapeutic effects of local treatment are unclear, these findings suggest that in human RA, efficacious therapy may be achieved by an adequate treatment of a limited number of involved joints.
In conclusion, data presented here demonstrate the crucial involvement of NF-κB in the pathogenesis of arthritis and establish a putative mechanism whereby inflammation facilitates the expansion of synovial stroma. Our data suggest that targeting NF-κB should not only alleviate inflammation, but may also inhibit hyperplasia in the RA synovium.
Acknowledgments
We thank R. Rajendran, O. Aprelikova, O. Sirenko, J. Karastoianova, R. Brown, and S. Anderle for technical assistance, D. McCarty for the gift of AdGFP and AdLacZ vectors, S. Neill and J. Bisi for help in production of the AdsrIκBα vector, T. Deaton for assistance in preparation of joint sections, K. Decker for the gift of rat TNFα cDNA, T. Collins for the gift of rat VCAM-1 cDNA, A. Shaw for the gift of rat IL-1β cDNA, J. Watson for help in editing the manuscript, and J. Schwab and P. Cohen for stimulating discussions. This work was supported by National Institutes of Health Grants AR/AI 44564 (to A.S.B. and S.S.M.), 5-P60-AR30701-14 (to S.S.M.), and AI 35098 (to A.S.B.), and by a grant from the Arthritis Foundation (to S.S.M.).
ABBREVIATIONS
- RA
rheumatoid arthritis
- SCW
streptococcal cell wall-induced arthritis
- Ad
adenoviral
- PG-APS
peptidoglycan–polysaccharide complex
- TNFα
tumor necrosis factor α
- FasL
Fas ligand
- IL
interleukin
- i.a.
intraarticular
- ODN
oligodeoxynucleotide
- CMV
cytomegalovirus
- wt
wild type
- mut
mutated
- EMSA
electrophoretic mobility-shift assay
- TUNEL
terminal deoxynucleotidyltransferase-mediated UTP end labeling
- Ab
antibody
- GFP
green fluorescent protein
References
- 1.Firestein G S. Arthritis Rheum. 1996;39:1781–1790. doi: 10.1002/art.1780391103. [DOI] [PubMed] [Google Scholar]
- 2.Zvaifler N J, Tsai V, Alsalameh S, von Kempis J, Firestein G S, Lotz M. Am J Pathol. 1997;150:1125–1138. [PMC free article] [PubMed] [Google Scholar]
- 3.Mountz J D, Wu J, Cheng J, Zhou T. Arthritis Rheum. 1994;37:1415–1420. doi: 10.1002/art.1780371002. [DOI] [PubMed] [Google Scholar]
- 4.Firestein G S, Nguyen K, Aupperle K R, Yeo M, Boyle D L, Zvaifler N J. Am J Pathol. 1996;149:2143–2151. [PMC free article] [PubMed] [Google Scholar]
- 5.Nishioka K, Hasunuma T, Kato T, Sumida T, Kobata T. Arthritis Rheum. 1998;41:1–9. doi: 10.1002/1529-0131(199801)41:1<1::AID-ART1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 6.Zhou T, Edwards C K, III, Yang P, Wang Z, Bluethmann H, Mountz J D. J Immunol. 1996;156:2661–2665. [PubMed] [Google Scholar]
- 7.Firestein G S, Yeo M, Zvaifler N J. J Clin Invest. 1995;96:1631–1638. doi: 10.1172/JCI118202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakajima T, Aono H, Hasunuma T, Yamamoto K, Shirai T, Hirohata K, Nishioka K. Arthritis Rheum. 1995;38:485–491. doi: 10.1002/art.1780380405. [DOI] [PubMed] [Google Scholar]
- 9.Asahara H, Hasunuma T, Kobata T, Inoue H, Muller-Ladner U, Gay S, Sumida T, Nishioka K. J Rheumatol. 1997;24:430–435. [PubMed] [Google Scholar]
- 10.Baldwin A S. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 11.Handel M L, McMorrow L B, Gravallese E M. Arthritis Rheum. 1995;38:1762–1770. doi: 10.1002/art.1780381209. [DOI] [PubMed] [Google Scholar]
- 12.Marok R, Winyard P G, Coumbe A, Kus M L, Gaffney K, Blades S, Mapp P I, Morris C J, Blake D R, Kaltschmidt C, et al. Arthritis Rheum. 1996;39:583–591. doi: 10.1002/art.1780390407. [DOI] [PubMed] [Google Scholar]
- 13.Beg A A, Baltimore D. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 14.Wang C-Y, Mayo M W, Baldwin A S., Jr Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 15.Van Antwerp D J, Martin S J, Kafri T, Green D R, Verma I M. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 16.Liu Z G, Hsu H, Goeddel D V, Karin M. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 17.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 18.Brockman J A, Scherer D C, McKinsey T A, Hall S M, Qi X, Lee W Y, Ballard D W. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makarov S S, Olsen J C, Johnston W N, Anderle S K, Brown R R, Baldwin A S, Jr, Haskill J S, Schwab J H. Proc Natl Acad Sci USA. 1996;93:402–406. doi: 10.1073/pnas.93.1.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheshire J C, Baldwin A S., Jr Mol Cell Biol. 1993;17:6746–6754. doi: 10.1128/mcb.17.11.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaltschmidt C, Kaltschmidt B, Henkel T, Stockinger H, Bauerle P. Biol Chem Hoppe-Seyler. 1995;376:9–16. doi: 10.1515/bchm3.1995.376.1.9. [DOI] [PubMed] [Google Scholar]
- 22.Schwab J H. In: Mechanisms and Models In Rheumatoid Arthritis. Henderson B, Pettipher R, Edwards J, editors. New York: Academic; 1995. pp. 431–446. [Google Scholar]
- 23.Vingsbo C, Sahlstrand P, Brun J G, Jonsson R, Saxne T, Holmdahl R. Am J Pathol. 1996;149:1675–1683. [PMC free article] [PubMed] [Google Scholar]
- 24.Fujisawa K, Aono H, Hasunuma T, Yamamoto K, Mita S, Nishioka K. Arthritis Rheum. 1996;39:197–203. doi: 10.1002/art.1780390205. [DOI] [PubMed] [Google Scholar]
- 25.Palombella V J, Rando O J, Goldberg A L, Maniatis T. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 26.Roessler B J, Allen E D, Wilson J M, Hartman J W, Davidson B L. J Clin Invest. 1993;92:1085–1092. doi: 10.1172/JCI116614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nita I, Ghivizzani S C, Galea-Lauri J, Bandara G, Georgescu H I, Robbins P D, Evans C H. Arthritis Rheum. 1996;39:820–828. doi: 10.1002/art.1780390515. [DOI] [PubMed] [Google Scholar]
- 28.Bielinska A, Shivdasani R A, Zhang L, Nabel G. Science. 1990;267:891–893. doi: 10.1126/science.2237444. [DOI] [PubMed] [Google Scholar]
- 29.Morishita R, Sugimoto T, Aoki M, Kida I, Tomita N, Moriguchi A, Maeda K, Sawa Y, Kaneda Y, Higaki J, et al. Nat Med. 1997;3:894–899. doi: 10.1038/nm0897-894. [DOI] [PubMed] [Google Scholar]
- 30.Ponton A, Clement M V, Stamenkovic I. J Biol Chem. 1996;271:8991–8995. doi: 10.1074/jbc.271.15.8991. [DOI] [PubMed] [Google Scholar]
- 31.Ghivizzani S C, Lechman E R, Kang R, Tio C, Kolls J, Evans C H, Robbins P D. Proc Natl Acad Sci USA. 1998;95:4613–4618. doi: 10.1073/pnas.95.8.4613. [DOI] [PMC free article] [PubMed] [Google Scholar]