

Abstract
Clostridium perfringens enterotoxin (CPE) causes the symptoms of a very common food poisoning. To assess whether CPE-induced cytotoxicity is necessary for enterotoxicity, a rabbit ileal loop model was used to compare the in vivo effects of native CPE or recombinant CPE (rCPE), both of which are cytotoxic, with those of the noncytotoxic rCPE variants rCPE D48A and rCPE168-319. Both CPE and rCPE elicited significant fluid accumulation in rabbit ileal loops, along with severe mucosal damage that starts at villus tips and then progressively affects the entire villus, including necrosis of epithelium and lamina propria, villus blunting and fusion, and transmural edema and hemorrhage. Similar treatment of ileal loops with either of the noncytotoxic rCPE variants produced no visible histologic damage or fluid transport changes. Immunohistochemistry revealed strong CPE or rCPE168-319 binding to villus tips, which correlated with the abundant presence of claudin-4, a known CPE receptor, in this villus region. These results support (i) cytotoxicity being necessary for CPE-induced enterotoxicity, (ii) the CPE sensitivity of villus tips being at least partially attributable to the abundant presence of receptors in this villus region, and (iii) claudin-4 being an important intestinal receptor for CPE. Finally, rCPE168-319 was able to partially inhibit CPE-induced histologic damage, suggesting that noncytotoxic rCPE variants might be useful for protecting against some intestinal effects of CPE.
Clostridium perfringens enterotoxin (CPE) is produced by ∼1 to 5% of all isolates of the gram-positive spore-forming anaerobic pathogen C. perfringens (25). CPE, a single 35-kDa polypeptide of 319 amino acids (6), is responsible for the gastrointestinal symptoms of C. perfringens type A food poisoning (25, 39), which is the second most common food-borne illness in the United States and the United Kingdom (36). In addition, CPE-producing C. perfringens isolates are increasingly recognized as a cause of antibiotic-associated or sporadic diarrhea (2, 3, 27).
Studies of CPE effects in experimental animals identified the ileum as the most sensitive region of the small intestine (31, 41). CPE treatment inhibits ileal fluid and electrolyte absorption (30, 31). However, with time, CPE-treated ileum develops gross fluid/electrolyte secretion and histologic damage that includes desquamation of the ileal epithelium, prominent villus blunting, and enterocyte plasma membrane blebbing (32, 33, 41). Recently, similar histopathologic changes were observed following ex vivo CPE treatment of human ileal tissues, along with increased transepithelial resistance and water permeability (9).
In vitro cell culture models have provided considerable information about the cellular and molecular actions of CPE (12, 13, 24, 26, 28, 34, 43). This enterotoxin first binds to certain members of the claudin family of tight junction proteins (19, 20, 29), which results in formation of an sodium dodecyl sulfate (SDS)-sensitive “small” complex of ∼90 kDa (48) that contains CPE and one or more claudins (38). At physiological temperatures, several small complexes interact to promote CPE oligomerization in a prepore state on the plasma membrane surface (45). This oligomerization results in formation of two very large, SDS-resistant CPE complexes, recently named CH-1 and CH-2, each containing a CPE hexamer and one or more claudins (38). In addition, the CH-2 complex contains occludin, another tight junction protein (38, 43, 49). The CH-1 (and possibly CH-2) prepore then inserts into the plasma membrane to form a functional pore that causes a strong Ca2+ influx (4, 5). The resultant elevated cytoplasmic Ca2+ levels then trigger host cell death by apoptosis or oncosis (4, 5).
Cell culture models have also proven valuable for studying the CPE structure-function relationship. By using these models, Vero cell and Caco-2 cell receptor binding activity has been mapped to the extreme C terminus of the enterotoxin. For example, a synthetic peptide corresponding to the 30 C-terminal amino acids was shown to exhibit strong competitive binding activity against native CPE. In addition, deleting even the five C-terminal amino acids from native CPE was found to completely ablate toxin binding and cytotoxic activities (14-17, 22). However, despite possessing the ability to bind to CPE receptors, the C-terminal half of CPE itself cannot kill cultured cells, which indicates that sequences in the N-terminal half of CPE are required for cytotoxicity (17, 18). Deletion and site-directed mutagenesis approaches have identified amino acids G47 to I51 in the N terminus of CPE as being important for CPE oligomerization and cytotoxicity (21, 22, 38, 44).
Despite recent progress in identifying the cellular action of CPE, several important questions remain regarding the in vivo activity of this enterotoxin. For example, CPE binding has not yet been localized within the ileum. Therefore, it remains unknown whether CPE binds uniformly to the villus or instead binds preferentially to specific regions (such as the villus tip or crypts). Localization of CPE binding in vivo has been difficult to assess using native CPE because the toxin can produce histologic damage within 15 min. A second unanswered question about the in vivo action of CPE concerns whether the cytotoxic and histopathologic activities of this toxin are required for the development of intestinal fluid transport alterations. Two observations support a role for cytotoxicity in CPE-induced enterotoxicity, i.e., in fluid and electrolyte alterations. First, only CPE doses capable of causing histologic damage are able to induce intestinal fluid secretion in rabbit ileal loops. Second, histologic damage develops concurrently with fluid transport alterations in CPE-treated rabbit ileal loops (31, 41). However, some recent studies (5, 19, 46) have opened the possibility of histologic damage not being necessary for CPE-induced intestinal fluid transport alterations. Those studies showed that receptor binding-proficient, but noncytoxic, C-terminal CPE fragments can remove proteins from tight junctions and affect transepithelial resistance in MDCK cells. Moreover, in the rat ileum, those C-terminal CPE fragments do not cause histologic damage but can alter transepithelial resistance and enhance intestinal dextran absorption. Collectively, those findings could indicate that CPE receptor occupancy is sufficient to induce increased paracellular permeability.
Therefore, our study tested, for the first time, the enterotoxic effects of two binding-capable, but noncytotoxic, CPE variants. These studies permitted localization of CPE binding in the ileum and also allowed us to test the relationship among CPE-induced cytotoxicity, intestinal histologic damage, and fluid secretion.
MATERIALS AND METHODS
Preparation of CPE or rCPE variants.
Native CPE was purified from C. perfringens strain NCTC8239 as described previously (35). Recombinant Escherichia coli strains producing recombinant CPE (rCPE) (full-length CPE with a N-terminal His6 tag), rCPE168-319 (amino acids 168 to 319 of native CPE with an N-terminal His6 tag), or rCPE D48A (rCPE with an aspartic acid-to-alanine substitution at CPE residue 48) (Fig. 1) were prepared using metal affinity chromatography, as previously described (44). Toxin was then aliquoted and stored at −80°C until use. All rCPE species were stable and did not show proteolytic degradation during enrichment, processing, or storage (not shown). Toxin yield was estimated by quantitative Western blotting as described previously (44).
FIG. 1.

rCPE constructs. (A) Native CPE was purified from C. perfringens as described in Materials and Methods. The N-terminal cytotoxicity core sequence (dark gray) needed for oligomerization and large complex formation is located between amino acids 47 and 51 (43), the putative transmembrane stem domain (light gray) lies between amino acids 81 and 106 (44), and the C-terminal receptor binding domain is shown in black. rCPE, the rCPE D48A variant, and rCPE168-319 were affinity enriched from E. coli lysates expressing these pTrcHis A-based constructs. rCPE is fully cytotoxic and is identical to CPE except for the addition of a vector-encoded 42-amino-acid N-terminal His6 tag (22). The rCPE D48A variant was constructed previously (43) and is comprised of rCPE with a single D-to-A point mutation at amino acid 48 that abrogates oligomerization and, thus, cytotoxicity. The deletion mutant rCPE168-319 (22) contains only amino acids 168 to 319 of native CPE and, like the rCPE D48A variant, is noncytotoxic but capable of receptor binding. (B) Western blot analysis of affinity-enriched 50-ng samples of CPE or each rCPE species. Migration of molecular mass markers run along with samples is identified with arrows to left of blot.
Cytotoxic and complex-forming phenotypes of rCPE and rCPE variants in Caco-2 cells.
The previously reported (22, 43) cytotoxic and complex-forming phenotypes of rCPE or the two rCPE variants in Caco-2 cells were confirmed prior to their in vivo use in the current study. Caco-2 cells were routinely cultured as described elsewhere (44), and confluent Caco-2 cell monolayers were washed once with Hanks' balanced salt solution (HBSS) containing Ca2+ and Mg2+ (Mediatech, Manassas, VA) before treatment with 2.5 μg/ml of toxin dissolved in a total of 2 ml of HBSS containing Ca2+ and Mg2+. After 1 h of incubation, cultures were photomicrographed using a Canon Powershot G5 digital camera attached to a Zeiss Axiovert 25 inverted microscope at a magnification of ×10.
The ability of CPE, rCPE, and the two rCPE variants to form the CH-1 and CH-2 complexes was assessed by treating isolated Caco-2 cells for 20 min at 37°C with 2.5 μg/ml of each CPE species in HBSS without Ca2+ and Mg2+ (Mediatech). The treated cells were centrifuged, washed with HBSS, and then lysed by resuspension in Laemmli buffer. Samples of these lysates were then separated on a 4% acrylamide SDS-polyacrylamide gel, electrotransferred to a nitrocellulose membrane, and Western blotted using rabbit polyclonal CPE antiserum. Horseradish peroxidase-conjugated goat anti-rabbit polyclonal antibody (Sigma) and Supersignal West Pico chemiluminescent substrate (Pierce) were used to visualize anti-CPE immunoreactivity.
Rabbit ileal loop assay.
Young adult, male or female, New Zealand White rabbits (Charles River, CA) were used to prepare ileal loops as previously described (40). One milliliter of Ringer's solution containing 50, 100, or 300 μg of CPE or an rCPE species was injected into each ileal loop of each experimental animal; additional loops (controls) received a similar volume of sterile Ringer's solution. The abdominal cavity was closed and the rabbits maintained under anesthesia for 6 h.
In another experiment, ileal loops were inoculated with 2 mg of rCPE168-319 diluted in Ringer's solution and incubated for 1 h; at that time 100 μg of CPE was injected and the loops were incubated for another 1-h period. rCPE168-319 was replaced by an equimolar solution of bovine serum albumin (BSA) in control loops for this experiment.
After the specified incubation period, the rabbits were euthanized with an overdose of sodium barbiturate (Beuthanasia; Schering-Plough Animal Health, Kenilworth, NJ), the abdominal cavity was reopened, and the small intestinal loops were excised sequentially in the same order that they had been inoculated. Fluid accumulation was measured by its weight in grams as a quantitative measurement of fluid accumulation, and loop length was recorded in centimeters. For each loop, a fluid weight/length ratio was calculated.
After gross analysis of each lesion, a thin slice of each loop was immediately embedded in an OCT freezing compound (Tissue Tech, St. Laurent, Quebec, Canada) and frozen at −80°C until processed for immunohistochemistry (see below). The remaining portion of each loop was fixed in 10% buffered pH 7.4 formalin for 24 to 48 h and then dehydrated through graded alcohol solutions to xylene and embedded in paraffin wax. Four-micrometer-thick sections were cut and stained conventionally with hematoxylin and eosin. Intestinal damage was then assessed microscopically by a blinded pathologist on a scale of 1 to 5 (with 5 being the most severe). All experimental procedures were approved by the Animal Care and Use Committee of the University of California, Davis (permit 11594).
Immunolocalization of CPE on rabbit intestinal epithelium.
Frozen or formalin-fixed tissue sections (see above) were processed by indirect immunoperoxidase techniques for the presence of CPE or rCPE species, as well as for E-cadherin and claudin-4. These techniques were performed using the Dako EnVision kit (Dako Corporation, Carpinteria, CA), according to the manufacturer's instructions. Briefly, 4-μm-thick sections were cut and mounted on glass slides. The endogenous peroxidase activity was then quenched by incubation in 3% hydrogen peroxide. Formalin-fixed tissue sections but not frozen sections were incubated with 10% protease for 10 min for antigen retrieval. Normal horse serum was applied to the sections for 20 min to block nonspecific binding, and the corresponding primary antiserum was applied before incubation for 40 min. After a washing cycle, the sections were incubated with horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G antibodies for 30 min, washed again, and incubated with nora red (Vector Laboratories, Burlingame, CA) for 5 min. Finally, the sections were dehydrated and counterstained with hematoxylin, and a coverslip was added. All incubations were carried out at 37°C.
Primary antiserum consisted of mouse monoclonal anti-CPE (monoclonal antibody 3C9) (50), anti-E-cadherin (Transduction Laboratories, Lexington, KY), or anti-claudin-4 (Zymed Laboratories, Carlsbad, CA) antibodies. Normal mouse serum was used to replace the primary antiserum in negative control slides. Before use in immunolocalization methods, each antibody was titrated by testing on positive control tissues. A rabbit ileal loop incubated for 30 min with purified CPE was used as a positive control for CPE immunohistochemistry. A loop of normal (untreated) ileum from the same rabbit was also used as a positive control for E-cadherin and claudin-4 localization. The sections were observed blindly by a pathologist under a conventional light microscope. A total of 50 intestinal villi per treatment were counted and classified as positive or negative for each immunohistochemical technique.
Statistical analyses.
Ileal loop fluid accumulation differences were evaluated by the Student t test.
RESULTS
In vitro phenotyping of rCPE species.
To test the relationships between the cytotoxic, histopathologic, and enterotoxic effects of CPE, we used affinity-enriched preparations of rCPE and two rCPE variants that had been generated in previous studies. One variant was an rCPE fragment containing only the C-terminal half of CPE (amino acids 168 to 319) and the second an rCPE D48A point variant (Fig. 1A). These two rCPE variants were previously reported to possess full CPE-like receptor binding activity but to lack cytotoxicity (at doses of >50 μg/ml) because they cannot form CH-1 or CH-2 (22, 44). Since the affinity-enriched rCPE D48A variant, rCPE168-319, and rCPE preparations contained some contaminating E. coli proteins in addition to their rCPE constructs, a mock enrichment was also performed with lysates from E. coli transformed with the pTrcHis A empty vector; this material (referred to as negative control enrichments) was used throughout the study as a negative control.
Consistent with previous reports (22, 38, 44), our current study confirmed that rCPE is fully cytotoxic for Caco-2 cells, where it forms both CH-1 and CH-2. As also expected from those previous studies, rCPE168-319 and rCPE D48A did not cause cytotoxic effects or form CH-1 or CH-2 complexes in Caco-2 cells, although both variants were capable of binding to those cultured cells.
In vivo phenotypes of CPE and rCPE species.
To evaluate whether cytotoxicity is required for CPE-induced histologic damage and fluid transport alterations, the in vivo phenotypes of our two binding-proficient (but noncytotoxic) rCPE variants, i.e., rCPE168-319 and the rCPE D48A variant, were assayed using the rabbit ileal loop assay.
Rabbit ileal loops incubated with either native CPE or rCPE for 6 h showed considerable fluid accumulation (Fig. 2), with both toxins producing a fluid accumulation (g/cm) ratio of ∼1.0 at 50, 100, or 300 μg (no statistically significant differences were observed in fluid accumulation between these doses). The ileal loops incubated with either CPE or rCPE were grossly dark red and flaccid. Histologically, they showed severe diffuse necrotizing enteritis, characterized by a complete loss of absorptive epithelial cells along the villi, and coagulation necrosis of the lamina propria, where large amounts of karyorrhectic debris were present (Fig. 3). The villi in these loops had almost completely disappeared and were replaced by a series of low denuded bumps that were occasionally lined by a pseudomembrane composed of fibrin, red blood cells, neutrophils, lymphocytes, plasma cells, necrotic epithelial cells, and cell debris (Fig. 3). Occasionally a few preserved crypts were observed, although most of the crypt epithelium was also necrotic and sloughed off. The submucosa showed severe edema, and the blood and lymphatic vessels were engorged with red blood cells and neutrophils. Fibrin thrombi were frequently observed in submucosal veins. No significant histologic differences were observed between loops incubated with CPE versus rCPE.
FIG. 2.

Fluid accumulation in rabbit ileal loops. Ligated ileal loops were constructed in New Zealand White rabbits as described in Materials and Methods. Experimental loops were then injected with 300 μg of the indicated toxin preparation and incubated for 6 h after reintroduction of the loops into the abdominal cavity. At the conclusion of the experiment, the loop fluid content was measured and the amount of accumulated fluid was related inversely to the length of each loop (g/cm). White bars for each toxin preparation represent the averages of duplicate ileal loop samples in a single rabbit, while black bars represent the combined average results for three rabbits. Error bars represent the standard deviations from the means. Statistical significance of <0.005 between a construct and rCPE is indicated by **, whereas * similarly indicates statistical significance of <0.05.
FIG. 3.
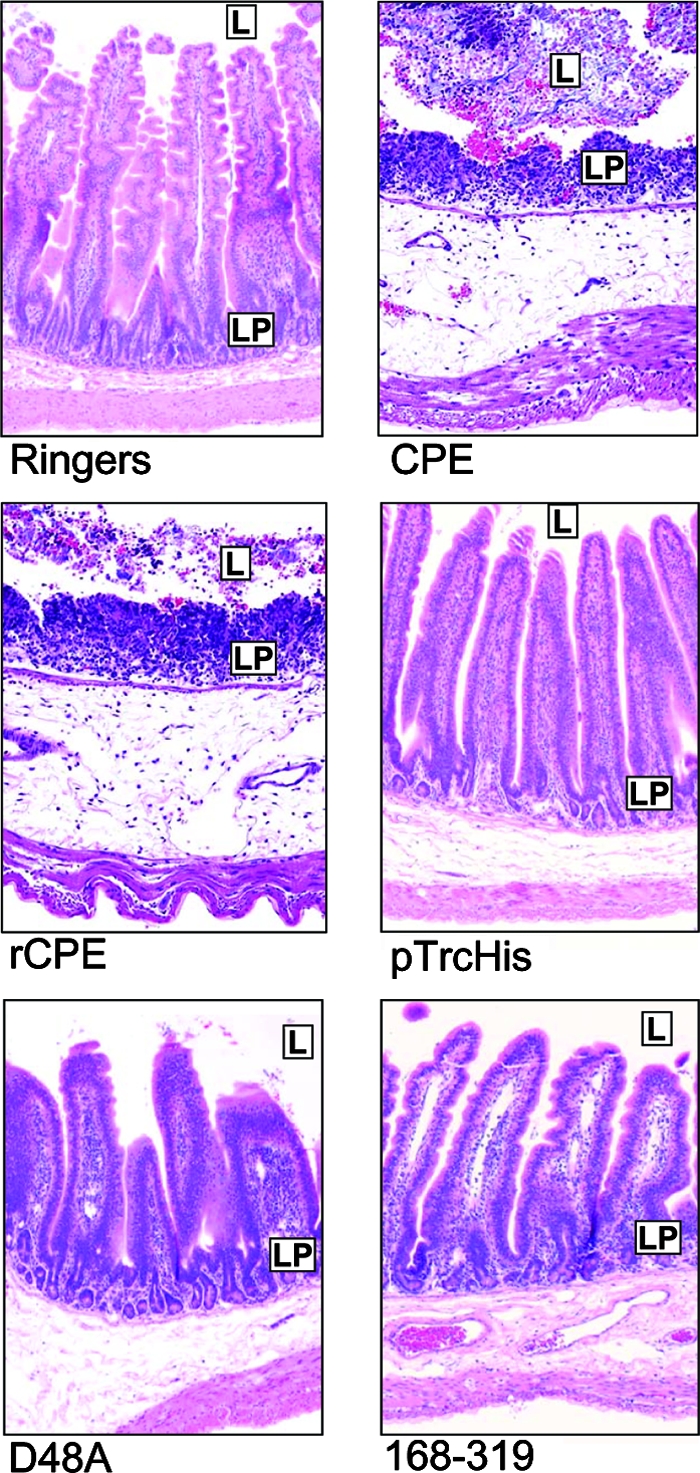
Histopathology induced by toxin preparations. After the 6-h fluid accumulation experiments, rabbit ileal loops selected for histologic examination were formalin fixed and embedded in paraffin. Hematoxylin and eosin was used to stain 4-μm-thick sections of intestinal tissue treated with the indicated construct. In each panel, L represents the intestinal lumen, while LP identifies the lamina propria. The preparation used to treat each loop is noted at the bottom of each panel. Final magnification of each photomicrograph, ×400.
In striking contrast, rabbit ileal loops treated for 6 h with up to 300 μg of the noncytotoxic rCPE168-319 or rCPE D48A variant exhibited the same low level of fluid accumulation as loops treated with Ringer's solution only or negative control enrichments (Fig. 2). In addition, no histologic damage was apparent in loops treated with the two noncytotoxic rCPE variants, as the histologic sections taken from these loops resembled sections taken from loops treated with Ringer's solution or the negative control enrichment (Fig. 3).
Assessment of the ability of noncytoxic rCPE variants to ameliorate CPE- or rCPE-induced histologic damage.
The inability of binding-capable noncytotoxic rCPE168-319 or rCPE D48A variants to induce intestinal effects in ileal loops suggested that these rCPE variants might be capable of acting as in vivo competitors of CPE or rCPE action. To begin testing this hypothesis, ileal loops were incubated for 1 h with Ringer's solution containing rCPE168-319 or an equivalent molarity of BSA before CPE was inoculated into those same loops. The severity of histologic changes in CPE loops pretreated with rCPE168-319 was scored as less (P, 0.07) than that observed for loops pretreated with BSA or loops treated with CPE alone (Fig. 4). Necrosis and desquamation of epithelium in loops pretreated with rCPE168-319 were significantly reduced (Fig. 4B), while epithelial necrosis at the tips of the villi was evident in loops pretreated with BSA or loops treated with CPE alone (Fig. 4A).
FIG. 4.
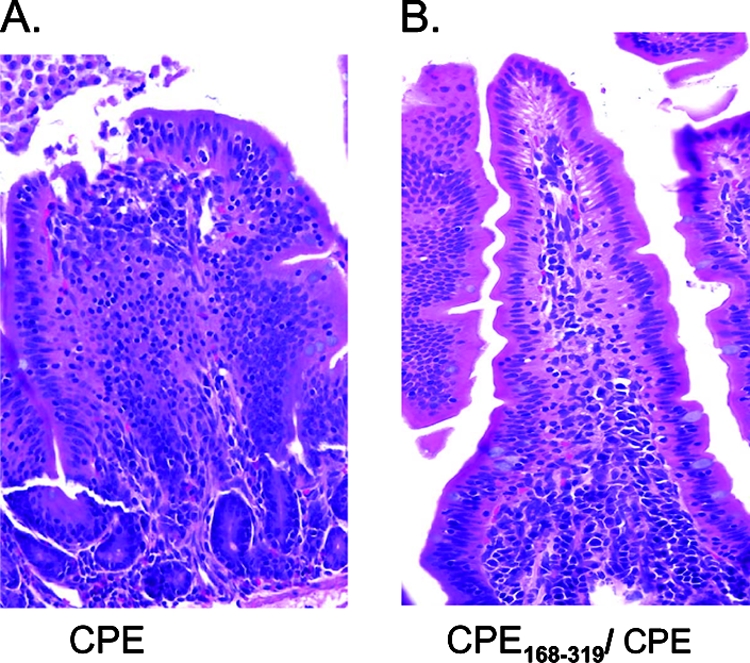
Protection of rabbit ileal loops against CPE effects by preincubation with rCPE168-319. After fluid accumulation was measured, rabbit ileal loops pretreated with rCPE168-319 or BSA were formalin fixed and embedded in paraffin. Hematoxylin and eosin was used to stain 4-μm-thick sections of intestinal tissue. The preparation(s) used to treat each loop is noted at the bottom of each panel. Final magnification of each photomicrograph, ×400.
Immunolocalization of CPE or rCPE variants in rabbit ileal loops.
As mentioned in the introduction, it has not yet been assessed whether CPE binds uniformly along the villus or to specific regions such as villus tips or crypts. Immunolocalization of CPE binding to the intestinal epithelium could address this issue but has not yet been attempted, in large part because of the rapid and extensive epithelial desquamation induced by the native enterotoxin. This contention was confirmed by poor-quality immunolocalization results obtained (data not shown) in a trial experiment where rabbit intestinal loops were treated with CPE or rCPE for 6 h, where there was severe blunting of villi (Fig. 3). Some very weak CPE staining of the few remaining crypt-localized enterocytes was apparent, but most immunostaining in these sections was of dead and detached cells that had sloughed into the intestinal lumen.
The current study therefore used two different approaches to immunolocalize CPE binding to less damaged, or undamaged, ileum. First, CPE immunostaining was performed using rabbit ileal loops treated with native CPE for only 15 min, a time point where much less histologic damage has developed compared to that at 6 h of CPE treatment (Fig. 3 and 5). In those 15-min CPE treatment loops (Fig. 5A), enterocytes extruded from the villus tips displayed heavy CPE immunostaining along all cell surfaces (Fig. 5A), and occasionally a thin brown line of positive CPE immunostaining was observed on the apical membranes of enterocytes located at the tips or upper third of the villi. This immunostaining pattern was observed in more than 95% of the villi examined. After a 15-min CPE treatment, no CPE immunostaining was detected in the crypts. No immunostaining was present when sections were incubated with Ringer's solution alone (Fig. 5B), verifying that the immunostaining in Fig. 5A was due to CPE treatment.
FIG. 5.

Immunolocalization of CPE or rCPE168-319. Rabbit ileal loops were treated for 15 min with 300 μg of native CPE (A), for 6 h with Ringer's solution (B), for 6 h with 300 μg of rCPE168-319 (C), or with equivalent volumes of negative control enrichments (D). Each loop was then processed for CPE immunolocalization as described in Materials and Methods.
To observe CPE immunostaining in the complete absence of any histologic damage, the CPE immunolocalization experiments were repeated using the noncytotoxic rCPE168-319 or rCPE D48A variants. When these noncytotoxic variants were incubated for 6 h in rabbit ileal loops (Fig. 5C and data not shown), both the rCPE168-319 and the rCPE D48A variants were localized predominantly to the upper part of the villi rather than to the crypts in more than 95% of the villi examined. Confirming that this immunostaining was due to bound rCPE species, no immunostaining was visible in loops treated for even 6 h with negative control enrichments (Fig. 5D). This immunolocalization of rCPE168-319 and rCPE D48A to the upper part of the villi also provided in vivo verification that these variants can bind to enterocytes despite lacking the ability to kill those cells.
Immunodetection of epithelial cell junctional proteins in rabbit ileal loops.
Since the results presented in Fig. 5 indicated that CPE binds preferentially to upper parts of the villi, we sought to determine whether this preference might be attributable to a greater abundance of CPE claudin receptors in this region. To address this question, intestinal tissues from treated loops were immunostained for the known CPE receptor claudin-4. Claudin-4 immunostaining of rabbit ileal loops treated with Ringer's solution for 15 min revealed very heavy staining on both the apical and basolateral surfaces of enterocytes at the upper parts of the villi (Fig. 6A) but only negligible staining in the villus crypts. This observation contrasted with results from immunostaining similar sections for E-cadherin, a protein found in the adherens junction of epithelial cells. E-cadherin immunostaining of Ringer's solution-treated rabbit ileal loops produced a uniform distribution of positive staining along the basolateral surfaces of enterocytes in both the villus tips and crypts (Fig. 5B). Treatment for 6 h with rCPE168-319 or rCPE D48A did not noticeably affect the villus distribution of either claudin-4 or E-cadherin (data not shown).
FIG. 6.
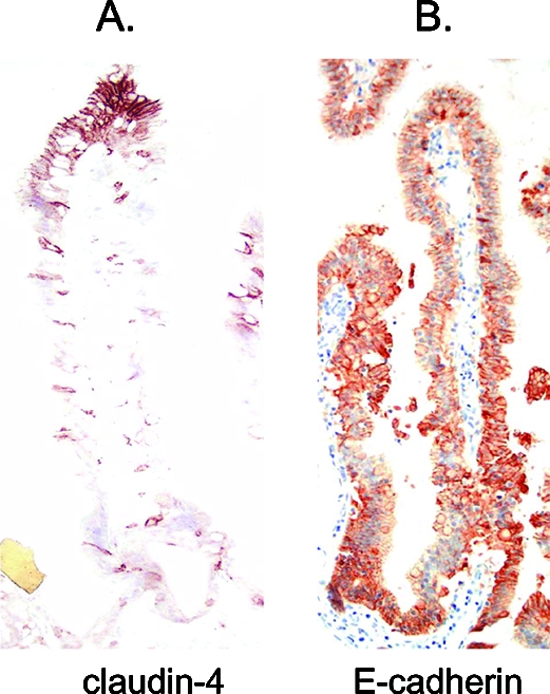
Immunolocalization of claudin-4 and E-cadherin. The presence of claudin-4 (A) or E-cadherin (B) on untreated villi in rabbit ileal loops was demonstrated by immunolocalization, as described in Materials and Methods.
DISCUSSION
Several older studies (31, 41) have suggested a possible linkage between CPE-induced cytotoxicity, intestinal histologic damage, and the onset of intestinal fluid transport alterations. However, more recent studies (19, 23, 46) showed that 4 to 6 h of treatment with noncytotoxic, but binding-capable, C-terminal CPE fragments can induce claudin internalization and alter transepithelial resistance in cultured cells. In addition, those C-terminal CPE fragments were unable to induce histologic damage in the ileum, but they did alter transepithelial resistance and dextran absorption in ileal loops. Those observations could have indicated that C-terminal CPE fragments can, independently of cytotoxicity or histologic effects, induce paracellular permeability changes that contribute to intestinal fluid transport changes. Therefore, our first goal was to test whether C-terminal CPE fragments cause ileal fluid transport changes, as this could elucidate whether CPE-induced cytotoxicity and histologic damage are required for ileal loop fluid accumulation.
Therefore, we compared the in vivo effects of rCPE (or native CPE) with those of two rCPE variants that are binding capable yet noncytotoxic in vitro (22, 44). As expected from previous studies (7, 33), pathophysiologically relevant doses of native CPE and rCPE (which are cytotoxic) caused both intestinal fluid accumulation (Fig. 2) and histologic damage (Fig. 3) in rabbit ileal loops. However, neither of those effects was observed when ileal loops were treated with pathophysiologically relevant doses (up to 100 μg/ml) of the rCPE168-319 or rCPE D48A variant, even though both those rCPE variants bound to the ileal epithelium (Fig. 4 and data not shown). Even artificially high doses (300 μg) of rCPE168-319 or rCPE D48A variants did not produce histologic changes or fluid accumulation in the ileum. These findings support cytotoxic activity as being important for the development of both the histologic and intestinal fluid transport effects of CPE in this model.
While some other cell culture models have also been used to study CPE action, the enterocyte-like Caco-2 cell line has recently become the standard in vitro model for analyzing both the CPE structure-function relationship and the cellular action of CPE (38, 42, 43, 45). By demonstrating that the cytotoxic phenotype of the rCPE168-319 or rCPE D48A variant for Caco-2 cells correlates very well with the histopathologic and fluid transport-altering properties of these rCPE variants in the rabbit ileum, our current findings provide important validation for the use of Caco-2 cells for in vitro modeling of CPE action.
Structure-function analyses have identified three major functional regions of the CPE protein: an N-terminal cytotoxicity core sequence extending from residue G47 to I51 (22, 44), a putative transmembrane stem domain including residues V81 to I106 (44), and a C-terminal binding domain consisting of residues S290 to F319 (14-17, 22). Our current results demonstrate that rCPE168-319 and rCPE D48A variants bind to the intestinal epithelium, as evident from their positive immunostaining with anti-CPE antibodies. This result is consistent with previous characterization of similar C-terminal CPE fragments in vitro (16-18, 22, 46, 47) and in vivo (8, 23). The inability of the oligomerization-deficient rCPE168-319 and rCPE D48A variants to cause enterotoxicity (Fig. 2 and 3) supports CPE oligomerization as being essential for CPE-induced enteric effects as well as for in vitro cytotoxicity.
Since CPE168-319 does not cause fluid accumulation or histologic damage yet still binds to the intestinal epithelium, this suggested that pretreating ileal loops with this variant might interfere with the subsequent development of CPE intestinal activity. As an initial proof of principle, our pilot experiment showed that rCPE168-319 pretreatment can inhibit the initial development of histologic damage in loops treated with CPE. Follow-up time course studies should be performed to assess whether rCPE168-319 pretreatment can afford longer-term intestinal protection against CPE-induced histologic damage and fluid accumulation (which does not occur within the 1-h time period of our initial experiment, which used a 50-μg CPE dose [data not shown]). Should those additional studies demonstrate substantial intestinal protection, future efforts might explore CPE168-319 as a potential CPE therapeutic.
In a prior electron microscopy study, CPE enterocyte damage and heavy cellular blebbing were found predominantly at villus tips of CPE-treated rabbit ileum (32). In addition, histologic analysis (33, 41; this study) showed that initial CPE-induced intestinal damage begins at villus tips in ileal loops. Our immunolocalization results now provide an explanation for this phenomenon; i.e., there is more CPE binding to villus tips than elsewhere on intestinal villi (Fig. 3). In part, this appears to be attributable to greater CPE receptor density in villus tips, as suggested by the claudin-4 immunolocalization results obtained during the current study (Fig. 6). The correlation between claudin-4 density and CPE binding at villus tips also suggests that this claudin, which is a known CPE receptor (19, 20, 38), plays an important role in intestinal CPE binding. However, since certain other claudins can also bind CPE in vitro (10), this correlation does not preclude the possibility that other claudins also contribute to CPE binding in vivo. This issue is currently under investigation by our laboratory.
The greater CPE sensitivity of villus tips may also involve at least two additional factors beyond the presence of higher receptor levels in this intestinal region. First, it has recently been established that senescent enterocytes in villus tips are expelled from the epithelium by an apoptotic extrusion process that exposes basolateral junctional proteins in adjacent cells (1, 11, 37). This effect could be important for CPE targeting of villus tips, since we have shown that in polarized cultures of enterocyte-like Caco-2 cells, (i) there are more claudin CPE receptors present on basolateral versus apical surfaces of those cells, and (ii) this polarized receptor distribution results in Caco-2 cells being much more sensitive to basolateral than to apical CPE challenge (41). Notably, Fig. 6 also demonstrates an abundant presence of claudin-4 on the basolateral surface of enterocytes at villus tips. Thus, the extrusion of cells in the villus tips could expose the abundant basolateral CPE receptors on adjacent enterocytes and render those cells highly CPE sensitive. It is interesting to note in this regard that other bacterial pathogens also exploit the apoptotic extrusion process at villus tips to penetrate the intestinal epithelium, with the most notable example being the binding of Listeria monocytogenes to E-cadherin exposed by removal of senescent enterocytes at villus tips (37).
A second possible contributing factor to the CPE sensitivity of villus tips is that enterocytes in the villus tip are in the process of becoming apoptotic (1, 11, 37). Since the Ca2+ influx caused by CPE pore formation can trigger an apoptotic cell death response (4, 5), preapoptotic enterocytes at the villus tip may be “primed” for cell death such that even low doses of, or brief exposure to, CPE quickly triggers cell death, further contributing to the high CPE sensitivity of this villus region.
Since CPE has been shown to cause, ex vivo, similar damage to the human ileum and rabbit ileum (9), our new results suggest the following model for CPE enterotoxicity. Once produced in the small intestine during sporulation, CPE first binds to certain claudin receptors at the villus tips because cells in that region have more abundant and probably more exposed (due to normal cellular extrusion processes) enterotoxin receptors. This CPE binding results in small complex formation, followed by formation of hexameric CH-1 pores on the enterocyte surface. These prepore CH-1 complexes then penetrate the plasma membrane to form a pore that permits a Ca2+ influx to drive death of villus tip cells, which are already primed for apoptosis. The resultant death of villus tip enterocytes exposes the basolateral membranes of adjacent enterocytes, allowing the process to repeat such that epithelial desquamation at the villus tips develops and impairs intestinal absorption. As the process continues, intestinal villi become increasingly blunted and denuded, causing net fluid secretion in the small intestine. These effects then manifest clinically as diarrhea and abdominal cramps. Additional investigations are currently under way to further test this model.
Acknowledgments
This work was generously supported by Public Health Service grant R37-AI019844 from the National Institute of Allergy and Infectious Diseases.
We thank Jim Carvota for the anesthesia of the experimental rabbits.
Editor: S. R. Blanke
Footnotes
Published ahead of print on 27 May 2008.
REFERENCES
- 1.Bojarski, C., J. Weiske, T. Schoneberg, W. Schroder, J. Mankertz, J. D. Schulzke, P. Florian, M. Fromm, R. Tauber, and O. Huber. 2004. The specific fates of tight junction proteins in apoptotic epithelial cells. J. Cell Sci. 1172097-2107. [DOI] [PubMed] [Google Scholar]
- 2.Borriello, S. P., H. E. Larson, A. R. Welch, F. Barclay, M. F. Stringer, and B. A. Bartholomew. 1984. Enterotoxigenic Clostridium perfringens: a possible cause of antibiotic-associated diarrhoea. Lancet i305-307. [DOI] [PubMed] [Google Scholar]
- 3.Carman, R. J. 1997. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev. Med. Microiol. 8S43-S45. [Google Scholar]
- 4.Chakrabarti, G., and B. A. McClane. 2005. The importance of calcium influx, calpain and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell. Microbiol. 7129-146. [DOI] [PubMed] [Google Scholar]
- 5.Chakrabarti, G., X. Zhou, and B. A. McClane. 2003. Death pathways activated in CaCo-2 cells by Clostridium perfringens enterotoxin. Infect. Immun. 714260-4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czeczulin, J. R., P. C. Hanna, and B. A. McClane. 1993. Cloning, nucleotide sequencing, and expression of the Clostridium perfringens enterotoxin gene in Escherichia coli. Infect. Immun. 613429-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duncan, C. L., H. Sugiyama, and D. H. Strong. 1968. Rabbit ileal loop response to strains of Clostridium perfringens. J. Bacteriol. 951560-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebihara, C., M. Kondoh, M. Harada, M. Fujii, H. Mizuguchi, S. Tsunoda, Y. Horiguchi, K. Yagi, and Y. Watanabe. 2007. Role of Tyr306 in the C-terminal fragment of Clostridium perfringens enterotoxin for modulation of tight junction. Biochem. Pharmacol. 73824-830. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez Miyakawa, M. E., V. Pistone Creydt, F. A. Uzal, B. A. McClane, and C. Ibarra. 2005. Clostridium perfringens enterotoxin damages the human intestine in vitro. Infect. Immun. 738407-8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujita, K., J. Katahira, Y. Horiguchi, N. Sonoda, M. Furuse, and S. Tsukita. 2000. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett. 476258-261. [DOI] [PubMed] [Google Scholar]
- 11.Gitter, A. H., K. Bendfeldt, J. D. Schulzke, and M. Fromm. 2000. Leaks in the epithelial barrier caused by spontaneous and TNF-alpha-induced single-cell apoptosis. FASEB J. 141749-1753. [DOI] [PubMed] [Google Scholar]
- 12.Giugliano, L. G., M. F. Stringer, and B. S. Drasar. 1983. Detection of Clostridium perfringens enterotoxin by tissue culture and double-gel diffusion methods. J. Med. Microbiol. 16233-237. [DOI] [PubMed] [Google Scholar]
- 13.Gyobi, Y., H. Kodama. 1978. Vero cell microcell culture technique for the detection of neutralization antibody against Clostridium perfringens enterotoxin and its application to sero-epidemiology. J. Food Hyg. Soc. Jap. 19294-298. [Google Scholar]
- 14.Hanna, P. C., and B. A. McClane. 1991. A recombinant C-terminal toxin fragment provides evidence that membrane insertion is important for Clostridium perfringens enterotoxin cytotoxicity. Mol. Microbiol. 5225-230. [DOI] [PubMed] [Google Scholar]
- 15.Hanna, P. C., T. A. Mietzner, G. K. Schoolnik, and B. A. McClane. 1991. Localization of the receptor-binding region of Clostridium perfringens enterotoxin utilizing cloned toxin fragments and synthetic peptides. The 30 C-terminal amino acids define a functional binding region. J. Biol. Chem. 26611037-11043. [PubMed] [Google Scholar]
- 16.Hanna, P. C., E. U. Wieckowski, T. A. Mietzner, and B. A. McClane. 1992. Mapping of functional regions of Clostridium perfringens type A enterotoxin. Infect. Immun. 602110-2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanna, P. C., A. P. Wnek, and B. A. McClane. 1989. Molecular cloning of the 3′ half of the Clostridium perfringens enterotoxin gene and demonstration that this region encodes receptor-binding activity. J. Bacteriol. 1716815-6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horiguchi, Y., T. Akai, and G. Sakaguchi. 1987. Isolation and function of a Clostridium perfringens enterotoxin fragment. Infect. Immun. 552912-2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katahira, J., N. Inoue, Y. Horiguchi, M. Matsuda, and N. Sugimoto. 1997. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J. Cell Biol. 1361239-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katahira, J., H. Sugiyama, N. Inoue, Y. Horiguchi, M. Matsuda, and N. Sugimoto. 1997. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J. Biol. Chem. 27226652-26658. [DOI] [PubMed] [Google Scholar]
- 21.Kokai-Kun, J. F., K. Benton, E. U. Wieckowski, and B. A. McClane. 1999. Identification of a Clostridium perfringens enterotoxin region required for large complex formation and cytotoxicity by random mutagenesis. Infect. Immun. 675634-5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kokai-Kun, J. F., and B. A. McClane. 1997. Deletion analysis of the Clostridium perfringens enterotoxin. Infect. Immun. 651014-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masuyama, A., M. Kondoh, H. Seguchi, A. Takahashi, M. Harada, M. Fujii, H. Mizuguchi, Y. Horiguchi, and Y. Watanabe. 2005. Role of N-terminal amino acids in the absorption-enhancing effects of the C-terminal fragment of Clostridium perfringens enterotoxin. J. Pharmacol. Exp. Ther. 314789-795. [DOI] [PubMed] [Google Scholar]
- 24.Matsuda, M., and N. Sugimoto. 1979. Calcium-independent and dependent steps in action of Clostridium perfringens enterotoxin on HeLa and Vero cells. Biochem. Biophys. Res. Commun. 91629-636. [DOI] [PubMed] [Google Scholar]
- 25.McClane, B. A. 2007. Clostridium perfringens, p. 423-444. In M. P. Doyle and L. R. Beuchat (ed.), Food microbiology: fundamentals and frontiers, 3rd ed. ASM Press, Washington, DC.
- 26.McClane, B. A., and J. L. McDonel. 1979. The effects of Clostridium perfringens enterotoxin on morphology, viability, and macromolecular synthesis in Vero cells. J. Cell Physiol. 99191-200. [DOI] [PubMed] [Google Scholar]
- 27.McClane, B. A., F. A. Uzal, M. E. Fernandez-Miyakawa, D. Lyerly, and T. Wilkins. 2006. The enterotoxic clostridia, p. 698-752. In M. Dworkin and S. Falkow (ed.), The prokaryotes: a handbook on the biology of bacteria, 3rd ed. Springer, New York, NY.
- 28.McClane, B. A., A. P. Wnek, K. I. Hulkower, and P. C. Hanna. 1988. Divalent cation involvement in the action of Clostridium perfringens type A enterotoxin. Early events in enterotoxin action are divalent cation-independent. J. Biol. Chem. 2632423-2435. [PubMed] [Google Scholar]
- 29.McDonel, J. L. 1980. Binding of Clostridium perfringens [125I]enterotoxin to rabbit intestinal cells. Biochemistry 194801-4807. [DOI] [PubMed] [Google Scholar]
- 30.McDonel, J. L. 1974. In vivo effects of Clostridium perfringens enteropathogenic factors on the rat ileum. Infect. Immun. 101156-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonel, J. L. 1986. Toxins of Clostridium perfringens (types A, B, C, D, and E), p. 477-506. In F. Dorner and J. Drews (ed.), Pharmacology of bacterial toxins. Pergamon Press, New York, NY.
- 32.McDonel, J. L., L. W. Chang, J. G. Pounds, and C. L. Duncan. 1978. The effects of Clostridium perfringens enterotoxin on rat and rabbit ileum: an electron microscopic study. Lab. Investig. 39210-218. [PubMed] [Google Scholar]
- 33.McDonel, J. L., and C. L. Duncan. 1975. Histopathological effect of Clostridium perfringens enterotoxin in the rabbit ileum. Infect. Immun. 121214-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonel, J. L., and B. A. McClane. 1979. Binding versus biological activity of Clostridium perfringens enterotoxin in Vero cells. Biochem. Biophys. Res. Commun. 87497-504. [DOI] [PubMed] [Google Scholar]
- 35.McDonel, J. L., and B. A. McClane. 1988. Production, purification, and assay of Clostridium perfringens enterotoxin. Methods Enzymol. 16594-103. [DOI] [PubMed] [Google Scholar]
- 36.Olsen, S. J., L. C. MacKinnon, J. S. Goulding, N. H. Bean, and L. Slutsker. 2000. Surveillance for foodborne-disease outbreaks—United States, 1993-1997. MMWR Surveill. Summ. 491-62. [PubMed] [Google Scholar]
- 37.Pentecost, M., G. Otto, J. A. Theriot, and M. R. Amieva. 2006. Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog. 2e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson, S. L., J. G. Smedley III, U. Singh, G. Chakrabarti, C. M. Van Itallie, J. M. Anderson, and B. A. McClane. 2007. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell. Microbiol. 92734-2755. [DOI] [PubMed] [Google Scholar]
- 39.Sarker, M. R., R. J. Carman, and B. A. McClane. 1999. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 33946-958. [DOI] [PubMed] [Google Scholar]
- 40.Sayeed, S., F. A. Uzal, D. J. Fisher, J. Saputo, J. E. Vidal, Y. Chen, P. Gupta, J. I. Rood, and B. A. McClane. 2008. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol. Microbiol. 6715-30. [DOI] [PubMed] [Google Scholar]
- 41.Sherman, S., E. Klein, and B. A. McClane. 1994. Clostridium perfringens type A enterotoxin induces tissue damage and fluid accumulation in rabbit ileum. J. Diarrhoeal Dis. Res. 12200-207. [PubMed] [Google Scholar]
- 42.Singh, U., L. L. Mitic, E. U. Wieckowski, J. M. Anderson, and B. A. McClane. 2001. Comparative biochemical and immunocytochemical studies reveal differences in the effects of Clostridium perfringens enterotoxin on polarized CaCo-2 cells versus Vero cells. J. Biol. Chem. 27633402-33412. [DOI] [PubMed] [Google Scholar]
- 43.Singh, U., C. M. Van Itallie, L. L. Mitic, J. M. Anderson, and B. A. McClane. 2000. CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J. Biol. Chem. 27518407-18417. [DOI] [PubMed] [Google Scholar]
- 44.Smedley, J. G., III, and B. A. McClane. 2004. Fine mapping of the N-terminal cytotoxicity region of Clostridium perfringens enterotoxin by site-directed mutagenesis. Infect. Immun. 726914-6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smedley, J. G., III, F. A. Uzal, and B. A. McClane. 2007. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect. Immun. 752381-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sonoda, N., M. Furuse, H. Sasaki, S. Yonemura, J. Katahira, Y. Horiguchi, and S. Tsukita. 1999. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: evidence for direct involvement of claudins in tight junction barrier. J. Cell Biol. 147195-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi, A., M. Kondoh, A. Masuyama, M. Fujii, H. Mizuguchi, Y. Horiguchi, and Y. Watanabe. 2005. Role of C-terminal regions of the C-terminal fragment of Clostridium perfringens enterotoxin in its interaction with claudin-4. J. Control Release 10856-62. [DOI] [PubMed] [Google Scholar]
- 48.Wieckowski, E. U., A. P. Wnek, and B. A. McClane. 1994. Evidence that an approximately 50-kDa mammalian plasma membrane protein with receptor-like properties mediates the amphiphilicity of specifically bound Clostridium perfringens enterotoxin. J. Biol. Chem. 26910838-10848. [PubMed] [Google Scholar]
- 49.Wnek, A. P., and B. A. McClane. 1989. Preliminary evidence that Clostridium perfringens type A enterotoxin is present in a 160,000-Mr complex in mammalian membranes. Infect. Immun. 57574-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wnek, A. P., R. J. Strouse, and B. A. McClane. 1985. Production and characterization of monoclonal antibodies against Clostridium perfringens type A enterotoxin. Infect. Immun. 50442-448. [DOI] [PMC free article] [PubMed] [Google Scholar]