

Abstract
Treatment of multidrug-resistant bacterial infections poses a therapeutic challenge to clinicians; combination therapy is often the only viable option for multidrug-resistant infections. A quantitative method was developed to assess the combined killing abilities of antimicrobial agents. Time-kill studies (TKS) were performed using a multidrug-resistant clinical isolate of Acinetobacter baumannii with escalating concentrations of cefepime (0 to 512 mg/liter), amikacin (0 to 256 mg/liter), and levofloxacin (0 to 64 mg/liter). The bacterial burden data in single and combined (two of the three agents with clinically achievable concentrations in serum) TKS at 24 h were mathematically modeled to provide an objective basis for comparing various antimicrobial agent combinations. Synergy and antagonism were defined as interaction indices of <1 and >1, respectively. A hollow-fiber infection model (HFIM) simulating various clinical (fluctuating concentrations over time) dosing exposures was used to selectively validate our quantitative assessment of the combined killing effect. Model fits in all single-agent TKS were satisfactory (r2 > 0.97). An enhanced combined overall killing effect was seen in the cefepime-amikacin combination (interactive index, 0.698; 95% confidence interval [CI], 0.675 to 0.722) and the cefepime-levofloxacin combination (interactive index, 0.929; 95% CI, 0.903 to 0.956), but no significant difference in the combined overall killing effect for the levofloxacin-amikacin combination was observed (interactive index, 0.994; 95% CI, 0.982 to 1.005). These assessments were consistent with observations in HFIM validation studies. Our method could be used to objectively rank the combined killing activities of two antimicrobial agents when used together against a multidrug-resistant A. baumannii isolate. It may offer better insights into the effectiveness of various antimicrobial combinations and warrants further investigations.
Acinetobacter baumannii is an emerging gram-negative bacillus associated with serious nosocomial infections; it is also associated with multiple mechanisms of resistance to various antimicrobial agents (4). Multidrug resistance in A. baumannii has been increasing over the past decades (11) and was shown to be associated with unfavorable clinical outcomes (13, 14). Treatment of multidrug-resistant Acinetobacter infections often represents a challenge to clinicians (7, 15), and there are very few agents in the advanced stage of development designed to target multidrug-resistant gram-negative bacteria. As a result, a task force from the Infectious Diseases Society of America (IDSA) has recently identified A. baumannii as a “particularly problematic pathogen” (22).
For infections caused by a multidrug-resistant pathogen, many available drug treatments are ineffective when used alone. Combination antimicrobial therapy is commonly used clinically for the management of these difficult-to-treat infections. Other than stringent infection control measures, combination therapy is currently our last line of defense in curbing the rising prevalence of multidrug resistance in gram-negative bacteria (28). The rationale is that when using two or more antimicrobial agents (often with different mechanisms of action) concurrently, an enhanced pharmacodynamic effect may be attained. It is hoped that a synergistic combination would provide an enhanced bactericidal effect in the treatment of infections due to multidrug-resistant strains. On the other hand, certain antimicrobial agents may negate one another when combined, resulting in a reduced overall effect (antagonism). Clearly, quantitative information about such synergistic and antagonistic relationships is both valuable and necessary for evaluating the effectiveness of various antimicrobial agent combinations.
However, a satisfactory methodology to evaluate combination therapy and to quantify the extent of pharmacodynamic drug interaction is currently not available. Consequently, it is difficult to compare different combinations in a rational manner. Conventional methods of studying the effect of antimicrobial agent combinations are associated with multiple limitations, and they have not been informative regarding the prediction of favorable clinical outcomes. In view of the numerous possible combinations (e.g., from six available agents, there are 15 possible two-agent combinations), antimicrobial agents to be used together are often selected empirically by clinicians under such circumstances (mostly by trial and error or based on prior personal experience). This approach is poorly guided and may not be optimal for patient care. The objective of this study was to develop a quantitative method to evaluate the combined killing abilities of two antimicrobial agents against a clinical strain of multidrug-resistant A. baumannii. It is hoped that this method could provide a more robust assessment of the activities of different antimicrobial agents when they are used in combination.
(This study was presented in part at the 47th Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, 17 to 20 September 2007 [15a].)
MATERIALS AND METHODS
Antimicrobial agents.
Cefepime was provided by the Bristol-Myers Squibb Research Institute (Princeton, NJ). Amikacin was purchased from LKT Laboratories, Inc. (St. Paul, MN). Levofloxacin was provided by Johnson & Johnson Pharmaceutical Research & Development (Raritan, NJ). A stock solution of each antimicrobial agent in sterile water was prepared, divided into aliquots, and stored at −70°C. Prior to each susceptibility test, an aliquot of the drug was thawed and diluted to the desired concentrations with cation-adjusted Mueller-Hinton II broth (Ca-MHB) (BBL, Sparks, MD).
Microorganism.
A clinical multidrug-resistant strain of A. baumannii (AB 1261, belonging to the ACB-20 clone) from a recent outbreak in Chicago was used in the study. Previous molecular investigations revealed that the isolate harbored OXA-40 (a carbapenem-hydrolyzing oxacillinase), a weak AmpC, and an unspecified TEM β-lactamase (16). The bacterium was stored at −70°C in Protect (Key Scientific Products, Round Rock, TX) storage vials. Fresh isolates were subcultured twice on 5% blood agar plates (Hardy Diagnostics, Santa Maria, CA) for 24 h at 35°C prior to each experiment.
Susceptibility studies.
Cefepime, amikacin, and levofloxacin MICs and minimum bactericidal concentrations (MBCs) were determined by a modified broth macrodilution method as described by the CLSI (8). The final concentration of bacteria in each macrodilution tube was approximately 5 × 105 CFU/ml of Ca-MHB. Serial twofold dilutions of the drugs were used. The MIC was defined as the lowest concentration of drug that resulted in no visible growth after 24 h of incubation at 35°C in ambient air. Samples (50 μl) from clear tubes and from the cloudy tube with the highest drug concentration were plated on Mueller-Hinton agar plates (Hardy Diagnostics). The MBC was defined as the lowest concentration of drug that resulted in ≥99.9% killing of the initial inoculum. The drug carryover effect was assessed by visual inspection of the distribution of colonies on medium plates. The studies were conducted in duplicate and were repeated at least once on a separate day.
Time-kill studies.
Time-kill studies (TKS) were conducted with cefepime, amikacin, and levofloxacin alone at different and escalating concentrations. An overnight culture of the isolate was diluted with prewarmed Ca-MHB and incubated further at 35°C until reaching log-phase growth. The bacterial suspension was diluted with Ca-MHB according to absorbance (at 630 nm); 15 ml of the suspension was transferred to 50-ml sterile conical flasks, each containing 1 ml of a drug dilution at 16 times the target concentration. The final concentration of the bacterial suspension in each flask was approximately 105 CFU/ml (ranging from 1 × 105 CFU/ml to 5 × 105 CFU/ml). The experiment was conducted in a shaker water bath set at 35°C. After 24 h of drug exposure, samples were obtained from each flask in triplicate and the bacterial population was determined by quantitative culture. Prior to being cultured, the bacterial samples (0.5 ml) were centrifuged at 10,000 × g for 15 min and reconstituted with sterile normal saline to their original volumes in order to minimize the drug carryover effect. Total bacterial populations were quantified by spiral plating 10× serial dilutions of the samples onto Mueller-Hinton agar plates (Spiral Biotech, Bethesda, MD). The medium plates were incubated in a humidified incubator (35°C) for 18 to 24 h, and the bacterial density from each sample was determined by visual inspection. The mean killing effect at 24 h was characterized by an inhibitory sigmoid Emax model using the ADAPT II program (9); fitting was weighted by the inverse of the observation variances.
Optimal design for combination studies.
Based on the concentration-effect relationship of each drug determined in the previous section, the optimal concentrations to capture parameter estimates describing the killing effect most precisely at 24 h were determined by using ADAPT II. The concentrations were constrained to the following clinically achievable ranges in human serum: cefepime (2 to 200 mg/liter), amikacin (4 to 80 mg/liter), and levofloxacin (0.5 to 9 mg/liter). D-optimality was employed to optimize the determinant of the inverse Fisher information matrix. Assay variance was based on the variance of the bacterial burden observations for each drug.
Combination TKS.
TKS similar to those described above were repeated using 25 concentration combinations in a five-by-five array for each two-agent combination. Specific concentrations for each agent used (including a placebo control) were as determined in the previous section. The total bacterial burden at 24 h (in triplicate) was determined by quantitative culture as described above, and the data were mathematically modeled using a three-dimensional response surface as described previously (25). Briefly, effect summation was used as the definition of additivity (null interaction) (5), as follows:
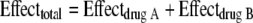 |
(1) |
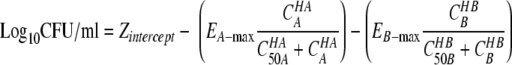 |
(2) |
Zintercept is the bacterial density at 24 h in the absence of the drug, EA−max and EB−max are the maximal effects of drug A and drug B, CA and CB are the concentrations of drug A and drug B, HA and HB are the sigmoidicities of drug A and drug B, and C50A and C50B are the concentrations of drug A and drug B to achieve 50% of the maximal effect, respectively.
Volumes under the plane (VUP) of the observed and expected surfaces were computed by interpolation and double integration, respectively (Mathematica 5.2; Wolfram Research, Inc., Champaign, IL). The overall combined killing activities of antimicrobial agents were assessed using the following interaction index: VUPobserved/VUPexpected. Synergy and antagonism were defined as interaction indices of <1 and >1, respectively. The confidence interval (95%) of VUPobserved was computed with mean data points ± 1.96 standard deviation.
Hollow-fiber infection model.
To selectively validate the quantitative assessment of combined killing with various antimicrobial agent combinations, a hollow-fiber infection model (HFIM) in which the bacteria were exposed to clinically relevant (fluctuating concentration over time due to repeated dosing and constant elimination) drug exposures was used. A schematic diagram of the HFIM has been described previously (23). The drug(s) was directly injected into the central reservoir to reach clinically achievable peak concentrations. Fresh (drug-free) growth medium (Ca-MHB) was continuously infused from the diluent reservoir into the central reservoir to dilute the drug in order to simulate drug elimination in humans. An equal volume of drug-containing medium was removed from the central reservoir concurrently to maintain an isovolumetric system. Bacteria were inoculated into the extracapillary compartment of the hollow-fiber cartridge (Fibercell Systems, Inc., Frederick, MD); they were confined in the extracapillary compartment but were exposed to the fluctuating drug concentration in the central reservoir by means of an internal circulatory pump in the bioreactor loop. The experimental setup was slightly modified if the elimination half-lives (t1/2) of the agents in a combination were considerably different (2).
Experimental setup.
For each study, the inoculum was prepared as described above. Twenty milliliters of A. baumannii suspension at approximately 105 CFU/ml was used. The experiment was conducted for 48 h in a humidified incubator set at 35°C. The infection models were subjected to different drug exposures simulating various steady-state pharmacokinetic profiles of unbound drugs (cefepime fCmax = 165 mg/liter, t1/2 = 2.5 h [1, 24]; amikacin fCmax = 60 mg/liter, t1/2 = 2.5 h [27]; and levofloxacin fCmax = 9 mg/liter, t1/2 = 6 h [18, 19]). Maintenance doses were given according to clinical dosing frequencies of the individual drugs to reattain the unbound maximum concentration (fCmax) targeted. Two (two-agent) combination regimens using the highest clinical doses (i.e., cefepime, 2 g every 8 h; amikacin, 1.5 g every 24 h; and levofloxacin, 750 mg every 24 h) were examined, as guided by the modeling results of combination TKS (the most and least synergistic combinations were used). For comparison, three single-drug regimens (at twice the highest clinical dose) and a placebo control were used. The effectiveness levels of these dosing regimens were compared based on the observed viable bacterial burden over time.
Pharmacokinetic validation.
Serial samples were obtained from the infection models and kept frozen at −20°C until analyses (within 2 months after the completion of the HFIM studies). The drug concentrations in these samples were assayed by validation methods as described below. The concentration-time profiles were modeled by fitting a one-compartment linear model to the observations by using ADAPT II.
Drug assays.
Cefepime and levofloxacin concentrations in Ca-MHB were assayed using a validated high-performance liquid chromatography (HPLC) method. The HPLC system consists of a Waters 2695 separation module with a 2487 UV detector and NovaPak C18 (4 μm) column (3.9 by 150 mm) (Waters Corporation, Milford, MA). The samples were spiked with an internal standard (levofloxacin for the cefepime assay; moxifloxacin for the levofloxacin assay). A gradient elution procedure with various proportions of acetonitrile, 0.1 M phosphoric acid (at pH 3.0), and 0.01 M n-octylamine (at pH 3.0) as the mobile phase (1 ml/min) was used, and detection was done at 292 nm. The cefepime assay was linear from 0.5 mg/liter to 100.0 mg/liter, and the inter-day coefficient of variation was <9%. The levofloxacin assay was linear from 0.5 mg/liter to 25.0 mg/liter, and the inter-day coefficient of variation was <6%.
On the other hand, amikacin concentrations in Ca-MHB were assayed using a validated liquid chromatography mass spectroscopy method. The same HPLC system was used with an EMD 1000 mass detector (Waters Corporation). After the samples were spiked with gentamicin as the internal standard, they were purified by solid-phase extraction using Oasis MCX cartridges (Waters Corporation). Amikacin and the internal standard were subsequently eluted with a mixture of methanol and ammonium hydroxide. A gradient elution procedure with various proportions of acetonitrile and 0.5% formic acid as the mobile phase (0.3 ml/min) was used. Amikacin and gentamicin were detected at m/z ratios of 264.0 and 322.0, respectively. The linear range of the amikacin assay was from 0.5 mg/liter to 10.0 mg/liter, and the inter-day coefficient of variation for the assay was <9%. Samples with drug concentrations expected to be outside of the linear assay ranges were diluted two- to 10-fold accordingly before the assay procedure.
Microbiologic response.
Serial samples were also obtained from each infection model at 0 (baseline), 4, 8, 12, 24 (predose), 28, 32, 36, and 48 h in duplicate. Bacterial burdens were determined in duplicate by quantitative culture as described above to examine the effects of various drug exposure(s) on the total bacterial population over time.
RESULTS
Susceptibility.
The isolate was resistant to all agents examined as anticipated. The MICs and MBCs of cefepime, amikacin, and levofloxacin were found to be 32 and 64 mg/liter, 256 and 256 mg/liter, and 32 and 32 mg/liter, respectively. In addition, the isolate was also resistant to imipenem, meropenem, and polymyxin B (data not shown).
TKS.
Model fits in all single-drug TKS were satisfactory (r2 > 0.97; Fig. 1). Optimal concentrations to define the killing effect most precisely at 24 h were found to be as follows: cefepime at 2, 14, 50, and 200 mg/liter; amikacin at 4, 40, 70, and 80 mg/liter; and levofloxacin at 0.5, 5, 8, and 9 mg/liter. In combination TKS, an enhanced overall combined killing effect was seen when cefepime was used together with amikacin and when cefepime was used concurrently with levofloxacin. In contrast, there was no significant increase in the overall combined killing effect in the levofloxacin-amikacin combination (Fig. 2). The quantitative assessment of the combined killing activities of various combinations is as shown in Table 1.
FIG. 1.

Model fits of the bacterial burden at 24 h in single-drug TKS. C, concentration. Data are shown as the mean ± the standard deviation.
FIG. 2.

Comparison of expected and observed killing activities of cefepime plus amikacin (A), amikacin plus levofloxacin (B), and cefepime plus levofloxacin (C). The red mesh surface is the expected killing activity of the antimicrobial agent combination, and the black dots are the observed killing. When a black dot is below the red mesh surface, the observed killing is more than the expected killing (synergism). On the other hand, when a black dot is above the red mesh surface, the observed killing is less than the expected killing (antagonism). The overall killing over the concentration ranges examined was assessed by computing the VUP. Synergy and antagonism were defined as interactive index (VUPobserved/VUPexpected) values of <1 and >1, respectively.
TABLE 1.
Assessment of the combined killing activities of various antimicrobial combinations
| Antimicrobial combination | Interactive index | 95% CI | Categorical interpretation |
|---|---|---|---|
| Cefepime plus amikacin | 0.698 | 0.675-0.722 | Synergism |
| Amikacin plus levofloxacin | 0.994 | 0.982-1.005 | Additivity |
| Cefepime plus levofloxacin | 0.929 | 0.903-0.956 | Synergism |
Pharmacokinetic validation.
All simulated drug exposures were satisfactory, and typical pharmacokinetic profiles observed are shown in Fig. 3.
FIG. 3.

Typical observed pharmacokinetic profiles in the following infection models: cefepime, 4 g every 8 h (A); amikacin, 3 g every 24 h (B); and cefepime, 2 g every 8 h, plus amikacin, 1.5 g every 24 h (C).
HFIM model studies.
The time courses of the bacterial burden associated with selected antimicrobial combinations are shown in Fig. 4. Overall, these observations were in general agreement with our assessment of combined killing activity (Table 1). As depicted in Fig. 4A, a considerable reduction (>99%) in the bacterial burden was observed after 4 h for both cefepime and amikacin when used alone. However, regrowth was apparent with both drugs after 12 h despite repeated dosing. In comparison, when cefepime was used concurrently with amikacin (the most synergistic combination evaluated), a more sustained suppression of the bacterial population was seen. The observed bacterial burdens with this combination were lower than for both single-drug regimens (despite being used at twice the dose) for up to 36 h. Furthermore, the growth rate of the surviving population was much lower than that of the parent isolate (data not shown), possibly implying a substantial biofitness deficit in this population. Repeat susceptibility testing of two random isolates obtained at the end of the experiment did not reveal a significant change (more than twofold) in susceptibilities to amikacin and levofloxacin. The cefepime MIC was elevated in one of the two isolates examined.
FIG. 4.

Microbiologic responses observed in the following infection models: cefepime plus amikacin (A) and amikacin plus levofloxacin (B). Data are shown as the mean ± the standard deviation.
Conversely, the benefit of adding levofloxacin to amikacin (being an additive combination) was not as evident, as illustrated in Fig. 4B. It was especially so when the reduction in the bacterial burden could hardly be achieved with levofloxacin alone (even at twice the clinical dose). The effect associated with the levofloxacin-amikacin combination was practically identical to that of amikacin alone, supporting our quantitative assessment of this combination.
DISCUSSION
Previous attempts to quantify arbitrary agent interactions are unsatisfactory as they are associated with multiple implicit assumptions of the interacting system; they have been reviewed in detail previously (5, 25). Briefly, the fractional inhibitory concentration index (the most widely used method for the in vitro interaction of antimicrobial agents) is based on Loewe additivity, in which linear concentration-effect relationships are implicitly mandated for all agents in a combination. This underlying assumption is clearly violated, as demonstrated by our data shown in Fig. 1. Also, the endpoint used in the fractional inhibitory concentration index is the suppression of bacterial growth rather than the killing of bacteria. On the other hand, conventional TKS are equally problematic, as usually only one concentration of each agent is used, thus providing limited insights into the combined effect when they are used clinically (fluctuating over a concentration range due to constant elimination and repeated dosing). Furthermore, in view of different endpoints and assumptions used in these conventional methods, another drawback is that the interpretations of results may not correlate with each other (3, 6), and they have not been useful in making reliable predictions in clinical studies regarding the effectiveness of various combinations (12, 20). Serious infections caused by multidrug-resistant pathogens are becoming more prevalent. There is a need for a better method to characterize the combined killing effect when antimicrobial agents are used together. If the most effective agent combination can be objectively identified, it is hoped that multidrug resistance can be suppressed (or controlled) until new agents become available.
Circumventing the limitations of widely accepted methods, we developed a relatively simple approach to quantitatively assess the activity of two antimicrobial agents when used together. Similar approaches have been developed in the past to characterize the combined antimicrobial activities of antifungal (17, 26) and antiretroviral (10, 21) agents. However, to the best of our knowledge, this is the first study in which a mathematical model could use limited data as inputs to make useful predictions regarding the relative levels of effectiveness of various antimicrobial agent combinations against a multidrug-resistant bacterial isolate. The utility of the model was exemplified by selective prospective validation of the predictions under experimental conditions in which drug concentrations vary over time.
The microbial response to single antimicrobial agents was first characterized. Based on these results, various agent combinations were then evaluated with respect to their killing effect when clinically achievable concentrations of these agents were used. The ability to objectively quantify the pharmacodynamic interaction between two antimicrobial agents offered the distinct advantage of ranking various agent combinations in terms of their relative levels of effectiveness against a specific pathogen using a numeric scale of the interactive index (i.e., cefepime plus amikacin would be a superior combination against AB 1261 compared to levofloxacin plus amikacin). In addition, the variances of the observed effects were used to compute a confidence interval of the parameter estimates for statistical comparison. Also, the analytic approach demonstrated in this study was not specific to any one pathogen and did not require prior knowledge of the mechanism(s) of resistance. To enhance the applicability of the model, predictions of the combined killing effect were subsequently selectively validated using an in vitro infection model in which humanlike (fluctuating) drug concentration profiles were simulated.
As pointed out previously, our model performed better when there was limited activity by individual drugs in a combination. This was due to our inability to measure the antibacterial effect that exceeded inoculum eradication (25). Therefore, we focused on a multidrug-resistant strain to which little activity was shown by the individual drug when used alone. In this study, we demonstrated a validated modeling approach using only one clinical strain of A. baumannii. The generalizability of the model would be more ensured with experimental validation using more antimicrobial agent combinations (with various magnitudes of pharmacodynamic interaction) and against a greater number of multidrug-resistant bacterial strains. The applicability to other pathogens and the in vivo relevance of the model are currently under investigation.
In conclusion, we developed a novel method to characterize and rank the levels of effectiveness of various antimicrobial agent combinations against a multidrug-resistant bacterial isolate. This method may provide better insights into their combined effectiveness than other approaches and warrants further investigations.
Acknowledgments
This study was supported in part by research grants from the Society of Infectious Diseases Pharmacists (Austin, TX) and SingHealth Foundation (Singapore).
Footnotes
Published ahead of print on 27 May 2008.
REFERENCES
- 1.Barbhaiya, R. H., S. T. Forgue, C. R. Gleason, C. A. Knupp, K. A. Pittman, D. J. Weidler, H. Movahhed, J. Tenney, and R. R. Martin. 1992. Pharmacokinetics of cefepime after single and multiple intravenous administrations in healthy subjects. Antimicrob. Agents Chemother. 36:552-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blaser, J. 1985. In-vitro model for simultaneous simulation of the serum kinetics of two drugs with different half-lives. J. Antimicrob. Chemother. 15(Suppl. A):125-130. [DOI] [PubMed] [Google Scholar]
- 3.Bonapace, C. R., R. L. White, L. V. Friedrich, and J. A. Bosso. 2000. Evaluation of antibiotic synergy against Acinetobacter baumannii: a comparison with Etest, time-kill, and checkerboard methods. Diagn. Microbiol. Infect. Dis. 38:43-50. [DOI] [PubMed] [Google Scholar]
- 4.Bonomo, R. A., and D. Szabo. 2006. Mechanisms of multidrug resistance in Acinetobacter species and Pseudomonas aeruginosa. Clin. Infect. Dis. 43(Suppl. 2):S49-S56. [DOI] [PubMed] [Google Scholar]
- 5.Boucher, A. N., and V. H. Tam. 2006. Mathematical formulation of additivity for antimicrobial agents. Diagn. Microbiol. Infect. Dis. 55:319-325. [DOI] [PubMed] [Google Scholar]
- 6.Cappelletty, D. M., and M. J. Rybak. 1996. Comparison of methodologies for synergism testing of drug combinations against resistant strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 40:677-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. 2004. Acinetobacter baumannii infections among patients at military medical facilities treating injured U.S. service members, 2002-2004. MMWR Morb. Mortal. Wkly. Rep. 53:1063-1066. [PubMed] [Google Scholar]
- 8.Clinical and Laboratory Standards Institute. 2007. Performance standards for antimicrobial testing: 17th informational supplement. CLSI M100-S17. Clinical and Laboratory Standards Institute, Wayne, PA.
- 9.D'Argenio, D. Z., and A. Schumitzky. 1997. ADAPT II user's guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource, University of Southern California, Los Angeles.
- 10.Drusano, G. L., D. Z. D'Argenio, W. Symonds, P. A. Bilello, J. McDowell, B. Sadler, A. Bye, and J. A. Bilello. 1998. Nucleoside analog 1592U89 and human immunodeficiency virus protease inhibitor 141W94 are synergistic in vitro. Antimicrob. Agents Chemother. 42:2153-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaynes, R., and J. R. Edwards. 2005. Overview of nosocomial infections caused by gram-negative bacilli. Clin. Infect. Dis. 41:848-854. [DOI] [PubMed] [Google Scholar]
- 12.Hilf, M., V. L. Yu, J. Sharp, J. J. Zuravleff, J. A. Korvick, and R. R. Muder. 1989. Antibiotic therapy for Pseudomonas aeruginosa bacteremia: outcome correlations in a prospective study of 200 patients. Am. J. Med. 87:540-546. [DOI] [PubMed] [Google Scholar]
- 13.Kuo, L. C., C. C. Lai, C. H. Liao, C. K. Hsu, Y. L. Chang, C. Y. Chang, and P. R. Hsueh. 2007. Multidrug-resistant Acinetobacter baumannii bacteraemia: clinical features, antimicrobial therapy and outcome. Clin. Microbiol. Infect. 13:196-198. [DOI] [PubMed] [Google Scholar]
- 14.Kwa, A. L., J. G. Low, E. Lee, A. Kurup, H. L. Chee, and V. H. Tam. 2007. The impact of multidrug resistance on the outcomes of critically ill patients with Gram-negative bacterial pneumonia. Diagn. Microbiol. Infect. Dis. 58:99-104. [DOI] [PubMed] [Google Scholar]
- 15.Levin, A. S., A. A. Barone, J. Penco, M. V. Santos, I. S. Marinho, E. A. Arruda, E. I. Manrique, and S. F. Costa. 1999. Intravenous colistin as therapy for nosocomial infections caused by multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Clin. Infect. Dis. 28:1008-1011. [DOI] [PubMed] [Google Scholar]
- 15a.Lim, T. P., K. R. Ledesma, K. T. Chang, A. L. Kwa, and V. H. Tam. 2007. Abstr. 47th Intersci. Conf. Antimicrob. Agents Chemother., abstr. A-8.
- 16.Lolans, K., T. W. Rice, L. S. Munoz-Price, and J. P. Quinn. 2006. Multicity outbreak of carbapenem-resistant Acinetobacter baumannii isolates producing the carbapenemase OXA-40. Antimicrob. Agents Chemother. 50:2941-2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meletiadis, J., J. W. Mouton, J. F. Meis, and P. E. Verweij. 2003. In vitro drug interaction modeling of combinations of azoles with terbinafine against clinical Scedosporium prolificans isolates. Antimicrob. Agents Chemother. 47:106-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Preston, S. L., G. L. Drusano, A. L. Berman, C. L. Fowler, A. T. Chow, B. Dornseif, V. Reichl, J. Natarajan, and M. Corrado. 1998. Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA 279:125-129. [DOI] [PubMed] [Google Scholar]
- 19.Rebuck, J. A., D. N. Fish, and E. Abraham. 2002. Pharmacokinetics of intravenous and oral levofloxacin in critically ill adults in a medical intensive care unit. Pharmacotherapy 22:1216-1225. [DOI] [PubMed] [Google Scholar]
- 20.Saballs, M., M. Pujol, F. Tubau, C. Pena, A. Montero, M. A. Dominguez, F. Gudiol, and J. Ariza. 2006. Rifampicin/imipenem combination in the treatment of carbapenem-resistant Acinetobacter baumannii infections. J. Antimicrob. Chemother. 58:697-700. [DOI] [PubMed] [Google Scholar]
- 21.Snyder, S., D. Z. D'Argenio, O. Weislow, J. A. Bilello, and G. L. Drusano. 2000. The triple combination indinavir-zidovudine-lamivudine is highly synergistic. Antimicrob. Agents Chemother. 44:1051-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talbot, G. H., J. Bradley, J. E. Edwards, Jr., D. Gilbert, M. Scheld, and J. G. Bartlett. 2006. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 42:657-668. [DOI] [PubMed] [Google Scholar]
- 23.Tam, V. H., A. Louie, T. R. Fritsche, M. Deziel, W. Liu, D. L. Brown, L. Deshpande, R. Leary, R. N. Jones, and G. L. Drusano. 2007. Impact of drug-exposure intensity and duration of therapy on the emergence of Staphylococcus aureus resistance to a quinolone antimicrobial. J. Infect. Dis. 195:1818-1827. [DOI] [PubMed] [Google Scholar]
- 24.Tam, V. H., P. S. McKinnon, R. L. Akins, G. L. Drusano, and M. J. Rybak. 2003. Pharmacokinetics and pharmacodynamics of cefepime in patients with various degrees of renal function. Antimicrob. Agents Chemother. 47:1853-1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tam, V. H., A. N. Schilling, R. E. Lewis, D. A. Melnick, and A. N. Boucher. 2004. Novel approach to characterization of combined pharmacodynamic effects of antimicrobial agents. Antimicrob. Agents Chemother. 48:4315-4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Te Dorsthorst, D. T. A., P. E. Verweij, J. Meletiadis, M. Bergervoet, N. C. Punt, J. F. G. M. Meis, and J. W. Mouton. 2002. In vitro interaction of flucytosine combined with amphotericin B or fluconazole against thirty-five yeast isolates determined by both the fractional inhibitory concentration index and the response surface approach. Antimicrob. Agents Chemother. 46:2982-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tod, M., O. Lortholary, D. Seytre, R. Semaoun, B. Uzzan, L. Guillevin, P. Casassus, and O. Petitjean. 1998. Population pharmacokinetic study of amikacin administered once or twice daily to febrile, severely neutropenic adults. Antimicrob. Agents Chemother. 42:849-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Urban, C., S. Segal-Maurer, and J. J. Rahal. 2003. Considerations in control and treatment of nosocomial infections due to multidrug-resistant Acinetobacter baumannii. Clin. Infect. Dis. 36:1268-1274. [DOI] [PubMed] [Google Scholar]