

Abstract
Under hypoxic conditions or upon exposure to low concentrations of nitric oxide, DevR transcriptional regulator mediates the activation of ∼50 genes that are believed to assist in dormancy development in Mycobacterium tuberculosis. Most of the strongly induced genes are characterized by the presence of one to four copies of a Dev box-like sequence at an upstream location. Among these are several gene pairs that are transcribed in opposite directions. This arrangement could provide for coordinated control of the adjacent genes under inducing conditions. In this work, we report the first detailed analysis of DevR-mediated hypoxic regulation of narK2-Rv1738 genes that are oppositely oriented in M. tuberculosis. Phosphorylated DevR interacts with intergenic sequences and protects ∼80 bp of DNA that contains three binding sites, designated Dev boxes D1, D2, and D3. The hypoxia-specific transcription start points of narK2 and Rv1738 were mapped, and it was noted that the −35 elements of both promoters overlapped with the proximally placed Dev box. DevR bound cooperatively to adjacently placed D2 and D3 boxes while binding to D1 was independent of DevR interaction with the D2 and D3 boxes. Mutational analysis and green fluorescent protein reporter assays established that the three Dev boxes function synergistically to mediate maximal transcriptional induction of both narK2 and Rv1738 in hypoxic cultures of M. tuberculosis. Analysis of narK2 promoter activity indicates that it is under negative regulation in addition to DevR-mediated positive regulation and also reveals differences between M. tuberculosis and Mycobacterium bovis BCG.
A hallmark of tuberculosis is the ability of Mycobacterium tuberculosis to persist in a dormant form in the host, sometimes even for decades, before reactivation to cause active disease. During latent infection, tubercle bacilli reside in granulomas and are likely to be growth restricted due to hypoxia, exposure to nitric oxide, and nutrient limitation leading to their adaptation to a nonreplicating persistent state. The in vitro hypoxia model of dormancy developed by Wayne and Sohasky and variations of it have been widely used to understand the molecular mechanisms underlying the process of bacterial adaptation (7, 9, 12, 15, 22). The DevR transcriptional regulator (Rv3133c, sometimes also called DosR) is believed to play a key role in bacterial adaptation and survival under hypoxic conditions (3, 10, 12, 20). Among the genes induced by DevR are those that are transcribed in opposite directions such as hspX-Rv2032, Rv2627c-Rv2628, Rv3130c-Rv3131, and narK2-Rv1738. The role of DevR interaction in the activation of one such gene pair, namely, narK2-Rv1738, is described in this work. Rv1738 encodes a conserved hypothetical protein of unknown but essential function (14), and narK2 codes for the nitrate/nitrite transporter protein.
Many bacteria use nitrate as the final electron acceptor in place of oxygen to support growth under anaerobic conditions. However, the increase in nitrate reduction in M. tuberculosis under hypoxic conditions is associated not with bacterial growth but with a switch to a state of nonreplicating persistence. Therefore, it was suggested that nitrate reduction may be required for maintaining redox balance or may serve as a temporary means to provide energy during the process of shiftdown (21). In M. tuberculosis, the increase in nitrate reduction under hypoxic conditions is attributed to an increase in nitrate transport through the nitrate/nitrite transporter protein, NarK2 (17). Transcription of narK2 along with that of the divergently transcribed Rv1738 gene is induced under hypoxic conditions (10) from a 286-bp intergenic region that is marked by the presence of four putative Dev boxes (Fig. 1). narK2-Rv1738 gene transcription is also induced after exposure of bacteria to nitric oxide in vitro (20) and during chronic infection of mice (16), which is suggestive of a crucial role for these genes during bacterial adaptation in vivo.
FIG. 1.

Schematic representation of narK2-Rv1738 genes in M. tuberculosis. Open reading frames in narK2-Rv1738 region. Putative Dev boxes (10) are indicated.
Toward understanding the mechanism of DevR-mediated transcription activation, we showed that phosphorylated DevR (DevR∼P) positively autoregulates its expression by binding to two Dev boxes located upstream of the Rv3134c-devRS operon (5). The positioning of these Dev boxes at −42.5 and −63.5 bp resembles the placing of cyclic AMP receptor protein (CRP) binding sites at CRP-regulated class III promoters (4). Here, we attempted to understand the mechanism by which DevR interacts with multiple binding sites (designated as Dev boxes 1, 2, 3, and 4) to regulate activation of narK2-Rv1738 genes in hypoxic cultures of M. tuberculosis. The transcription start points (TSPs) of both narK2 and Rv1738 under hypoxic conditions were mapped to nonoverlapping sites. The work reported here demonstrates that DevR∼P interacts with three out of four putative Dev boxes in the narK2-Rv1738 intergenic region. Binding of DevR to two Dev boxes located upstream of Rv1738 was interdependent but independent of DevR interaction with the third Dev box placed upstream of narK2. Mutational analysis and green fluorescent protein (GFP) reporter assays indicate that the three Dev boxes function synergistically to mediate the transcriptional activation of narK2-Rv1738 genes. To the best of our knowledge, this represents the first report of the regulation of natural divergent nonoverlapping promoters, each of which has class III promoter architecture. A comparative assessment of select DevR-regulated class III promoters suggests that rapid and powerful gene activation is associated with the presence of two or more appropriately positioned interacting Dev boxes in the target promoter. Lastly, deletion analysis reveals that narK2 promoter regulation is significantly different in M. tuberculosis and Mycobacterium bovis BCG.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
M. tuberculosis H37Rv and M. tuberculosis recombinant strains were cultured in Dubos medium containing 0.05% Tween-80 plus 0.5% albumin-0.75% dextrose-0.085% NaCl at 37°C. GFP reporter assays were conducted under aerobic shaking and standing conditions as described previously (5). Briefly, standing cultures were established by dispensing 200-μl culture aliquots in triplicate into 96-well black, clear-bottom microtiter plates (Becton Dickinson, United Kingdom) and incubating the sealed plates at 37°C (plate format). Aerobic promoter activity was measured in cultures that were simultaneously grown in 50-ml tubes (5 ml of culture) with shaking at 220 rpm. For reporter assays under hypoxic conditions, stock cultures of M. tuberculosis frozen at −80°C were aerobically subcultured twice to mid-logarithmic phase (A595 of ∼0.3 to 0.4); the cultures were diluted to an A595 of 0.05, and 3-ml culture aliquots were dispensed in 4-ml Vacutainer tubes (Becton Dickinson) and allowed to stand for various time periods at 37°C (tube format). The gradual decrease in oxygen concentration was monitored through the fading and decolorization of methylene blue, which occurred on day 5 and day 10 of incubation, respectively. At specified time points cultures were withdrawn from dedicated tubes, and GFP fluorescence was measured as described previously (1). Rv3134c, hspX, Rv1738, and narK2 promoter activities were compared in the plate format assay. Escherichia coli strains and culture conditions were as described earlier (1). When required, antibiotics were used at the following concentrations: ampicillin at 100 μg/ml, kanamycin at 50 μg/ml in E. coli and 25 μg/ml in M. tuberculosis, and hygromycin at 50 μg/ml in M. tuberculosis and 200 μg/ml in E. coli. All the plasmids used in this study are listed in Table 1.
TABLE 1.
Plasmids used in this study
| Plasmid | Relevant feature(s)a | Reference or source |
|---|---|---|
| pSC-DevR | pGEX4T1 overexpressing DevR with a glutathione S-transferase N-terminal tag | 1 |
| pDevR D54V | pSC-DevR containing aspartic acid to valine mutation at amino acid residue 54 of DevR | K. Kaur and J. S. Tyagi |
| pFPV27 | E. coli mycobacterial shuttle plasmid with promoterless gfp; Kmr | 19 |
| pnarK2 | pFPV27 containing narK2 promoter (−220 to +57); Hygr | This study |
| p1738 | pFPV27 containing Rv1738 promoter (−203 to +74); Hygr | This study |
| pAmutD1 | pnarK2 containing mutated D1 DevR binding site; Hygr | This study |
| pAmutD2 | pnarK2 containing mutated D2 DevR binding site; Hygr | This study |
| pAmutD3 | pnarK2 containing mutated D3 DevR binding site; Hygr | This study |
| pAmutD1 D3 | pnarK2 containing mutated D1 and D3 DevR binding sites; Hygr | This study |
| pBmutD1 | p1738 containing mutated D1 DevR binding site; Hygr | This study |
| pBmutD2 | p1738 containing mutated D2 DevR binding site; Hygr | This study |
| pBmutD3 | p1738 containing mutated D3 DevR binding site; Hygr | This study |
| pBmutD1 D3 | p1738 containing mutated D1 and D3 DevR binding sites; Hygr | This study |
| pnarK2Δup | pFPV27 containing narK2 promoter (−121 to +57); Hygr | This study |
| pnarK2Δdn | pFPV27 containing narK2 promoter (−220 to +13); Hygr | This study |
| phspX | pFPV27 containing hspX promoter (−132 to +48); Hygr | D. K. Saini and J.S. Tyagi |
| p3134c-3 | pFPV27 containing Rv3134c promoter (−348 to +90); Hygr | 5 |
| psigA | pFPV27 containing sigA promoter (−238 to + 80); Hygr | N. K. Taneja and J.S. Tyagi |
The coordinates of the promoters (in parentheses) are with reference to the transcriptional start site except for the sigA promoter, which is with respect to translational start site.
RNA isolation and primer extension.
RNA was isolated from M. tuberculosis H37Rv cultures grown in Dubos medium (see above) under aerobic shaking and standing (48 h) conditions as described previously (5). TSPs were mapped using primer extension as described previously (5, 13), using 32P-labeled narK2R3 or Rv1738R primers (Table 2) and 30 μg of RNA from aerobic and standing cultures (two separate lots). The reactions were run alongside the sequence ladder generated using the same primer and M. tuberculosis DNA. The gel was dried and visualized by phosphorimager (Bio-Rad).
TABLE 2.
Primers used in this study
| Primer | Sequence (5′ → 3′)a | Application |
|---|---|---|
| narK2F | GACACGATCCGGGGTCTCG | Reporter assay, gel shift assay, DNase I footprinting |
| narK2R | TCCCCTTTCCAGTGGCGACC | Reporter assay, gel shift assay, DNase I footprinting |
| narK2F1 | GGGGAATTCCGGACAGCCATGCGCTAACCC | Reporter assay |
| narK2R1 | GGGGAATTCCCCTCCGATAGGGCCTGAAGT | Reporter assay |
| narK2R3 | GTTCCACGCCCAGAAGTTGAC | Primer extension |
| Rv1738R | CGCCCGAGTCAATCCTTCGTGTTC | Primer extension |
| PD1 | ATGGCGCCGGCTCAGTTACCCCCGGAAGTCCCC | Mutational analysis |
| PD2 | CACTGCCCTGATTCGTCCGCCAGGGGC | Mutational analysis |
| PD3 | AGCCCTCCGATAGGGCCTGAAGTGGGGGCCACTGGGGTC | Mutational analysis |
EcoR1 sites are underlined, and mutated residues are in boldface.
Construction of reporter plasmids.
The narK2-Rv1738 intergenic sequence was amplified using primers narK2F and narK2R and cloned first in pGEMT-Easy and then into the promoterless GFP reporter plasmid, pFPV27 (19), in both orientations as described previously (5) to generate pnarK2 and p1738. The 5′ truncated narK2 promoter (pnarK2Δup) was amplified using primers narK2F and narK2R1. The 3′ truncated narK2 promoter (pnarK2Δdn) was amplified using primers narK2R and narK2F1. These PCR products were cloned in pFPV27 at the EcoR1 site. All of the cloned inserts were verified by DNA sequencing.
Dev box mutants were generated by the mega-primer method as described previously (5, 13). Mutations were confirmed by DNA sequencing. The mutated plasmids were electroporated in M. tuberculosis H37Rv. The primers used in this study are listed in Table 2.
Gel shift assay and DNase I footprinting.
DevR and phosphorylation-defective DevR D54V proteins were purified as described previously (1). Gel shift and DNase I footprinting assays were performed with purified DevR and the phosphorylation-defective protein DevR D54V. Gel shift assays were carried out as described previously (1, 5) except for minor changes in binding buffer [25 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 20 mM KCl, 6 mM MgCl2, 5% glycerol, 1 μg of poly(dI-dC), 100 μg/ml of bovine serum albumin, 1 mM dithiothreitol, and 2% polyethylene glycol]. The gel was analyzed using Quantity One software (Bio-Rad). The fraction of bound DNA was estimated by subtracting free DNA from input DNA. DNase I footprinting assays were performed as described earlier (5).
DNA pull-down assay.
The narK2-Rv1738 intergenic region was amplified by PCR and biotinylated as described previously (6). DevR protein was phosphorylated using acetyl phosphate as described previously (5). DevR and DevR∼P (10 μM) were incubated separately with the biotinylated PCR fragment (250 ng) in binding buffer devoid of polyethylene glycol and bovine serum albumin (see above) for 20 min at 25°C. A mock reaction having DevR∼P (10 μM) and no DNA was carried out in parallel. The reaction contents were then mixed with 200 μl of streptavidin MagneSphere paramagnetic particles/beads (Promega) for 30 min at 25°C with constant mixing. The magnetic beads were allowed to settle, supernatant was removed, and the beads were washed four times with binding buffer to remove unbound protein. The bound proteins were eluted with binding buffer containing 500 mM NaCl and subjected to Western blotting with polyclonal anti-DevR antibody.
RESULTS
Mapping of in vivo transcription initiation sites.
In order to map the promoters and examine the influence of hypoxia on Rv1738 and narK2 promoter activities, the TSPs of both genes were identified by primer extension using RNA extracted from aerobic and 48-h standing cultures of M. tuberculosis. One nonoverlapping hypoxia-inducible TSP was observed for each of the genes narK2 and Rv1738 (Fig. 2a and b). No TSP was detected for either gene under aerobic conditions, which indicates that these promoters are inactive in aerobic cultures, as reported earlier (7). The narK2 TSP was located at −58 bp upstream of the translation start site at a C nucleotide (Fig. 2a), and the Rv1738 TSP was mapped at a T nucleotide present at −82 bp upstream of the translation start site (Fig. 2b). Sequences having a partial match with SigC −10 and −35 consensus sequences (18) were detected upstream of both the narK2 and Rv1738 TSPs (Fig. 2c), suggesting the possibility that these promoters are utilized by RNA polymerase containing SigC.
FIG. 2.

TSP mapping. (a and b) Primer extension with M. tuberculosis RNA isolated from a 48-h standing culture (lanes 1) and from aerobic culture (lanes 2); narK2 TSP was mapped with the primer narK2R3 and Rv1738 TSP was mapped with the primer Rv1738R. Dideoxy sequencing reactions using the same primers are shown to the left. (c) Alignment of narK2 and Rv1738 sequences from −40 to +20 bp (with respect to TSP). (d) Putative −10 and −35 promoter sequences of Rv1738 and narK2 which resemble SigC consensus sequences. The nucleotides which do not match the consensus sequence are shown in lowercase letters.
Interaction of DevR∼P with the narK2-Rv1738 intergenic region.
Interaction assays were performed to assess DevR binding to the narK2-Rv1738 intergenic region, which was predicted to contain four putative Dev boxes, namely, D1, D2, D3, and D4 (10). The intergenic DNA probe strongly bound to DevR∼P (Fig. 3a) and very weakly to the DevR D54V phosphorylation-defective protein (Fig. 3b) in gel shift assays. At a 150 nM DevR∼P concentration, ∼50% of the DNA was present in free form, and at twice the protein concentration nearly all the DNA was present in one bound species of extremely low mobility (Fig. 3a and c). This is suggestive of cooperative binding of DevR∼P to DNA. DNA pull-down assays further confirmed that only DevR∼P protein interacted with DNA (Fig. 3d).
FIG. 3.

Analysis of DevR binding with narK2-Rv1738 intergenic region. 32P-labeled narK2-Rv1738 intergenic DNA (amplified with narK2F and narK2R primers (Fig. 5a) was incubated with increasing concentrations of DevR ∼P (a) or DevR D54V protein (b). The horizontal dashed line indicates the positions of wells. (c) Fraction of bound DNA (Fig. 3a) versus DevR∼P concentration (see Materials and Methods). (d) Western blot of eluted fractions from DNA pull-down assay developed with anti-DevR polyclonal antibody. Lane 1, purified glutathione S-transferase-tagged DevR protein; lane 2, eluate of reaction with DevR and acetyl phosphate without DNA; lanes 3 and 4, eluate of reactions in which biotinylated narK2-Rv1738 intergenic DNA was incubated with DevR in the absence or presence of acetyl phosphate.
DevR∼P binds 80 bp of DNA in the narK2-Rv1738 intergenic region.
The precise position of DevR binding sites was determined by DNase I footprinting analysis of independently labeled strands of the intergenic region. An ∼80-bp-long footprint was observed consistently (with both strands) spanning the D1, D2, and D3 boxes and also included 20 bp of DNA located between the D1 and D2 binding sites (Fig. 4 and 5). The intervening sequence does not bear much resemblance to a Dev box sequence, and its protection may be aided by DevR interaction with the three Dev boxes present in this region. The proximal Dev box (D1 or D3) is at an equidistant location (−32 bp) from the TSPs of both the narK2 and Rv1738 promoters while the D2 box is placed between the D1 and D3 motifs. The fourth putative binding site, D4, did not interact with DevR although its sequence appeared to be reasonably well conserved. A hypersensitive site was reproducibly observed exactly halfway between the D1 and D2 boxes (Fig. 4 and 5a) when the Rv1738 coding strand was labeled. This may be due to a conformational change in DNA resulting from DevR binding. This observation is consistent with the finding that DNA bending is associated with protein binding in the DevR C-terminal domain DNA crystal structure (23). No footprint was observed with the DevR D54V protein even at a 3.2 μM concentration (Fig. 4) or with unphosphorylated DevR (not shown).
FIG. 4.
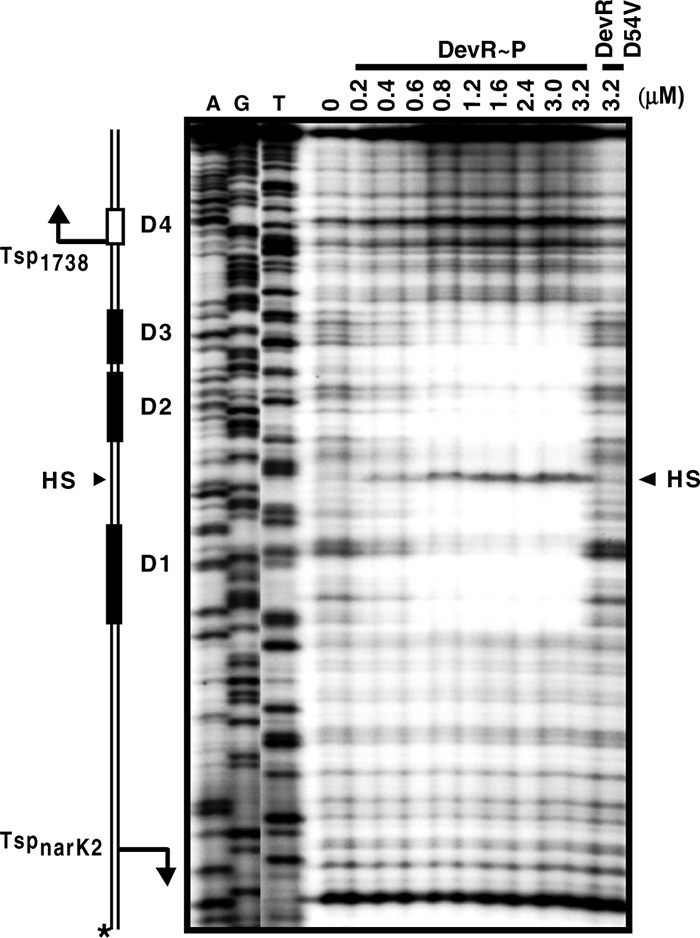
DevR∼P interacts with three binding sites in narK2-Rv1738 intergenic region. DNase I footprinting was performed with narK2-Rv1738 intergenic DNA (with the Rv1738 coding strand labeled with 32P) and either increasing concentrations of DevR∼P or the DevR D54V protein at highest concentration. D4 does not interact with DevR∼P. Bent arrows indicate the positions of the TSPs. The arrowhead indicates the hypersensitive sites (HS). Dideoxy sequencing reactions using the same primer and DNA template are also shown. The asterisk indicates the strand that is 32P labeled.
FIG. 5.

(a) Nucleotide sequence of narK2-Rv1738 intergenic region. TSPs (+1) are indicated by bent arrows. The putative −10 and −35 promoter elements are indicated by dashed boxes. The interacting Dev boxes, D1, D2, and D3, are indicated by gray rectangular boxes, and D4, which does not interact with DevR, is indicated by a white box. The positions of Dev boxes D1, D2, and D3 are indicated in parentheses with respect to the Rv1738 TSP above the sequence and with respect to the narK2 TSP below the sequence. The T→C nucleotide difference between M. tuberculosis and BCG is indicated by a vertical arrow. The upstream activating and the downstream inhibiting regions of the narK2 promoter in BCG (8) that were deleted in the present study (pnarK2Δup and pnarK2Δdn, respectively) are indicated by horizontal bars filled with diagonal lines and dots, respectively. The positions of primers are indicated by half-arrows. The arrowhead indicates the hypersensitive site. The bold underlined letters in the Dev boxes are the nucleotides that were mutated to their complementary nucleotides for various studies. (b) Map of the intergenic region. D1, D2, and D3 DevR binding sites are represented by shaded rectangular boxes. Box D4 (white box) does not interact with DevR. TSPs (+1) mapped in this study are shown by bent arrows. Putative −10 and −35 promoter elements are indicated by small black boxes. The distances between the DevR boxes and TSPs and between the binding sites are indicated (bp).
Effect of Dev box mutations on in vitro DevR binding.
It was previously suggested that bases G4, G5, G6, and C8 in the DevR binding motif could be crucial for interaction with DevR (10, 23). In order to understand the role of each Dev box in transcription, mutations were made at these conserved positions, as indicated in Fig. 6a, and interaction between mutated Dev boxes and DevR was examined by DNase I footprinting assays. When D1 was mutated, no footprint was observed on itself, but the D2 and D3 boxes were protected on both strands (Fig. 6b and c). When D2 was mutated, a footprint was observed at D1 on both strands (6b and c), and faint protection was observed at D3 at the highest protein concentration only when the narK2 coding strand was labeled (Fig. 6c). When D3 was mutated, a footprint was observed at D1, but no protection was observed at either the D3 or D2 box when either DNA strand was labeled (Fig. 6b; also data not shown). A hypersensitive site located exactly between D1 and D2 was observed on DevR binding irrespective of mutation in D1, D2, or D3 only when the Rv1738 coding strand was labeled (Fig. 6b). No discrete footprint was observed when D1 and D3 were doubly mutated although a faint footprint at the D2 box and a weak hypersensitive site were visible at the highest protein concentration tested (Fig. 6b). DNase I footprinting results suggest that DevR binding to the adjacently placed D2 and D3 sites is interdependent while binding at the D1 box and 20 bp of adjacent sequence is independent of protein occupancy at the D2 and D3 sites. Since the 20-bp region is protected on DNA having D2 or D3 site mutations and not protected on DNA mutated at the D1 site (Fig. 6b), it is likely that the binding of DevR to the intervening region is dependent on occupancy of the D1 site.
FIG. 6.

DNase I footprinting analysis of mutated Dev boxes. (a) Mutations in individual Dev boxes are shown in bold letters. DNase I footprinting was performed with wild type (wt) and various Dev box-mutated DNAs. (b) 32P-labeled Rv1738 coding strand. (c) 32P-labeled narK2 coding strand. mut-D1, mut-D2, and mut-D3 represent DNA harboring mutated Dev boxes D1, D2, and D3, respectively; mut-D1 D3 represents a double mutant of the D1 and D3 boxes. The hypersensitive sites are indicated by an arrowheads.
Hypoxic induction of narK2 and Rv1738 promoters.
The Rv1738-narK2 intergenic region (286 bp) was fused in both orientations upstream of the promoterless gfp gene in the reporter vector pFPV27, and the constructs were individually introduced into M. tuberculosis. pnarK2 plasmids contained the region from −220 to +57 of the narK2 promoter, and p1738 plasmids contained the region from −203 to +74 of the Rv1738 promoter (Table 1). Promoter activity was assessed by measurement of GFP fluorescence under aerobic and hypoxic conditions. Both promoters were completely inactive under aerobic conditions until stationary phase (data not shown). These results were consistent with the failure to detect TSPs in aerobic cultures (Fig. 2). Both the genes were highly upregulated in hypoxic cultures in both plate and tube formats (Fig. 7 and Fig. S1 in the supplemental material). The Rv1738 promoter (p1738) was ∼10-fold more active than the narK2 promoter (pnarK2) under inducing conditions (12,198 versus 1,250 relative fluorescence units/optical density unit [RFU/OD unit] in 48-h standing cultures) (Fig. 7). Transcription activation of both genes was observed to be DevR dependent (data not shown) as shown previously (10).
FIG. 7.

Wild-type promoter activity and effect of Dev box mutations on transcriptional activation of narK2 and Rv1738. GFP fluorescence (RFU/OD unit at 595 nm) of M. tuberculosis standing cultures after subtraction of background fluorescence derived from pFPV27. GFP fluorescence is constant in bacteria harboring the pFPV27 control vector. Percent expression with respect to the wild-type promoter is given in parentheses. Relative induction is a ratio of RFU/OD unit of standing cultures versus that of aerobic shaking cultures at 48 h (without subtracting background fluorescence derived from pFPV27). Shown are the average values of four experiments, each performed in triplicate. The number of RFU varied <15% between the experiments.
Synergistic transcriptional activation by Dev boxes.
The effect of Dev box mutations on narK2 and Rv1738 promoter activation was assessed next by means of a GFP reporter assay. In 48-h hypoxic cultures, a massive induction of 273-fold was measured from p1738 that harbors the wild-type Rv1738 promoter. For this promoter, DevR mediated 6-fold induction from the D3 box-mutated template, 73-fold induction from the D2 box-mutated template, and 216-fold induction from the D1 box-mutated template. The magnitude of induction that was observed in the presence of wild-type D2 and D3 boxes (216-fold) is much greater than the additive effects of DevR working through either the D2 (6-fold) or D3 (73-fold) site. Thus, maximal induction occurred when all three DevR binding sites were intact (Fig. 7) and suggests a functional synergism for activation. Transcription activation of Rv1738 was mainly dependent on DevR occupancy at the D2 and D3 sites while binding at the upstream D1 site mediated an additional ∼25% increase to fully induce the Rv1738 promoter.
Like the Rv1738 promoter, narK2 promoter activation (pnarK2) was also maximal when all three DevR binding sites were intact. Inducible expression was completely abolished when the proximal D1 box was mutated (Fig. 7, pAmutD1). Mutating either D2 or D3 diminished transcription activation by ∼ 25% in pAmutD2 or pAmutD3, respectively. When D1 and D3 sites were doubly mutated (pAmutD1 D3 and pBmutD1 D3), transcription activation of both genes was almost completely abolished (Fig. 7). The observed effects of binding site mutations on the activity of both promoters were confirmed in both plate and tube formats over a period of time (see Fig. S1 in the supplemental material).
Relation between the architecture and strength of DevR-dependent promoters.
CRP-dependent promoters are grouped into three classes based on the placement of the CRP binding sites relative to the TSP (4). At a class I promoter, the CRP binding site can be centered near position −93, −83, −72, or position −62. At class II promoters CRP binds to a site that overlaps the promoter −35 element, apparently replacing it, and is typically centered near position −42. Class III promoters are characterized by the presence of two or more binding sites either at different class I positions or at class II and class I positions (4). The placement of Dev boxes in some DevR-regulated promoters was examined (Fig. 8a). A Dev box is always centered at −42.5/−43.5 upstream of the TSP, and this site is essential for DevR-mediated hypoxic induction at all four promoters that were examined. The second Dev box at three promoters (Rv1738, Rv3134c, and hspX) is likely to be associated with cooperative interaction of DevR with boxes placed in tandem (at −42.5/−43.5 and −63.5/−65.5). At the narK2, Rv1738, and hspX promoters, a third Dev box located further upstream centered at −100.5/−104.5 is necessary for maximal activation under hypoxia. In all cases the boxes are placed so as to align them on the same helical phase of DNA. In these respects, the promoters appear to resemble the class III CRP-regulated promoters described above.
FIG. 8.

Comparative assessment of M. tuberculosis DevR-dependent promoters. (a) Alignment of Rv1738, hspX, Rv3134c, and narK2 promoters. Dev boxes are represented by gray rectangles and their positions with respect to TSPs are indicated in parentheses below the boxes. (b) The activity of Rv1738, hspX, Rv3134c, and narK2 promoters was assessed in M. tuberculosis cultures over time in standing cultures (plate format). The mean RFU/OD unit of triplicate cultures is shown.
To investigate the relation between architecture and the strength of select DevR-regulated class III promoters, their activities were analyzed using the plate format assay (Fig. 8b). Maximal induction was observed with the Rv1738 promoter (plasmid p1738; 15,705 RFU/OD unit), followed by the hspX promoter (plasmid phspX; 13,183 RFU/OD unit). The promoters exhibit similar patterns of induction, which may be a consequence of similarity in their architecture where three Dev boxes might activate the transcription synergistically. Rv3134c promoter activity (plasmid p3134c-3) was less than that of the Rv1738 and hspX promoters. The narK2 promoter was the least induced among the promoters studied, and one reason could be that its transcription is primarily controlled by a single Dev box (D1); additionally, its activity is also negatively regulated (see below). Taken together, these results suggest that there is a good correlation between promoter structure and the extent of hypoxic gene activation. However, basic features of the individual promoter sequences and the involvement of additional cis/trans regulators (besides DevR) could also control the expression of individual genes according to the cellular requirement.
The narK2 promoter is regulated differently in M. tuberculosis and M. bovis BCG.
In M. bovis BCG hypoxic induction of the narK2 promoter is dependent on the “upstream activating region,” and deletion of this region results in the loss of hypoxic upregulation (8). It was also shown that removal of a “downstream inhibiting region” results in a massive induction of narK2 promoter activity (8). Except for a single nucleotide difference (T to C at −5 bp with respect to narK2 TSP), the nucleotide sequences of the narK2-Rv1738 intergenic region in BCG and M. tuberculosis H37Rv are identical (Fig. 5a). Therefore, the role of the upstream activating and downstream inhibiting regions was studied in M. tuberculosis. Deletion of the upstream activating region in M. tuberculosis (pnarK2Δup) did not adversely affect narK2 promoter induction; on the contrary, the promoter was ∼1.5-fold more active than the wild-type promoter under inducing conditions (Fig. 7). When the downstream inhibiting region was deleted (in pnarK2Δdn), activity was approximately twofold greater than that of the wild-type promoter(Fig. 7). In contrast, deletion of exactly the same region in BCG was associated with a ∼20-fold increase in the induction ratio (8). Further investigation is necessary to determine whether deletion of the downstream region affects processes after transcription initiation (such as mRNA stability and translational efficiency) rather than events occurring at the promoter. The underlying mechanism for the observed differences between the two species also remains to be elucidated.
DISCUSSION
Results of this study establish that DevR interaction with all three Dev boxes is necessary for full activation of divergently transcribed narK2-Rv1738 genes under hypoxia. Under inducing conditions, compared to the narK2 promoter that is activated 35-fold, the Rv1738 promoter is powerfully activated 273-fold (Fig. 7). narK2 activation is primarily mediated by DevR interaction with the D1 Dev box, which overlaps with the −35 promoter element; however, the D2 and D3 boxes were also required for full induction. Similarly, Rv1738 transcription is chiefly activated by two DevR∼P molecules placed in tandem at the D2 and D3 sites where D3 overlaps with the −35 promoter sequence. Here, too, binding to a third box, D1, is necessary for full activation.
The crucial importance of the proximally located Dev box, D1 or D3, in transcription activation of the narK2 and Rv1738 promoters, respectively, was established by mutational analysis (Fig. 7). A similar role for DevR at the Rv3134c promoter was recently shown (5). Since these interactions occur between DevR and binding sites that overlap the −35 promoter element, it is highly likely that DevR interacts with RNA polymerase at these promoters. However, unlike the scenario at the Rv3134c promoter, where the distal Dev box mutation completely abolished hypoxic promoter activation (5), 73-fold activation of the Rv1738 promoter was noted (26% of maximal relative activation) when the corresponding D2 binding site was mutated. In this situation it is likely that weak interaction of DevR with the D3 site is stabilized by its interaction with the 20-bp intergenic sequence and D1 box (Fig. 6c), which thus compensates partially for the missing D2 box-DevR interaction.
The sequences of several Dev boxes whose interactions with DevR were analyzed by DNase I footprinting were compared in Table 3. It was previously suggested that bases G4, G5, G6, and C8 in the 20-bp palindromic box could be crucial for interaction with DevR and that G4 was essential (10, 23). Three out of eight Dev boxes that bind to DevR do not have a G nucleotide at position 4, which suggests that mutation at position G can be tolerated. By contrast, G5, G6, and C8 nucleotides were conserved in all of the interacting boxes. Interestingly, a C-to-G mutation at position 8 in either DNA strand was detected in boxes that do not interact with DevR, which points to the importance of C8 at this position.
TABLE 3.
Sequence alignment and interaction status of Dev boxes
| Dev box | Dev box sequencea | Interaction with DevR∼P | Reference or source |
|---|---|---|---|
| D1 | TTAGGGCCGGAAGTCCCCAA | + | This study |
| D2 | TGGCGGACGAATGACCCCAG | + | This study |
| D3 | GCCGGGACTTCAGGCCCTAT | + | This study |
| D4 | TTGGGGTGTAACCTCCCCAG | − | This study |
| Rv3134c D | ATAAGGACTAACGGCCCTCA | + | 5 |
| Rv3134c P | GTGGGGACCAACGCCCCTGG | + | 5 |
| Rv3134c 1 | ACCTGGACGAGCCACCCGTG | − | 5 |
| Rv3134c 4 | ACGGGATGTATCCGCCCCAG | − | 5 |
| hspX D | ACAGGGTCAATGGTCCCCAA | + | 5 |
| hspX C | GCGCGGACAAATGGCCCGCG | + | 5 |
| hspX P | TCGGGGACTTCTGTCCCTAG | + | 5 |
| DevR consensus motif | TTSGGGACTWWAGTCCCSAA | 10 |
The conserved G4, G5, G6, and C8 nucleotides are shown in boldface. S represents G or C, and W represents A or T. C, D, and P are abbreviations for Dev box designations in Rv3134c and hspX genes.
The narK2 promoter sequences in M. tuberculosis and M. bovis BCG are identical except for a single nucleotide alteration (Fig. 5a). However, several disparities were noted in narK2 transcription regulation between the two species. First, in BCG the reported narK2 TSP is at −47 (8), while in M. tuberculosis the TSP maps at −58. Second, in BCG narK2 hypoxic induction is totally dependent on a far-upstream region (8), whereas in M. tuberculosis deletion of the same sequence modestly increases hypoxic induction by ∼1.5- to 2-fold (Fig. 7, pnarK2Δup; see also Fig. S1 in the supplemental material). Third, in BCG deletion of a 47-bp sequence downstream of the TSP increases hypoxic induction by ∼20-fold, whereas in M. tuberculosis deletion of the same sequence leads to only approximately a two- to threefold increase in hypoxic induction (Fig. 7, pnarK2Δdn; see also Fig. S1 in the supplemental material). This result indicates that the narK2 promoter is also under negative regulation that is yet to be deciphered, and there appear to be significant differences in the negative regulation between the two species.
Cooperative DNA binding is a well-characterized mechanism for synergistic activation. Thus, DevR bound to one site would strengthen the interaction of a second DevR molecule with a second site and subsequently activate transcription from the downstream promoter. The binding of DevR to the adjacent D2 and D3 sites represents a simple mechanism that may account for a proportion of the transcriptional synergy observed in this study. However, some of our observations are inconsistent with activation's being entirely due to cooperative DNA binding. First, the presence of a mutated D2 or D3 site does not influence binding of DevR to the D1 box. Second, in spite of efficient binding to D1, which overlaps the −35 promoter element, DevR binding to the upstream sites (D2 and D3) mediates a ∼25% increase in narK2 transcription (Fig. 7, compare pAmutD2 or pAmutD3 with pnarK2). Similarly, mutating the D1 site does not influence DevR binding to the D2 and D3 boxes. Binding of DevR to these two sites leads to an impressive induction of Rv1738 transcription (216-fold). In this scenario binding to the third upstream binding site, D1, enhances activation by an additional 25% to 273-fold (Fig. 7). Therefore, it seems likely that there must be alternate mechanism(s) of synergy beyond cooperative binding that permits full activation of the Rv1738 and narK2 promoters. We suggest that all three DNA-bound DevR molecules may interact with the transcription machinery to provide synergistic activation of narK2 and Rv1738 transcription. This type of activation was shown for an artificial promoter, where a pair of CRP dimers bound to CRP binding sites at a class II position (−41.5) and where one of a number of class I positions (from −74.5 to −102.5) interacts with RNA polymerase to activate transcription synergistically in E. coli (2). This study and our previous one (5) have provided glimpses into DevR interactions at promoters having two or three Dev boxes and the consequences of these interactions on transcription activation. Further studies are necessary to decipher the mechanism of transcription activation of DevR-regulated promoters.
Supplementary Material
Acknowledgments
This work was financially supported by a grant to J.S.T. from the Department of Biotechnology, Government of India. S.C. is grateful to CSIR for a Senior Research Fellowship.
We acknowledge the facilities of the Biotechnology Information Systems, Department of Biotechnology, and Government of India. We acknowledge A. Baisantry, D. Bakshi, and N. K. Taneja for preliminary work on narK2 transcription.
Footnotes
Published ahead of print on 23 May 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Bagchi, G., S. Chauhan, D. Sharma, and J. S. Tyagi. 2005. Transcription and autoregulation of the Rv3134c-devR-devS operon of Mycobacterium tuberculosis. Microbiology 1514045-4053. [DOI] [PubMed] [Google Scholar]
- 2.Belyaeva, T. A., V. A. Rhodius, C. L. Webster, and S. J. Busby. 1998. Transcription activation at promoters carrying tandem DNA sites for the Escherichia coli cyclic AMP receptor protein: organisation of the RNA polymerase alpha subunits. J. Mol. Biol. 277789-804. [DOI] [PubMed] [Google Scholar]
- 3.Boon, C., and T. Dick. 2002. Mycobacterium bovis BCG response regulator essential for hypoxic dormancy. J. Bacteriol. 1846760-6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busby, S., and R. H. Ebright. 1999. Transcription activation by catabolite activator protein (CAP). J. Mol. Biol. 293199-213. [DOI] [PubMed] [Google Scholar]
- 5.Chauhan, S., and J. S. Tyagi. 21 March 2008. Cooperative binding of phosphorylated DevR to upstream sites is necessary and sufficient for activation of the Rv3134c-devRS operon in Mycobacterium tuberculosis: implication in the induction of DevR target genes. J. Bacteriol. doi: 10.1128/JB.01308-07. [DOI] [PMC free article] [PubMed]
- 6.Chowdhury, R. P., S. Gupta, and D. Chatterji. 2007. Identification and characterization of the dps promoter of Mycobacterium smegmatis: promoter recognition by stress-specific extracytoplasmic function sigma factors σH and σF. J. Bacteriol. 1898973-8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Florczyk, M. A., L. A. McCue, A. Purkayastha, E. Currenti, M. J. Wolin, and K. A. McDonough. 2003. A family of acr-coregulated Mycobacterium tuberculosis genes shares a common DNA motif and requires Rv3133c (dosR or devR) for expression. Infect. Immun. 715332-5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hutter, B., and T. Dick. 2000. Analysis of the dormancy-inducible narK2 promoter in Mycobacterium bovis BCG. FEMS Microbiol. Lett. 188141-146. [DOI] [PubMed] [Google Scholar]
- 9.Kendall, S. L., F. Movahedzadeh, S. C. Rison, L. Wernisch, T. Parish, K. Duncan, J. C. Betts, and N. G. Stoker. 2004. The Mycobacterium tuberculosis dosRS two-component system is induced by multiple stresses. Tuberculosis 84247-255. [DOI] [PubMed] [Google Scholar]
- 10.Park, H. D., K. M. Guinn, M. I. Harrell, R. Liao, M. I. Voskuil, M. Tompa, G. K. Schoolnik, and D. R. Sherman. 2003. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol. 48833-843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reference deleted.
- 12.Saini, D. K., V. Malhotra, D. Dey, N. Pant, T. K. Das, and J. S. Tyagi. 2004. DevR-DevS is a bona fide two-component system of Mycobacterium tuberculosis that is hypoxia-responsive in the absence of the DNA-binding domain of DevR. Microbiology 150865-875. [DOI] [PubMed] [Google Scholar]
- 13.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 14.Sassetti, C. M., D. H. Boyd, and E. J. Rubin. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 4877-84. [DOI] [PubMed] [Google Scholar]
- 15.Sherman, D. R., M. Voskuil, D. Schnappinger, R. Liao, M. I. Harrell, and G. K. Schoolnik. 2001. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha-crystallin. Proc. Natl. Acad. Sci. USA 987534-7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi, L., C. D. Sohaskey, B. D. Kana, S. Dawes, R. J. North, V. Mizrahi, and M. L. Gennaro. 2005. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. USA 10215629-15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sohaskey, C. D., and L. G. Wayne. 2003. Role of narK2X and narGHJI in hypoxic upregulation of nitrate reduction by Mycobacterium tuberculosis. J. Bacteriol. 1857247-7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun, R., P. J. Converse, C. Ko, S. Tyagi, N. E. Morrison, and W. R. Bishai. 2004. Mycobacterium tuberculosis ECF sigma factor sigC is required for lethality in mice and for the conditional expression of a defined gene set. Mol. Microbiol. 5225-38. [DOI] [PubMed] [Google Scholar]
- 19.Valdivia, R. H., A. E. Hromockyj, D. Monack, L. Ramakrishnan, and S. Falkow. 1996. Applications for green fluorescent protein (GFP) in the study of host-pathogen interactions. Gene 17347-52. [DOI] [PubMed] [Google Scholar]
- 20.Voskuil, M. I., D. Schnappinger, K. C. Visconti, M. I. Harrell, G. M. Dolganov, D. R. Sherman, and G. K. Schoolnik. 2003. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198705-713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wayne, L. G., and L. G. Hayes. 1998. Nitrate reduction as a marker for hypoxic shiftdown of Mycobacterium tuberculosis. Tuber. Lung Dis. 79127-132. [DOI] [PubMed] [Google Scholar]
- 22.Wayne, L. G., and C. D. Sohaskey. 2001. Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 55139-163. [DOI] [PubMed] [Google Scholar]
- 23.Wisedchaisri, G., M. Wu, A. E. Rice, D. M. Roberts, D. R. Sherman, and W. G. Hol. 2005. Structures of Mycobacterium tuberculosis DosR and DosR-DNA complex involved in gene activation during adaptation to hypoxic latency. J. Mol. Biol. 354630-641. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.