

Abstract
Anaerobic ammonium oxidation is a recent addition to the microbial nitrogen cycle, and its metabolic pathway, including the production and conversion of its intermediate hydrazine, is not well understood. Therefore, the effect of hydroxylamine addition on the hydrazine metabolism of anaerobic ammonium-oxidizing (anammox) bacteria was studied both experimentally and by mathematical modeling. It was observed that hydroxylamine was disproportionated biologically in the absence of nitrite into dinitrogen gas and ammonium. Little hydrazine accumulated during this process; however, rapid hydrazine production was observed when nearly all hydroxylamine was consumed. A mechanistic model is proposed in which hydrazine is suggested to be continuously produced from ammonium and hydroxylamine (possibly via nitric oxide) and subsequently oxidized to N2. The electron acceptor for hydrazine oxidation is hydroxylamine, which is reduced to ammonium. A decrease in the hydroxylamine reduction rate, therefore, leads to a decrease in the hydrazine oxidation rate, resulting in the observed hydrazine accumulation. The proposed mechanism was verified by a mathematical model which could explain and predict most of the experimental data.
The anammox process is the oxidation of ammonium with nitrite as an electron acceptor—in the absence of oxygen—resulting in the formation of dinitrogen gas (N2) (33). This process has so far only been shown for certain organisms belonging to the order Planctomycetales (29). Because of its unique ability to combine two different nitrogen compounds to form dinitrogen gas (34) and its relevance to the global nitrogen cycle (4, 14, 15, 19, 23, 25), the anammox process has gained much attention lately. Furthermore, the process was successfully applied in wastewater treatment (8, 18, 35, 39).
Although catabolic pathways of anammox bacteria have been proposed (31, 34), no definite catabolic scheme is available since none of the intermediates has been detected under physiological conditions (when converting ammonium and nitrite). Hydrazine is suggested to be an intermediate because of its production upon the addition of hydroxylamine (34) or nitric oxide (NO) (B. Kartal, personal communication) in enrichment cultures. Whether hydroxylamine and/or nitric oxide are intermediates in the anammox process as well is unclear. The presence of nirS (which encodes an enzyme that converts nitrite exclusively to nitric oxide) in the genome of “Candidatus Kuenenia stuttgartiensis,” however, implies that NO is an intermediate (31).
The production of hydrazine upon the addition of hydroxylamine in microorganisms is unique, and thus, hydrazine measurements following the hydroxylamine addition can be regarded as a “benchmark” for anammox bacteria (9). Therefore, hydroxylamine addition experiments were used during several enrichment studies (5, 11, 12, 22, 36, 40), and the subsequent production of hydrazine was regarded as an indicator of successful enrichment.
The unavailability of pure anammox cultures makes research into the metabolism particularly challenging. Several complementary approaches have been used over the years to elucidate the metabolism. First, enzymes were purified from anammox enrichment cultures (3, 20, 26); second, anammox proteins were expressed in Escherichia coli (7); third, the genetic blueprint of an anammox bacterium was unraveled in an environmental genome project (31); and finally, batch experiments were performed with physically purified cells (10, 30).
Additionally, valuable information can also be obtained by the disturbance of the metabolism of anammox bacteria by the sudden addition of relevant chemicals. Perturbations lead to an imbalance in the metabolism and, therefore, to measurable changes in the concentration of intermediates. In this study, we have used this perturbation approach by adding hydroxylamine and monitoring the concentration of ammonium and hydrazine in time. This dynamic response under different conditions was used to evaluate the possible interaction between different metabolic reactions in anammox cells and to evaluate the role of hydroxylamine in anammox enrichments. Based on the data, a metabolic model was constructed that was able to predict most of the observed phenomena.
MATERIALS AND METHODS
Biomass origin.
Two different anammox species were used in this study. Most of the experiments used a biomass collected from a lab-scale anammox reactor, containing a granular biomass from an earlier enrichment (37). The reactor volume was 15 liters, with a liquid volume of 8 liters. The pH was not controlled but was stable at 7.5 ± 0.4. The temperature was controlled at 38°C. The hydraulic retention time was 2.5 days. The reactor was fed with a medium which was similar to the one described by Van de Graaf et al. (33) but with increased ammonium and nitrite levels to obtain a total nitrogen load of 100 mM/day. The reactor contained granules with an average diameter of 0.3 mm and a total biomass concentration of 5 g C/liter. To maintain anoxic conditions and to provide a pH buffer, the reactor was flushed at 25 ml/min with 95% Ar-5% CO2 (vol/vol). Fluorescence in situ hybridization analysis (hybridization with probes AMX-820 and KST-1273) showed that the enriched organism was “Candidatus Kuenenia stuttgartiensis” in a proportion of about 70% of the total biomass. To assess whether the findings were strain specific, a second set of experiments was performed with a “Candidatus Brocadia fulgida” enrichment from a sequencing batch reactor under similar conditions (13).
Biomass preparation.
Thirty-milliliter samples from the enrichment reactor were taken and washed with a nitrogen-free medium similar to that of Van de Graaf et al. (33) but containing no ammonium, nitrate, or nitrite. Granules were washed until the nitrate concentration was below 0.1 mM. The biomass was transferred to 100-ml bottles, and the liquid volume was set to 50 ml with the nitrogen-free medium solution. The bottles contained septa for sample taking and were made anaerobic by sparging with 95% Ar-5% CO2 (vol/vol). The bottles were placed in a thermostated shaker (37°C, 150 rpm), and 1 ml of a 100 mM solution of ammonium [as (NH4)2SO4] was added to achieve a starting concentration of about 2 mM. After acclimatizing for 2 h, experiments were started with the addition of NH2OH.
Hydroxylamine addition tests.
A total of 0.25 to 5 ml of freshly prepared 100 mM NH2OH·HCl solutions were added to the test bottles to achieve initial hydroxylamine concentrations of 0.5 to 10 mM. Samples were taken from the bottles after short (5-s) settling of the granular biomass and immediately filter-sterilized (0.2-μm filters). The filtrate was analyzed for hydroxylamine, hydrazine, ammonium, and—for selected samples—nitrate and nitrite. The concentrations were corrected for the volume removed by the sampling. The biomass content was determined after the experiment by the measurement of the dry weight (DW). As a negative control, the same experiment was performed with the nitrogen-free medium in the absence of the biomass.
Influence of ammonium, short-chain fatty acids, and oxygen.
Freshly prepared hydroxylamine solution (2 ml, 100 mM) was added to the test bottles (prepared as described above) to achieve an initial concentration of 4 mM. Ammonium, formate, acetate, or propionate was added immediately to achieve a concentration of 2 mM in the test bottles. In the aerobic experiment, the sample bottles were flushed with air (rather than with Ar/CO2 gas) during preparation. During this experiment, the headspace was in direct contact with the atmosphere. The oxygen level was not measured, but in view of the low conversion rates, the liquid was supposed to be saturated with oxygen throughout the experiment.
Effect of hydroxylamine on NO, N2O, and NO2.
Because of the small volume of the sample bottles, NO, N2O, and NO2 could not be measured in the batch tests, and therefore, the hydroxylamine addition experiments were also conducted directly in the 15-liter enrichment reactor which contained the “Candidatus Kuenenia stuttgartiensis” biomass. The experiment was started by stopping the influent and effluent pumps for 3 h, followed by the addition of hydroxylamine (50 ml of 1 M NH2OH, giving a final concentration of 6 mM). Samples were taken regularly for hydroxylamine, ammonium, nitrite, nitrate, and N2O. The pH was measured online, and off-gas was collected continuously in sample bags. From these sample bags, the NO and NO2 levels were determined automatically every 20 min.
Analytical procedures.
The hydroxylamine level (30 to 100 μM) was measured spectrophotometrically using the method of Frear and Burrell (6). Briefly, 100 μl of 0.5 M phosphate buffer (pH 7), 200 μl of 12% (wt/wt) trichloroacetic acid, and 200 μl of 1% (wt/wt) quinolinol in absolute ethanol were added to 2.7 ml of (diluted) sample. After shaking, 1 ml of 10.6% (wt/wt) (1 M) Na2CO3 solution was added. The mixture was heated for 1 min in a water bath at 100°C. After cooling (10 min), the hydroxylamine level was determined spectrophotometrically at 705 nm (Novaspec 4049; Biochrom, Cambridge, United Kingdom). Hydrazine detection (3 to 30 μM) was performed using a modification of the method of Watt and Chrisp (38). A total of 100 μl of hydrazine reagent (4 g para-dimethylaminobenzaldehyde dissolved in 20 ml ethanol and 2 ml concentrated HCl) was added to 900 μl of the sample. After 20 min, the sample was measured spectrophotometrically at 458 nm (Novaspec 4049; Biochrom, Cambridge, United Kingdom).
Ammonium (1 to 143 μM), nitrite (43 to 429 μM), and nitrate (16 to 964 μM) were detected using commercial test kits (Dr. Lange kits LCK304, LCK 341 and LCK339, respectively; Hach Lange GmbH, Düsseldorf, Germany) and determined on a designated spectrophotometer (CADAS 50S). The standard deviations of the measurements were estimated to be 5% of the measured value. Acetate and propionate levels were determined by gas chromatography (Chrompack CP 9001 equipped with a Hewlett Packard HP INNOWAX column). The pH was measured at the beginning and end of each experiment and was always in the optimum pH range for anammox activity (32).
NO and NO2 levels were measured in the gas phase using a chemiluminescence analyzer (CLD 700EL; Ecophisics, Ann Arbor, MI). The N2O level in the liquid was measured off-line with a reversed Clark-type sensor (Unisense; Århus, Denmark).
Mathematical modeling.
Simulations with the proposed kinetic model were performed with AQUASIM software (version 2.0; P. Reichert, Swiss Federal Institute for Environmental Science and Technology, Dübendorf, Switzerland). The reaction volume was modeled as one homogeneously mixed compartment. Rate equations were of Monod-type saturation for all relevant substrates. Reaction stoichiometry and rate equations that were used are presented in the Discussion section. The initial solute and biomass concentrations in the simulated batch tests were set to the measured values.
Thermodynamic calculations.
The Gibbs energy of the reaction (ΔGR0′) was always calculated under standard conditions in the biological frame of reference (temperature, 298 K; pressure, 1 bar; concentration of reactants and products, 1 M, pH 7). Values for the Gibbs energy of formation (ΔGf°) of H2O, N2, NH4+, and NO were taken from the CRC Handbook of Chemistry and Physics (17). For hydroxylamine and hydrazine, ΔGf° values of −23.4 and 127.8 kJ/mol, respectively, were used. These values were calculated from standard reduction potentials (16).
RESULTS
Hydrazine and hydroxylamine evolution in time.
The results of the batch experiments in which hydroxylamine was added to the anammox cells in the absence of nitrate and nitrite are shown in Fig. 1A. In all hydroxylamine addition tests, little hydrazine accumulated (maximum concentration, ∼5 to 10 μM) as long as hydroxylamine was still present. When hydroxylamine was nearly completely converted, the hydrazine concentration rose within 5 min (up to 10-fold more than the starting concentration) and then gradually decreased. Ammonium production was significant in all experiments until the appearance of the hydrazine peak. Thereafter, no significant ammonium production could be detected. In the negative control (in which no biomass was present), a hydroxylamine conversion rate of only 0.1 mM/h was estimated, and no hydrazine could be detected at all. Thus, the rate of “chemical” hydroxylamine conversion was too low to explain the observed behavior, indicating that the process was biological in nature.
FIG. 1.

Measured evolutions of different nitrogen compounds after hydroxylamine addition to anammox enrichments in small (100-ml) bottles (A) and a large (15-liter) reactor (B, C). Hydroxylamine addition leads first to ammonium production and a slight hydrazine accumulation. A transient hydrazine peak appears upon the near completion of hydroxylamine conversion. The experimental conditions were as follows: (A) 100-ml bottle, 3.7 g DW/liter biomass, and 4 mM initial hydroxylamine concentration added at 0 h; (B, C) 15-liter cultivation reactor after a stop in the feed (t = 0) and hydroxylamine added after 5.7 h. Symbols for panels A and B: ♦, NH4+; □, NH2OH; ○, NO3−; ___, pH; ▴, N2H4. Symbols for panel C: •, NO(L); ✻, NO(G); ▴, N2O. a.u., arbitrary units; subscripts (L) and (G), liquid phase and gas phase, respectively.
From the experiments with various biomass and hydroxylamine concentrations, it can be seen that larger amounts of the biomass in the experiment resulted in a shorter time before the hydrazine peak appeared (Table 1) and the more rapidly hydroxylamine was converted. Higher initial concentrations of hydroxylamine resulted in a later appearance of the hydrazine peak with the same amount of biomass (Table 1 and Fig. 2).
TABLE 1.
Experimental conditions and main results of the hydroxylamine addition batch tests
| Experiment | Initial NH2OH concn (mM) | Time to N2H4 peak (h) | Height of N2H4 peak (μM) | NH4+ produced at N2H4 peak (mM) | −ΔNH4+/ΔNH2OH (mol/mol) | DW (g/liter) | NH2OH conversion rate (mmol/g DW/h) | N2H4 disappearance rate after N2H4 peak (mmol/g DW/h) |
|---|---|---|---|---|---|---|---|---|
| Different initial NH2OH concns | 0.5 | 0.32 | 15 | NDd | ND | 5a | 0.31 | 0.0086 |
| 1 | 0.47 | 26 | ND | ND | 5a | 0.42 | 0.0070 | |
| 2 | 0.43 | 62 | 0.26 | 0.23 | 5.1 | 0.91 | 0.010 | |
| 2 | 0.75 | 57 | 0.39 | 0.20 | 4.8 | 0.71 | 0.017 | |
| 4 | 1.5 | 53 | 1.3 | 0.31 | 5.2 | 0.51 | 0.010 | |
| 4b | 1.9 | 69 | 1.9 | 0.48 | 3.7 | 0.43 | 0.017 | |
| 6 | 2.8 | 61 | 1.1 | 0.19 | 4.5 | 0.47 | 0.024 | |
| 10 | 5.9 | 98 | 2.8 | 0.28 | 3.0 | 0.57 | 0.024 | |
| Different biomass concns | 4c | 0.8 | 82 | 0.9 | 0.22 | 7.8 | 0.66 | 0.012 |
| 4 | 4.3 | 59 | 1.0 | 0.27 | 2.1 | 0.41 | 0.006 | |
| Acetate addition (2 mM) | 4 | 3.5 | 62 | 1.0 | 0.25 | ND | n.a.f | n.a. |
| Different NH4+ concns | ||||||||
| 6 mM | 4 | 3.1 | 96 | ND | ND | ND | n.a. | n.a. |
| 9 mMe | 6 | 2.2 | 152 | 2.5 | 0.41 | 5.1 | 0.52 | 0.021 |
| “Ca. Brocadia fulgida” enrichment | 4 | 4.0 | 28 | 0.5 | 0.25 | ND | n.a. | n.a. |
| Presence of oxygen | 4 | No peak | No peak | n.a. | ND | ND | n.a. | n.a. |
| Control (no biomass) | 4 | No N2H4 | No N2H4 | n.a. | n.a. | n.a. | n.a.g | n.a. |
Estimate based on the amount of biomass in the reactor and the sample size.
Experimental results were also depicted in Fig. 1A.
Experimental results were used for model fitting and depicted in Fig. 5.
ND, not determined.
This experiment was performed with 8 liters of biomass within the biomass cultivation reactor. Results are also shown in Fig. 1B and C.
n.a., not applicable.
The conversion rate was estimated at 0.1 mM/h (at 4 mM NH2OH).
FIG. 2.

Measured evolution of the hydrazine concentration after the addition of various amounts of hydroxylamine. The initial hydroxylamine concentrations (mM) are denoted in circles above the hydrazine maximum. The higher initial hydroxylamine levels led to a delayed apparition of the hydrazine peak.
N2O and NO production.
Experiments in which NO and N2O levels were measured (Fig. 1C) were performed directly in the 15-liter reactor. The evolution of hydroxylamine, ammonium, and hydrazine in time in this reactor (Fig. 1B) showed similar trends with the experiments in small (100-ml) bottles (Fig. 1A). The N2O level in the liquid rose to 2 μM after the hydroxylamine addition and remained constant until all hydroxylamine was converted. After the conversion of hydroxylamine, the N2O level dropped again. The NO level in the gas phase initially increased to 100 ppm (4 μM; this value corresponds to about 2 μM in the liquid phase at equilibrium) and decreased again after the hydrazine peak had reached its maximum. The slow logarithmic decrease of NO in the gas phase after the hydrazine peak can be explained by the slow gas dilution rate (the headspace had a volume of 7 liters, which is a large amount compared to the gas flow rate of 25 ml/min). Therefore, NO production can be assumed to have already stopped when all hydroxylamine had been converted. NO2 in the gas phase was always below the detection level (<1 ppm or <5% of the measured NO level).
Measurements of nitrite seemed to show an accumulation (up to 34 μM) upon the hydroxylamine addition. This level decreased again after the conversion of hydroxylamine. However, a critical evaluation of the analytical procedure showed that the measured nitrite value can be fully attributed to the interference of nitric oxide on the analytical determination of nitrite. Control measurements with water (containing no nitrite) sparged with 100 ppm NO (therefore, containing 2 μM NO in the liquid) showed already a value of 10 μM nitrite. Although clearly not a measure for nitrite, these measurements are nevertheless useful as a qualitative indication of the NO concentration in the liquid and are therefore shown in Fig. 1C. Corroborated with the NO measurements in the gas phase, these measurements confirm the observation that NO accumulated slightly upon the hydroxylamine addition but disappeared again directly after the full conversion of the hydroxylamine.
Influence of the presence of acetate, formate, and propionate.
It has been shown that anammox bacteria can convert short-chain fatty acids to CO2 as an additional energy source while nitrite is reduced to N2 via ammonium. The produced ammonium is immediately turned over to combine with nitrite to form dinitrogen gas (10). Formate, acetate, or propionate was added together with hydroxylamine in a series of batch tests. However, none of these acids were found to affect the conversion of hydroxylamine (Table 1), indicating that the conversion of NH2OH could neither be further stimulated nor inhibited by these organic acids. The other general trends, such as the ammonium production and the sharp hydrazine peak, were also observed when the organic acids were added.
Influence of ammonium, nitrate, and oxygen.
Varying the ammonium concentration in the batch tests did not affect the overall behavior. The initial concentrations of 2, 6, and 9 mM (the latter not in the 100-ml vessels but directly in the 15-liter reactor) all resulted in hydroxylamine conversion, ammonium production, and sudden hydrazine accumulation (Table 1). As the experiment in the 15-liter reactor, which gave results similar to those from the 100-ml batch tests, was performed in the presence of nitrate, nitrate (at least up to 12 mM) also seemed not to influence the general behavior. Also, in the presence of oxygen, hydroxylamine conversion occurred at a rate very similar to the rate of conversion in the absence of oxygen. Hydrazine production, however, was not observed at all under these conditions (Table 1).
Influence of anammox species.
To determine whether the sudden appearance of hydrazine was a specific effect for the employed enrichment (consisting of “Candidatus Kuenenia stuttgartiensis”), a batch experiment was also performed with an anammox enrichment containing “Candidatus Brocadia fulgida.” A qualitatively similar behavior was observed (the sudden appearance of hydrazine and about 25% ammonium production), but the hydroxylamine conversion was slower and thus the hydrazine peak appeared later than in similar experiments with “Candidatus Kuenenia stuttgartiensis.” Also, the height of the hydrazine peak was lower (Table 1).
DISCUSSION
General trends.
In our experiments, hydroxylamine was converted in the absence of added electron acceptors by anammox bacteria. The control experiments in the absence of a biomass (both in this study as, in the presence of nitrite, in the study by Van de Graaf et al. [34]) did show a very low conversion of hydroxylamine, indicating that the observed conversion was not chemical in nature. When hydroxylamine is converted by anammox bacteria, the main products are ammonium (about 25% of the converted hydroxylamine) and N2. The nitrogen gas was not measured, but it is most likely the main end product because no other gaseous nitrogen compounds (such as N2O, NO, and NO2) were detected in appreciable amounts. The conversion into ammonium and dinitrogen gas indicates that the overall hydroxylamine conversion is a disproportionation,
 |
with a ΔGR0′ of −228 kJ/mol NH2OH. According to this disproportionation, the yield of ammonium from hydroxylamine is 0.33 mol/mol. This value is close to the experimentally determined one of about 0.25 mol/mol.
Although hydrazine accumulation upon hydroxylamine conversion has also been detected by others (5, 21, 22, 34), the hydrazine peak has never appeared as suddenly as in this study. The number of hydrazine samples taken from each experiment in this study is substantially larger than the number taken from the hydroxylamine addition experiments in most other studies. It is therefore possible that the hydrazine accumulation had also occurred in the previously reported experiments just as suddenly as in this study but that the sampling frequency was too low to notice this behavior. The maximum concentrations of hydrazine reported in other studies are markedly higher than in this study (up to 2.8 mM [5] compared with 100 μM in this study). It was not possible, however, to correlate these higher values in hydrazine level to differences in experimental conditions or to a different type of anammox bacteria that was used in these experiments.
The observation that the rate of hydroxylamine conversion in the presence of oxygen was similar to the rate in the absence of oxygen was quite unexpected, since oxygen is inhibiting the conversion of ammonium and nitrite by anammox bacteria. Hydrazine accumulation, however, could not be detected upon (near) hydroxylamine depletion in experiments conducted in the presence of oxygen (Table 1). This indicated that hydrazine was not formed during this experiment or that the formed hydrazine was immediately oxidized.
Influence of initial biomass and initial hydroxylamine concentrations.
The relationship between the (initial) hydroxylamine/biomass ratio and the time until the hydroxylamine peak is linear. Therefore, the kinetics of hydroxylamine consumption fit well with a zero-order reaction rate (Fig. 3). This indicates that in the chosen range of the initial hydroxylamine/biomass ratio, neither hydroxylamine limitation nor inhibition took place. Also, the time to the hydrazine peak is inversely proportional to the initial biomass concentration (Fig. 3), which reflects a first-order dependency of the hydroxylamine conversion rate on the biomass concentration. The initial hydroxylamine conversion rate was 0.6 (±0.2) mmol NH2OH/g DW/h on average. From the linear part of the decreasing slope of the hydrazine peak, a net hydrazine conversion rate of 0.012 (±0.006) mmol N2H4/g DW/h was estimated (see Table 1). This rate is 20 to 80 times lower than the initial hydroxylamine conversion rate. The hydrazine peak height reached a maximum of about 70 to 100 μM (Fig. 2) for an initial hydroxylamine/biomass ratio of 0.4 mmol NH2OH/g DW. Essentially, the behavior of the experiments at higher initial hydroxylamine/biomass ratios were, after lower hydroxylamine biomass ratios were reached during the experiment due to the conversion of hydroxylamine, similar to the experiments started at those lower ratios. Only for those experiments which were started at an initial hydroxylamine/biomass ratio lower than 0.4 mmol NH2OH/g DW was the hydrazine peak significantly lower.
FIG. 3.

Results from batch tests in 100-ml bottles at various initial hydroxylamine and biomass concentrations show that the time from hydroxylamine addition until the time of appearance of the hydrazine peak is proportional to the biomass-specific hydroxylamine concentration (▪). In contrast, the maximum hydrazine concentration in each experiment (▴) is stable at a hydroxylamine/biomass ratio above 0.4 mmol NH2OH/g DW.
Influence of the anammox species.
The behavior of the “Candidatus Kuenenia stuttgartiensis” enrichment employed in this study was qualitatively similar to the behavior of the “Candidatus Brocadia fulgida” enrichment. We cannot exclude that the quantitative differences between the two enrichments (in conversion rate and peak height) could be species specific. It is, however, also possible that the mode of enrichment, level of enrichment, differences in sample preparation, or time delay between sample taking (in Nijmegen, The Netherlands) and performance of the test (in Delft, The Netherlands) 16 h later have led to the measured differences in peak height and hydroxylamine conversion rate.
Influence of other community members.
Because no pure anammox culture is available for experiments at this moment, information regarding the anammox process is generally obtained using enrichment cultures. The possible contribution of other members of the anammox community to the hydroxylamine conversion was not determined in this study. Other possible community members are denitrifiers, nitrifiers, and sulfate-reducing bacteria. However, to our knowledge, no hydrazine accumulation upon hydroxylamine addition by bacteria other than anammox bacteria has been reported. Furthermore, since hydroxylamine conversion and hydrazine production were always observed in anammox cultures (at several levels of enrichment and probably with different community compositions), it is unlikely that the other community members played a significant role in the hydroxylamine conversion.
Source of NO and N2O.
N2 gas was the main oxidation product of hydroxylamine conversion. From the nitrogen mass balance during the experiment in the 15-liter reactor, the amount of hydroxylamine which was converted into nitrous and nitric oxide was estimated to be at maximum 0.01%. However, small amounts of N2O and NO did accumulate during hydroxylamine conversion (Fig. 1C). This could indicate that (i) nitrous oxide and/or nitric oxide were intermediates in the hydroxylamine disproportionation, (ii) they were the end products of side reactions, or (iii) they were produced by other community members than anammox bacteria. The different options are discussed below.
(i) If nitric oxide or nitrous oxide were intermediates in the hydrazine formation reaction (the first reaction; see the mechanism in the next paragraph), this implies the oxidation of hydroxylamine to NO/N2O, followed by reduction to form hydrazine as proposed on the basis of the genome data (31). Since NO is probably an intermediate in the anammox process—more specifically, a substrate in the hydrazine formation, as is suggested based on the genome information (31)—this is indeed a possible mechanism. We are not aware of any indications that N2O can be converted by anammox bacteria, and it is at the moment unlikely that N2O is an intermediate.
(ii) Hydroxylamine can also be converted to NO and N2O by the hydroxylamine oxidoreductase enzyme present in substantial amounts in anammox bacteria (20), and this side reaction could be the source of the NO and/or N2O. Furthermore, nitrous oxide production via one of several potential nitric oxide detoxification enzymes identified from the anammox genome (10) is another possible anammox-related source of nitrous oxide.
(iii) Given the small amounts of N2O and NO produced during the experiment, the possibility that other community members (see the previous paragraph) are producing the nitrogen oxides cannot be excluded. These other bacteria would then start to produce NO/N2O upon hydroxylamine addition. Denitrification by heterotrophs is improbable, as there is no known reaction of denitrification for hydroxylamine. Aerobic ammonium-oxidizing bacteria, present in small quantities in anammox enrichments, are known to produce NO and N2O, especially under anoxic conditions (28), and this could be a source of NO and N2O.
Possible mechanism of hydrazine accumulation.
The sudden accumulation of hydrazine upon the full conversion of hydroxylamine in the batch tests could be caused by: (i) a quick increase in the production of hydrazine and/or (ii) a sharp decrease in its conversion. The measured concentration changes presented in Fig. 1 and 2 show that hydrazine concentrations increased gradually before they abruptly increased. Possibly, at a hydroxylamine concentration of >0.2 mM, hydrazine is continuously turned over—that is, produced and consumed at the same rate in different reactions—with only a very small excess in its production rate. This would also be consistent with the production of 15N ☰ N14 when 15N-labeled hydroxylamine or ammonium is employed in hydroxylamine addition experiments (34).
A sudden rise in the net hydrazine production upon the near completion of hydroxylamine conversion could only be explained by assuming a toxic or inhibitive effect of hydroxylamine on the hydrazine production rate, as postulated by Shimamura et al. (26). This mechanism could potentially explain the hydrazine production upon hydroxylamine addition in the presence of nitrite and ammonium, as when hydrazine is already being produced from ammonium and nitrite, inhibition of the hydrazine oxidation reaction would immediately lead to hydrazine accumulation. Our work (in the absence of nitrite) does not suggest any inhibition by hydroxylamine, because if hydroxylamine were to inhibit hydrazine oxidation, a stronger inhibition at higher concentrations is likely. Therefore, the addition of hydroxylamine would have led to the accumulation of hydrazine immediately after the hydroxylamine addition (when the hydroxylamine concentration was highest). The delayed hydrazine production observed in this study, taking place only upon the nearly complete consumption of the hydroxylamine, could therefore not be explained by hydroxylamine inhibition.
A sudden stop in hydrazine conversion could, however, be explained by a limited amount of electron acceptor in the later stage of the experiments. A possible reduction reaction in the system is the conversion of hydroxylamine to ammonium. When coupled with hydrazine oxidation, the ΔGR0′ of this reaction is −318 kJ/mol NH2OH, and thus, the reaction is thermodynamically favorable. If this hydroxylamine reduction reaction would cease or significantly slow down (e.g., because of the depletion of one of the electron carriers or because of the low affinity for its substrate, hydroxylamine), the oxidation of hydrazine would suddenly stop as well.
Two possible schemes, based on such a sudden stop in hydrazine conversion upon the nearly complete conversion of hydroxylamine, that could explain the observed disproportionation and sudden accumulation of hydrazine are shown in Fig. 4A and B. Hydrazine conversion after the hydrazine peak could run via a generalized (possibly multiple-enzyme) hydrazine disproportionation reaction (Fig. 4A) or could involve the reverse of the hydrazine formation step, thus producing hydroxylamine and ammonium (Fig. 4B). The small amounts of hydroxylamine produced in this way can be converted again to ammonium with the remaining hydrazine (which is converted to N2). Both mechanisms thus result in the same overall reaction (see the first reaction equation below). However, the actual reversibility of the hydrazine-producing reaction is not known, since the enzyme capable of this conversion has not been purified and the plausibility of the reversibility assumption thus cannot be tested.
FIG. 4.

Two reaction mechanisms (A and B) can explain the response of ammonium and hydrazine toward hydroxylamine additions in anammox enrichments. Three regimens can be distinguished in each mechanism. Regimen I (at high hydroxylamine levels) involves a continuous turnover of hydrazine with ammonium and N2 production. Regimen II (at low hydroxylamine levels) is a stop in hydroxylamine reduction, which ceases hydrazine oxidation, leading to sudden hydrazine accumulation. Regimen III (at relatively high hydrazine levels and low hydroxylamine levels) in mechanism A is the disproportionation of hydrazine into ammonium and dinitrogen gas; in mechanism B, regimen III is the slow removal of hydrazine via oxidation to N2 and the reversal of the hydrazine combination reaction. The possible place of NO as an additional intermediate in hydrazine formation, and the resulting electron flows, are shown with dotted lines.
In both schemes in Fig. 4, the following three metabolic regimens can be distinguished from the evolution of the concentrations of nitrogen compounds in the hydroxylamine addition batch experiments.
(i) For regimen I, the hydroxylamine combination with ammonium to form hydrazine is the rate-limiting step. Hydrazine oxidation to N2 is coupled with hydroxylamine reduction to ammonium. In this regimen, the hydroxylamine concentration falls and ammonium increases, while the hydrazine concentration remains at very low levels.
(ii) For regimen II, the ammonium production is strongly slowed down when hydroxylamine is nearly depleted. Therefore, hydrazine oxidation (which is coupled with hydroxylamine reduction) is stopped, and as a result, hydrazine accumulates.
(iii) For regimen III, the hydrazine concentration falls due to its removal via two possible mechanisms: direct disproportionation into ammonium and N2 (Fig. 4A) or disproportionation into ammonium and hydroxylamine, the latter further reacting to ammonium while the remaining hydrazine is converted to N2 (Fig. 4B).
These reaction schemes involving NH2OH do not exclude NO participation in the hydroxylamine disproportionation or in the anammox metabolism, as both alternatives (Fig. 4A and B) could possibly be extended to have NO as an additional intermediate (as is shown in Fig. 4). The hydroxylamine combination with ammonium to form hydrazine is still the overall reaction in these schemes, but hydroxylamine is first converted to nitric oxide. Nitric oxide (instead of hydroxylamine) subsequently combines with ammonium to form hydrazine. Since the overall reaction is the same, the electron balance also remains unaffected. The three-electron oxidation of hydroxylamine to NO is balanced by the three-electron reduction from the combination of NO and ammonium to form hydrazine.
Mathematical model.
A mathematical model was constructed to test the hypothetical metabolic schemes proposed for the hydroxylamine degradation. The model could describe the sudden hydrazine accumulation and its subsequent consumption. The reaction equations were based on alternatives A and B and were constructed in such a way that in all reactions the electron and element balances were satisfied. Ammonium (NH4+) was considered in all reactions instead of ammonia (NH3), because at the working pH (7.5), this is the dominant species, at least in the extracellular solution. The pH effects were not taken into account in this model. The notations and parameters used in the model equations are presented in Table 2.
TABLE 2.
Parameters for the kinetic model of the hydroxylamine disproportionation in the experiment shown in Fig. 5
| Parameter | Description | Unit | Valuea
|
|
|---|---|---|---|---|
| Model A | Model B | |||
| k1 | Rate constant of hydrazine formationb | mmol/g DW/h | 0.25 | 0.29 |
| k2 | Rate constant of ammonificationb | mmol/g DW/h | 0.30 | 0.31 |
| k3A | Rate constant for hydrazine disproportionationb | mmol/g DW/h | 0.024 | n.a.d |
| k3B | Rate constant for reversed hydrazine formationb | mmol/g DW/h | 0.080 | |
| K1,NH2OH | Half-saturation constant for hydroxylamine conversion with ammonium into hydrazineb | mM | 0.0011 | 0.010 |
| K2,N2H4 | Half-saturation constant for hydrazine oxidation to N2b | mM | 0.00063 | 0.00050 |
| K2,NH2OH | Half-saturation constant for hydroxylamine reduction to ammoniumb | mM | 0.14 | 0.20 |
| K3A,N2H4 | Half-saturation constant for hydrazineb | mM | 0.044 | n.a. |
| K3B,N2H4 | Half-saturation constant for hydrazineb | mM | n.a. | 0.0061 |
| Cx | Biomass concnc | g DW/liter | 7.8 | 7.8 |
| C0,N2H4 | Initial hydrazine concnc | mM | 0.0001 | 0.0001 |
| C0,NH4 | Initial ammonium concnc | mM | 2.0 | 2.0 |
| C0,NH2OH | Initial hydroxylamine concnc | mM | 4.1 | 4.1 |
Model A involved a separate hydrazine disproportionation reaction, whereas with model B, the hydrazine formation reaction was considered to be reversible. The two reaction schemes are presented in Fig. 4.
Parameters were fitted to the measured nitrogen species concentrations.
Initial conditions were based on measured values.
n.a., not applicable.
The first reaction is the hydrazine-forming step from ammonium and hydroxylamine,
 |
with a ΔGR0′ of −47 kJ/mol NH2OH and rate r1 as follows:
 |
The rate dependency of the ammonium concentration was neglected because (i) the measured ammonium levels (2 to 3 mM) are much higher than the half-saturation coefficients (Km) for ammonium in bacteria (which are generally in the 10 to 100 μM range) and (ii) batch experiments did not indicate sensitivity of the reaction rate to the ammonium concentration (Table 1).
In a second reaction, hydrazine oxidation to N2 is coupled with hydroxylamine reduction to NH4+ as
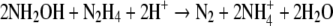 |
with a ΔGR0′ of −318 kJ/mol NH2OH and rate r2 as follows:
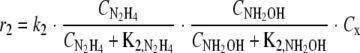 |
The application of the combination of these first two reactions in a mathematical model is capable of reproducing the hydroxylamine consumption and the swift hydrazine accumulation (results not shown). However, for the hydrazine consumption when all hydroxylamine is depleted (regimen III), a separate reaction has to be introduced.
Hydrazine disappears in this third reaction, which can be either hydrazine disproportionation (Fig. 4A) or the reverse of reaction 1 with the production of ammonium and hydroxylamine (Fig. 4B). In the case of the direct disproportionation of hydrazine, reaction 3A is employed as
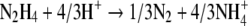 |
with a ΔGR0′ of −180 kJ/mol N2H4 and rate r3A as follows:
 |
In the second alternative (reaction 3B), the reaction is the reverse of reaction 1, that is,
 |
with a ΔGR0′ of +47 kJ/mol N2H4 and rate r3B as follows:
 |
The ΔGR0′ for reaction 3B is close enough to zero to make the assumption of reversibility plausible, since at the actual concentrations, after the hydrazine peak, of ammonium (∼10−3 M), hydroxylamine (∼10−5 M), and hydrazine (∼10−4 M), the ΔGR′ is much closer to zero (∼20 kJ/mol).
The kinetic model both in reaction 3A as well as in reaction 3B satisfactorily could explain the experimentally determined evolution of hydrazine concentration over time (Fig. 5). The model parameters (i.e., the maximum rate constants and half-saturation coefficients) were experimentally determined from the batch experiment with 7.8 g DW/liter biomass and 4.1 mM hydroxylamine. All the parameters for this kinetic model are shown in Table 2.
FIG. 5.

The experimental results of a batch experiment with a hydroxylamine addition (□, hydroxylamine; ▴, hydrazine; ⧫, ammonium) are fitted with the kinetic models A (---) and B (___). The models are both capable of describing the observed phenomena quantitatively. The experimental conditions (NH2OH, 4.1 mM; biomass, 7.8 g DW/liter) and all other parameters were taken from Table 2. N2 is represented as a cumulative production of mmol N2 per mol liquid volume.
The kinetic model with parameters thus obtained in one batch experiment can be used to predict the effect of various biomass and initial hydroxylamine levels in the other experiments. However, the model is not capable of predicting exactly the time at which the hydrazine peak occurs. The differences between the model and the experiments can be fully attributed to variances in the measured biomass-specific hydroxylamine reduction rate. Because the overall reaction of the model is the disproportionation of hydroxylamine in ammonium and N2, the final ammonium production is fixed at one-third of the converted hydroxylamine. Since the measured production is actually about 25% lower, this shows a systematic error in the prediction of the ammonium level.
In the described mathematical model, the affinity for hydroxylamine is much lower than the affinity for hydrazine (that is, 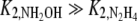 ). A lower difference in affinities in the model will result in a more gradual accumulation of hydrazine (because the hydroxylamine reduction and thus the hydrazine oxidation are slowed down earlier), which would not be in agreement with the observed behavior. In anammox bacteria, the enzyme responsible for ammonification is unknown, but based on genomic (31) and enzymological (10, 20) information, pentaheme nitrite reductase (NrfA), hybrid cluster protein (HCP), and the cd1 nitrite reductase (NirS) are candidates for this conversion. The reported half-saturation constants of these enzymes, purified from bacteria other than anammox bacteria, with hydroxylamine as a substrate are high, ranging from 0.6 mM to 30 mM (1, 2, 27). The values reported in these studies are more than two orders of magnitude higher than the Km value of these enzymes for nitrite (which is in the μM range). The model requirement of a low affinity for hydroxylamine in the ammonification reaction, therefore, seems to be also plausible based on the nature of the enzyme(s) performing this conversion.
). A lower difference in affinities in the model will result in a more gradual accumulation of hydrazine (because the hydroxylamine reduction and thus the hydrazine oxidation are slowed down earlier), which would not be in agreement with the observed behavior. In anammox bacteria, the enzyme responsible for ammonification is unknown, but based on genomic (31) and enzymological (10, 20) information, pentaheme nitrite reductase (NrfA), hybrid cluster protein (HCP), and the cd1 nitrite reductase (NirS) are candidates for this conversion. The reported half-saturation constants of these enzymes, purified from bacteria other than anammox bacteria, with hydroxylamine as a substrate are high, ranging from 0.6 mM to 30 mM (1, 2, 27). The values reported in these studies are more than two orders of magnitude higher than the Km value of these enzymes for nitrite (which is in the μM range). The model requirement of a low affinity for hydroxylamine in the ammonification reaction, therefore, seems to be also plausible based on the nature of the enzyme(s) performing this conversion.
Implications of the mechanism for the “normal” anammox metabolism.
The production of hydrazine in the presence of excess hydroxylamine was originally considered a strong indication that hydrazine and hydroxylamine were both intermediates in the anammox process (34). However, recent evidence points toward the reduction of nitrite to nitric oxide (not hydroxylamine) as the fist step in anammox metabolism, suggesting that nitric oxide and hydrazine are the intermediates in the anammox process (24, 31). The subsequent conversion of nitric oxide and ammonium to hydrazine involves the coupling of two nitrogen atoms, in combination with a three-electron reduction reaction. If hydroxylamine were an additional intermediate, there would be a separation of the three-electron reduction (NO to hydroxylamine) from the formation of the N-N bond (combination of hydroxylamine and ammonium to hydrazine). If the combination of hydroxylamine and ammonium via the mechanism that was proposed in this study takes place directly (and not via NO [see Fig. 4]), the possibility exists that also under physiological conditions hydroxylamine is an (additional) intermediate in the anammox process. However, the direct measurement of hydroxylamine during the conversion of ammonium and nitrite is required to validate this hypothesis.
Conclusions.
Hydroxylamine was disproportionated by anammox enrichments into ammonium and N2. Hydrazine accumulated slightly during the conversion of hydroxylamine but rose sharply when nearly all of the hydroxylamine had been consumed. NO and N2O were also produced in small amounts. It was suggested that hydrazine was an intermediate in the hydroxylamine disproportionation. In this hypothesis, the sudden accumulation of hydrazine was caused by the sudden stop in the conversion of hydrazine, while the production still continued. Although the experiments were mostly performed with one anammox species (“Candidatus Kuenenia stuttgartiensis”), comparable results in a test with “Candidatus Brocadia fulgida” indicate that the described phenomenon is a general anammox characteristic. Two simple kinetic models were capable of explaining the observed behavior. Both models predict a very low hydroxylamine affinity for the hydroxylamine reduction to ammonium, compared to the conversion of hydroxylamine with ammonium to form hydrazine.
Acknowledgments
Caspar Jonker is thanked for conducting part of the experiments. Marc Strous, J. Gijs Kuenen, and Marlies J. Kampschreur are gratefully acknowledged for support and discussions.
This research was sponsored by Technology Foundation STW (project number 05987).
Footnotes
Published ahead of print on 30 May 2008.
REFERENCES
- 1.Bamford, V. A., H. C. Angove, H. E. Seward, A. J. Thomson, J. A. Cole, J. N. Butt, A. M. Hemmings, and D. J. Richardson. 2002. Structure and spectroscopy of the periplasmic cytochrome c nitrite reductase from Escherichia coli. Biochemistry 41:2921-2931. [DOI] [PubMed] [Google Scholar]
- 2.Cabello, P., C. Pino, M. F. Olmo-Mira, F. Castillo, M. D. Roldan, and C. Moreno-Vivian. 2004. Hydroxylamine assimilation by Rhodobacter capsulatus E1F1. Requirement of the hcp gene (hybrid cluster protein) located in the nitrate assimilation nas gene region for hydroxylamine reduction. J. Biol. Chem. 279:45485-45494. [DOI] [PubMed] [Google Scholar]
- 3.Cirpus, I. E. Y., M. De Been, H. J. M. Op den Camp, M. Strous, D. Le Paslier, G. J. Kuenen, and M. S. M. Jetten. 2005. A new soluble 10 kDa monoheme cytochrome c-552 from the anammox bacterium Candidatus “Kuenenia stuttgartiensis.” FEMS Microbiol. Lett. 252:273-278. [DOI] [PubMed] [Google Scholar]
- 4.Dalsgaard, T., D. E. Canfield, J. Petersen, B. Thamdrup, and J. Acuna-Gonzalez. 2003. N2 production by the anammox reaction in the anoxic water column of Golfo Dulce, Costa Rica. Nature (London) 422:606-608. [DOI] [PubMed] [Google Scholar]
- 5.Egli, K., U. Fanger, P. J. J. Alvarez, H. Siegrist, J. R. Van der Meer, and A. J. B. Zehnder. 2001. Enrichment and characterization of an anammox bacterium from a rotating biological contactor treating ammonium-rich leachate. Arch. Microbiol. 175:198-207. [DOI] [PubMed] [Google Scholar]
- 6.Frear, D. S., and R. C. Burrell. 1955. Spectrophotometric method for determining hydroxylamine reductase activity in higher plants. Anal. Chem. 27:1664-1665. [Google Scholar]
- 7.Huston, W. M., H. R. Harhangi, A. P. Leech, C. S. Butler, M. S. M. Jetten, H. J. M. Op den Camp, and J. W. B. Moir. 2007. Expression and characterisation of a major c-type cytochrome encoded by gene kustc0563 from Kuenenia stuttgartiensis as a recombinant protein in Escherichia coli. Protein Expr. Purif. 51:28-33. [DOI] [PubMed] [Google Scholar]
- 8.Jardin, N., A. Hippen, C. F. Seyfried, K.-H. Rosenwinkel, and F. Greulich. 2001. Deammonification of sludge liquor at wastewater treatment plant Hattingen with moving bed system. GWF, Wasser/Abwasser 142:479-484. [Google Scholar]
- 9.Jetten, M., M. Schmid, K. Van de Pas-Schoonen, J. Sinninghe Damsté, and M. Strous. 2005. Anammox organisms: enrichment, cultivation, and environmental analysis. Methods Enzymol. 397:34-57. [DOI] [PubMed] [Google Scholar]
- 10.Kartal, B., M. M. M. Kuypers, G. Lavik, J. Schalk, H. J. M. Op den Camp, M. S. M. Jetten, and M. Strous. 2007. Anammox bacteria disguised as denitrifiers: nitrate reduction to dinitrogen gas via nitrite and ammonium. Environ. Microbiol. 9:635-642. [DOI] [PubMed] [Google Scholar]
- 11.Kartal, B., J. Rattray, L. Van Niftrik, J. L. Van de Vossenberg, M. C. Schmid, J. A. Fuerst, J. Sinninghe Damsté, M. S. M. Jetten, and M. Strous. 2007. Candidatus “Anammoxoglobus propionicus” gen. nov., sp. nov., a new propionate oxidizing species of anaerobic ammonium oxidizing bacteria. Syst. Appl. Microbiol. 30:39-49. [DOI] [PubMed] [Google Scholar]
- 12.Kartal, B., L. van Niftrik, J. Rattray, J. L. C. M. van de Vossenberg, M. C. Schmid, J. Sinninghe Damsté, M. S. M. Jetten, and M. Strous. 2008. Candidatus ‘Brocadia fulgida’: an autofluorescent anaerobic ammonium oxidizing bacterium. FEMS Microbiol. Ecol. 63:46-55. [DOI] [PubMed] [Google Scholar]
- 13.Kartal, B., L. A. Van Niftrik, A. O. Sliekers, M. Schmid, I. Schmidt, K. Van de Pas-Schoonen, I. Cirpus, W. R. L. Van der Star, M. C. M. Van Loosdrecht, W. Abma, J. G. Kuenen, J. W. Mulder, M. S. M. Jetten, H. Op den Camp, M. Strous, and J. Van de Vossenberg. 2004. Application, eco-physiology and biodiversity of anaerobic ammonium-oxidizing bacteria. Rev. Environ. Sci. Bio/Technol. 3:255-264. [Google Scholar]
- 14.Kuypers, M. M. M., G. Lavik, D. Woebken, M. Schmid, B. M. Fuchs, R. Amann, B. B. Jorgensen, and M. S. M. Jetten. 2005. Massive nitrogen loss from the Benguela upwelling system through anaerobic ammonium oxidation. Proc. Natl. Acad. Sci. USA 102:6478-6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuypers, M. M. M., A. O. Sliekers, G. Lavik, M. Schmid, B. B. Jorgensen, J. G. Kuenen, J. S. Sinninghe Damsté, M. Strous, and M. S. M. Jetten. 2003. Anaerobic ammonium oxidation by anammox bacteria in the Black Sea. Nature (London) 422:608-611. [DOI] [PubMed] [Google Scholar]
- 16.Latimer, W. 1952. The oxidation states of the elements and their potentials in aqueous solutions, 2nd ed. Prentice Hall, New York, NY.
- 17.Lide, D. R. (ed.) 2006. CRC handbook of chemistry and physics, 87th ed., p. 5.85-5.88. CRC Press, Boca Raton, FL.
- 18.Pynaert, K., B. F. Smets, S. Wyffels, D. Beheydt, S. D. Siciliano, and W. Verstraete. 2003. Characterization of an autotrophic nitrogen-removing biofilm from a highly loaded lab-scale rotating biological contactor. Appl. Environ. Microbiol. 69:3626-3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rysgaard, S., R. N. Glud, N. Risgaard-Petersen, and T. Dalsgaard. 2004. Denitrification and anammox activity in Arctic marine sediments. Limnol. Oceanogr. 49:1493-1502. [Google Scholar]
- 20.Schalk, J., S. De Vries, J. G. Kuenen, and M. S. M. Jetten. 2000. Involvement of a novel hydroxylamine oxidoreductase in anaerobic ammonium oxidation. Biochemistry 39:5405-5412. [DOI] [PubMed] [Google Scholar]
- 21.Schalk, J., H. Oustad, J. G. Kuenen, and M. S. M. Jetten. 1998. The anaerobic oxidation of hydrazine: a novel reaction in microbial nitrogen metabolism. FEMS Microbiol. Lett. 158:61-67. [DOI] [PubMed] [Google Scholar]
- 22.Schmid, M., K. Walsh, R. Webb, W. Irene, C. Rijpstra, K. van de Pas-Schoonen, M. J. Verbruggen, T. Hill, B. Moffett, J. Fuerst, S. Schouten, J. S. Sinninghe Damsté, J. Harris, P. Shaw, M. Jetten, and M. Strous. 2003. Candidatus “Scalindua brodae,” sp. nov., Candidatus “Scalindua wagneri,” sp. nov., two new species of anaerobic ammonium oxidizing bacteria. Syst. Appl. Microbiol. 26:529-538. [DOI] [PubMed] [Google Scholar]
- 23.Schmid, M. C., N. Risgaard-Petersen, J. van de Vossenberg, M. M. M. Kuypers, G. Lavik, J. Petersen, S. Hulth, B. Thamdrup, D. Canfield, T. Dalsgaard, S. Rysgaard, M. K. Sejr, M. Strous, H. J. M. Op den Camp, and M. S. M. Jetten. 2007. Anaerobic ammonium-oxidizing bacteria in marine environments: widespread occurrence but low diversity. Environ. Microbiol. 9:1476-1484. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt, I., C. Hermelink, K. Van de Pas-Schoonen, M. Strous, H. J. op den Camp, J. G. Kuenen, and M. S. M. Jetten. 2002. Anaerobic ammonia oxidation in the presence of nitrogen oxides (NOx) by two different lithotrophs. Appl. Environ. Microbiol. 68:5351-5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schubert, C. J., E. Durisch-Kaiser, B. Wehrli, B. Thamdrup, P. Lam, and M. M. M. Kuypers. 2006. Anaerobic ammonium oxidation in a tropical freshwater system (Lake Tanganyika). Environ. Microbiol. 8:1857-1863. [DOI] [PubMed] [Google Scholar]
- 26.Shimamura, M., T. Nishiyama, H. Shigetomo, T. Toyomoto, Y. Kawahara, K. Furukawa, and T. Fujii. 2007. Isolation of a multiheme protein with features of a hydrazine-oxidizing enzyme from an anaerobic ammonium-oxidizing enrichment culture. Appl. Environ. Microbiol. 73:1065-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh, J. 1974. Cytochrome oxidase from Pseudomonas aeruginosa. III. Reduction of hydroxylamine. Biochim. Biophys. Acta 333:28-36. [DOI] [PubMed] [Google Scholar]
- 28.Sliekers, A. O., N. Derwort, J. L. C. Gomez, M. Strous, J. G. Kuenen, and M. S. M. Jetten. 2002. Completely autotrophic nitrogen removal over nitrite in one single reactor. Water Res. 36:2475-2482. [DOI] [PubMed] [Google Scholar]
- 29.Strous, M., J. J. Heijnen, J. G. Kuenen, and M. S. M. Jetten. 1998. The sequencing batch reactor as a powerful tool for the study of slowly growing anaerobic ammonium-oxidizing microorganisms. Appl. Microbiol. Biotechnol. 50:589-596. [Google Scholar]
- 30.Strous, M., J. G. Kuenen, and M. S. M. Jetten. 1999. Key physiology of anaerobic ammonium oxidation. Appl. Environ. Microbiol. 65:3248-3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strous, M., E. Pelletier, S. Mangenot, T. Rattei, A. Lehner, M. W. Taylor, M. Horn, H. Daims, D. Bartol-Mavel, P. Wincker, V. Barbe, N. Fonknechten, D. Vallenet, B. Segurens, C. Schenowitz-Truong, C. Medigue, A. Collingro, B. Snel, B. E. Dutilh, H. J. M. Op den Camp, C. Van der Drift, I. Cirpus, K. T. Van de Pas-Schoonen, H. R. Harhangi, L. Van Niftrik, M. Schmid, J. Keltjens, J. Van de Vossenberg, B. Kartal, H. Meier, D. Frishman, M. A. Huynen, H.-W. Mewes, J. Weissenbach, M. S. M. Jetten, M. Wagner, and D. Le Paslier. 2006. Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature (London) 440:790-794. [DOI] [PubMed] [Google Scholar]
- 32.Strous, M., E. Van Gerven, P. Zheng, J. G. Kuenen, and M. S. M. Jetten. 1997. Ammonium removal from concentrated waste streams with the anaerobic ammonium oxidation (anammox) process in different reactor configurations. Water Res. 31:1955-1962. [Google Scholar]
- 33.Van de Graaf, A. A., P. De Bruijn, L. A. Robertson, M. S. M. Jetten, and J. G. Kuenen. 1996. Autotrophic growth of anaerobic ammonium-oxidizing microorganisms in a fluidized bed reactor. Microbiology 142:2187-2196. [Google Scholar]
- 34.Van de Graaf, A. A., P. De Bruijn, L. A. Robertson, M. S. M. Jetten, and J. G. Kuenen. 1997. Metabolic pathway of anaerobic ammonium oxidation on the basis of 15N studies in a fluidized bed reactor. Microbiology 143:2415-2421. [DOI] [PubMed] [Google Scholar]
- 35.Van der Star, W. R. L., W. R. Abma, D. Blommers, J. W. Mulder, T. Tokutomi, M. Strous, C. Picioreanu, and M. C. M. Van Loosdrecht. 2007. Startup of reactors for anoxic ammonium oxidation: experiences from the first full-scale anammox reactor in Rotterdam. Water Res. 41:4149-4163. [DOI] [PubMed] [Google Scholar]
- 36.Van Dongen, L. G. J. M., M. S. M. Jetten, and M. C. M. Van Loosdrecht. 2001. The combined SHARON/anammox process. IWA Publishing, London, United Kingdom.
- 37.Van Dongen, U., M. S. M. Jetten, and M. C. M. Van Loosdrecht. 2001. The SHARON-Anammox process for treatment of ammonium rich wastewater. Water Sci. Technol. 44(1):153-160. [PubMed] [Google Scholar]
- 38.Watt, G. W., and J. D. Chrisp. 1952. A spectrophotometric method for the determination of hydrazine. Anal. Chem. 24:2006-2008. [Google Scholar]
- 39.Wett, B. 2006. Solved upscaling problems for implementing deammonification of rejection water. Water Sci. Technol. 53(12):121-128. [DOI] [PubMed] [Google Scholar]
- 40.Zheng, P., X. Feng, M. S. M. Jetten, and J. G. Kuenen. 1997. Study on substrate conversion characteristics of mixed microbial culture for anaerobic ammonium oxidation. Zhejiang Nongye Daxue Xuebao 23:409-413. [Google Scholar]