

Abstract
In vitro reaction conditions using HIV reverse transcriptase (RT) and nucleocapsid protein (NC) that allowed efficient synthesis of single-stranded DNA products over a thousand nucleotides in length from genomic HIV RNA were characterized. Consistent with previous reports, the reactions required high concentrations of NC and RT. Long products were produced as a result of frequent strand transfer between RNA templates, averaging at least one transfer per 300 nucleotides synthesized. No change in RT processivity was observed in the reactions in the presence versus absence of NC. Synthesis of long products required formation of a high molecular mass aggregate between NC and nucleic acids. The aggregate formed rapidly and pelleted with low speed centrifugation. The aggregate was accessible to RT as pre-formed aggregates synthesized long products when RT was added. NC finger mutants lacking either finger one or two or with the finger positions switched were all effective in promoting long products. This suggests that the aggregation/condensation but not helix-destabilizing activity of NC was required. We propose that these high molecular mass aggregates promote synthesis of long reverse transcription products in vitro by concentrating nucleic acids, RT enzyme and NC to close proximity, thereby mimicking the role of the capsid environment within the host cell.
Keywords: nucleocapsid protein, NC, RNase H, recombination, cDNA synthesis
Introduction
Human immunodeficiency virus (HIV-1), like all retroviruses, undergoes reverse transcription during its replication cycle. This involves copying of its single-stranded RNA genome into double-stranded DNA that later gets integrated into the host chromosome. Although some viral particles may initiate reverse transcription before entering cells,1–5 the process generally begins when the viral particle enters the cytoplasm of the host cell and occurs exclusively in the host cytoplasm. Although HIV cores are disrupted shortly after virus-cell fusion,6 reverse transcription occurs within a confined environment in capsid-like structures derived from the virion core permeable to dNTPs.7,8 Reverse transcription intermediates and products identical to those made in infected cells can be synthesized in the purified virions. This requires application of mild detergents to permeabilize the envelope and addition of deoxyribonucleoside triphosphates and a divalent cation as the metal cofactor (referred to as endogenous reverse transcription or ERT).9–14 This process can even occur in the absence of added detergent (referred to as natural endogenous reverse transcription or NERT).15–17 However, the yield of completed products in ERT and NERT reactions is extremely low. It can be concluded that the virion environment is not sufficient to allow efficient replication or that certain cellular components or structural alterations are essential for efficient replication.
Reverse transcription can also be carried out in vitro in totally reconstituted reactions including only a primed RNA template and purified reverse transcriptase (RT). Several effects of the viral nucleocapsid protein (NC) have been demonstrated using in vitro or cell culture based approaches including: increasing the processivity (average number of nt added to the primer in a single binding event between primer-template and enzyme) of RT18,19, enhancing binding of the host tRNA primer to the viral primer binding site (as part of the Gag precursor protein),20–23 promoting dimerization between the two genomes in the viral capsid22,24,60 and stimulating strong-stop minus and plus strand transfer and viral recombination in general.25–33 In fact, all the reactions and steps required to produce complete double-stranded DNA appear to reside in RT and viral NC enhances many of these reactions. It has also been suggested that other viral proteins (Tat, for example) may play a role, but are probably not major constituents of the replication complex.34 Also, both RTand NC are highly stable in vitro. Therefore, given enough time, one would predict the production of completely processed double-stranded DNA from genomic RNA in in vitro systems. However, this is not observed and even fully synthesized single-stranded DNAs (minus strand) are not generated from genome length RNA. Most products are only a few hundred and at most a few thousand nt in length when templates of several thousand bases are used,35 indicating that these reactions are even less efficient than NERT or ERT reactions.
In the presence of specific amounts of NC and high RT concentrations an increase in reverse transcription efficiency in in vitro reactions can be observed. Results showed that at greater than or equal to 50% saturation of NC binding sites (one NC coats approximately 7 nt36–38), there is up to a 90% decrease in total DNA synthesis, as measured by incorporation of dNTPs.39 However, the cDNA products made are almost exclusively full-length, although in this case an RNA template of only 874 bases was used.
A series of reports have demonstrated that under the right conditions NC can stimulate the rapid formation of large aggregate complexes that contain NC, RT, and RNA.40–43 The complexes are functional as isolated complexes can synthesize DNA in the presence of dNTPs, use tRNA to prime viral RNA synthesis, and catalyze minus strand strong-stop strand transfer.40
As part of a longstanding effort in our laboratory to produce an in vitro system that more closely mimics cellular replication, we describe here conditions that produced DNA products up to 4 kb from genomic RNA of HIV at relatively high efficiency. These reactions included high concentrations of RT and enough NC to completely coat all the nucleic acids in the reaction. Synthesis occurred in large rapidly formed aggregates similar to those described above and product formation required strand transfer. The characterization of this system is described. In addition to enhancing our understanding of the replication process, an in vitro system that mimics cellular replication could potentially be used to screen reverse transcription inhibitors.
Results
Optimization of reaction conditions for the synthesis of long DNA products in vitro
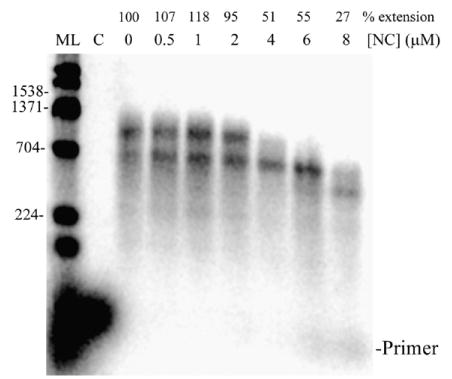
The general approach used for reverse transcription assays is depicted in Figure 1(a). Reaction conditions were optimized first for the synthesis of a 1538 nt DNA product in vitro. This was done by performing the reverse transcription assay with different concentrations of NC (Figure 1(b)) and also with different concentrations of HIV-RT (data not shown). The template to primer ratio was 4:1 in these reactions (see Materials and Methods). In the absence of NC, primer extension resulted in synthesis of smaller DNAs but no full-length products. A small amount of fully extended products were observed with 0.5 and 1 μM NC and full-length products were substantially increased when 2, 4, 6, or 8 μM NC was used. There was also a large decrease in smaller products at these NC concentrations and a decrease in total primer extension relative to reactions without NC (as indicated above each lane in Figure 1(b)). The level of extension with NC varied to some extent depending on the RNA preparation used and was typically between 50–80% with 4 μM NC and 4:1 template: primer. The decrease in primer extension with NC should be accompanied by a corresponding increase in unextended primer, but this is not usually seen because a large but variable portion of the small primers diffuse out of the alkaline agarose gel during electrophoresis and processing. The unextended primers were evident on denaturing polyacrylamide gels although large products cannot be resolved on these gels (see Figure 8, −trap+ NC lane). Note that in all presented experiments, the level of primer extension was calculated relative to the control reaction in the absence of NC, which was set to 100%. Since some of the smaller extension products diffuse from the gels the actual level of extended primers would be underestimated by this approach, especially for reactions without NC where there were more small products. Although this would have some effect on the quantitative accuracy of the results, it did not impact any of the important conclusions.
Figure 1.
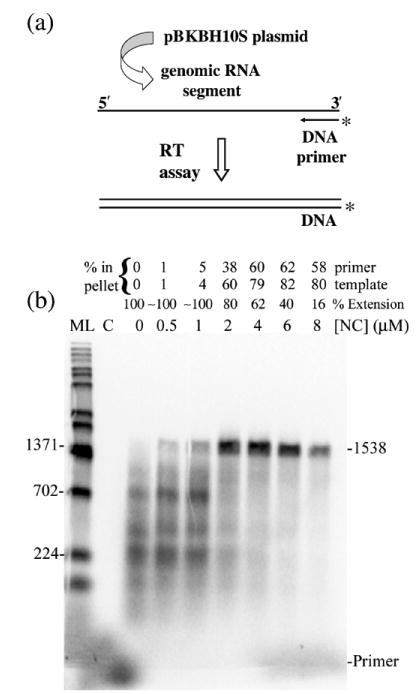
(a) and (b) Reverse transcription assay with different concentrations of wild-type NC shows that NC stimulates long product formation. (a) Schematic representation of the reverse transcription assay. Genomic RNA used as template for the reverse transcription assay was derived from pBKBH10S plasmid by run-off transcription. The RNA was hybridized to a radiolabeled DNA primer and the assay carried out by adding RT. The DNA products obtained were run on a 1% alkaline agarose gel. The template lengths were 1857 (1.9 kb) and 4007 (4 kb) nt and fully extended DNA products were 1538 and 4007 nt for the short and long templates, respectively. (b) Shown is an autoradiogram of an assay with 1.9 kb RNA using different NC concentrations (as indicated) or in the absence of NC (0 μM) performed as indicated in Materials and Methods. The percentage of primer extension (% Extension) in reactions with NC with respect to primer extension in reactions without NC (set to 100%) is indicated. The percent primer and template in the pellet fraction is also indicated above each lane (see Materials and Methods and Figures 6 and 7 and accompanying description). Size markers (ML, in nt) are shown on the left and full-length products are indicated on the right (1538) along with the primer location. Lane C, control reaction without RT. The experiment was repeated with similar results.
Figure 8.
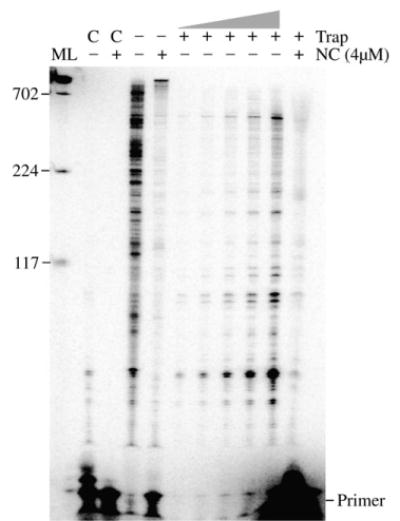
Reverse transcription assay indicates that enhanced processivity of RT plays no role in the synthesis of long DNA products in vitro. Shown is an autoradiogram of a trap assay using the 1.9 kb RNA segment of HIV as template. Control reactions (C) were performed in the presence (+) or absence (−) of 4 μM NC to test the effectiveness of the trap (see Materials and Methods). Reactions without trap were also performed with or without NC as indicated. In reactions with poly(rA)-oligo (dT) trap, RT was pre-incubated with the primer-template in the presence or absence of NC, and synthesis was initiated by the addition of divalent cation and dNTPs along with trap. The trap sequesters RT molecules that dissociate from the primer-template limiting extension to a single binding event. The trap reactions without NC were serially diluted 1:2 from right to left with the far right lane corresponding to a single reaction and the far left 1/16th of a reaction. The trap reaction with NC is ten reactions combined after extraction and precipitation, then loaded in a single lane. The samples were run on a 5% polyacrylamide gel. All markings are as in Figure 1(b). A repeat experiment yielded similar results.
In addition to NC concentration, other reaction conditions were also tested (data not shown). Increasing the [RT] beyond 80 nM did not significantly affect synthesis while a clear reduction in full-length products was observed below this level. Reactions worked best using synthesis conditions that were optimal for HIV-RT (6 mM MgCl2, 100 μM dNTPs, pH 8). Reactions were not very sensitive to salt concentrations, showing little difference at 10 versus 80 mM KCl. Concentrations of MgCl2 between 2 and 10 mM produced essentially the same results while a clear reduction in long products was observed with 1 or 0.5 mM MgCl2. This probably resulted from a slower rate of incorporation as these conditions are sub-optimal for RT polymerization. Experiments where NC was added 5 or 10 min after initiating primer extension with RT looked essentially like the no NC controls, indicating that prior incubation with NC was required for long products to form. This was likely due to the requirement of complex formation for long products (see below).
Reverse transcription was then performed using a longer RNA on which fully extended primers produced products of approximately 4 kb. Various experiments were performed to characterize these reactions and to optimize reaction conditions. Figure 2 shows a time-course experiment, where the reactions were carried out in the presence of 8 μM NC and stopped at time points of 0.5, 1, 2, and 3 h. Fully extended products were only observed after 2 h in the presence of NC, although there was a substantial decrease in total primer extension (27% extension relative to no NC). Increasing the amount of template in these reactions modestly increased the level of primer extension but the effect was less dramatic than with the smaller template (see Figure 7). Unlike reactions with the 1.9 kb template (Figure 1(b)), fully extended products made up just a small fraction of the total products at the end of the reactions. Reactions with the longer template were also less reproducible as not all preparations of the RNA yielded long products. Due to the long time period required to synthesize 4 kb products it was possible that the lower level of full length products resulted from a loss of RT activity during the reaction. However, addition of more RT after 1 h did not change the profile (data not shown). Attempts to synthesize even longer products were made using a 7 kb HIV-derived RNA. Inhibition of primer extension by NC was observed in these reactions but no full-length products were made (data not shown).
Figure 2.
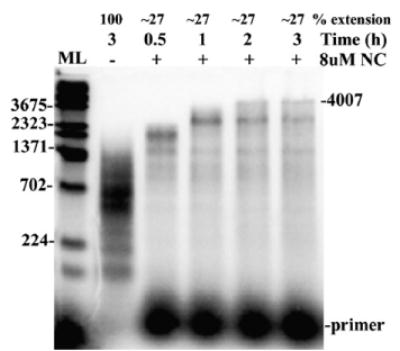
Reverse transcription assay using 4 kb genomic RNA segment as template shows that 4 kb products can be made in the reactions. Shown is an autoradiogram of a time-course assay using 4 kb genomic RNA template. The reactions were carried out in the presence of 8 μM NC for time points 0.5, 1, 2 and 3 h. The reaction without NC was carried out for 3 h. The position of the full-length products is indicated (4007) along with the primer location. ML denotes lane with molecular mass marker (in nt). Other markings are as indicated in Figure 1(b). A repeat experiment yielded similar results.
Figure 7.
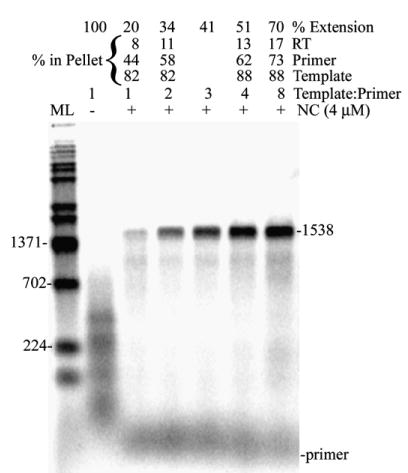
Reverse transcription assay with increasing concentrations of template shows that template, primer, and RT migrate with different fractions depending on the template concentration. Shown is an autoradiogram of an assay using 1.9 kb RNA as template with no excess template (1:1 template:primer) and with increasing concentrations of template (2, 3, 4, and 8 template:primer). Lanes with – indicate reactions without NC and those with +, with NC (4 μM). The percentages of RT activity, primer and template in the pellet at different concentrations of template are indicated and were determined as described in Materials and Methods (not calculated for 3:1 template: primer). Other markings are as in Figure 1(b). A repeat experiment yielded similar results.
Synthesis of long DNA products involves strand transfer
One possible mechanism for producing long DNA products is copy-choice type strand transfer, which is used by retroviruses for recombination. This mechanism does not occur efficiently on DNA templates.44 Therefore a strand transfer mechanism that produces long DNAs with RT and NC should not function efficiently on a DNA template. Experiments were performed to test whether the above conditions could also stimulate synthesis on a DNA template. Asymmetrical PCR was used to produce a 1.5 kb DNA template that was homologous to the 1.9 kb RNA over the region where primer extension occurred. Increasing concentrations of NC (0, 0.5, 1, 2, 4, 6, and 8 μM) and 80 nM RT were included in the reactions, which were stopped after 75 min (Figure 3). Many of the products produced on the DNA were nearly full length even in the absence of NC, although full-length (1538 nucleotides) products were not observed. The average length of extension products in the absence of NC was clearly greater than was observed with RNA (see Figure 1 (b)). The greater efficiency was probably due to the DNA not being susceptible to RNase H activity. This activity can cause the nascent DNA and RNA template to dissociate, making extension more difficult on RNA. The major effect of NC was to inhibit extension as the level of extended products clearly decreased at high NC concentrations. No increase in longer products was observed with the DNA template. The average length of extended products actually showed some decrease in the presence of 4, 6, and 8 μM NC. Overall the results show that the mechanism responsible for producing long products with RNA template does not function on DNA, making strand transfer a likely possibility.
Figure 3.
Reverse transcription assay using DNA template shows that NC does not stimulate long product production on DNA. Shown is an autoradiogram of an assay using 1.5 kb DNA as template in the presence of increasing concentrations of NC (0, 0.5, 1, 2, 4, 6 and 8 μM) and 80 nM RT. C denotes the lane with reaction carried out without RT. ML denotes lane with molecular mass marker (in nt). Other markings are as in Figure 1(b). A repeat experiment yielded similar results.
To directly test the possibility that long products result from strand transfer, a time-course assay that simulated strand transfer events occurring during minus strand synthesis was performed. Donor RNA (1394 bases long), the template on which DNA synthesis initiated and threefold excess acceptor RNA (1518 bases long, see Materials and Methods), the template to which DNAs initiating on the donor can potentially transfer, were mixed in reactions. DNA synthesis was initiated from a 5′ end-labeled DNA primer that was designed to bind only to the 3′ end of the donor RNA (see Figure 4(a)). Strand transfer can occur at any point after primer extension occurs on the donor, since the donor and acceptor are homologous over this region. The acceptor contains 144 additional bases at the 5′ end and products transferring to and subsequently extended on the acceptor will be 1538 nt as compared to 1394 for extension on the donor. Shown in Figure 4(b) is an autoradiogram of a strand transfer time-course assay in the presence of 4 μM NC. A reaction with just donor template is also shown to mark the position of products made on the donor (far right lane). Transfer products, migrating slightly higher than full-length donor-directed products, were observed in the reactions by 45 min and increased up till 60 min. In reactions with acceptor, no products consistent with full-length donor-directed products were observed at any time point. This suggests that transfer to the acceptor occurs before the end of the donor is reached (internal strand transfer). A time-course reaction in which excess acceptor was replaced by excess donor showed essentially the same time-frame for appearance of full-length products as shown in Figure 4(b) (data not shown).
Figure 4.
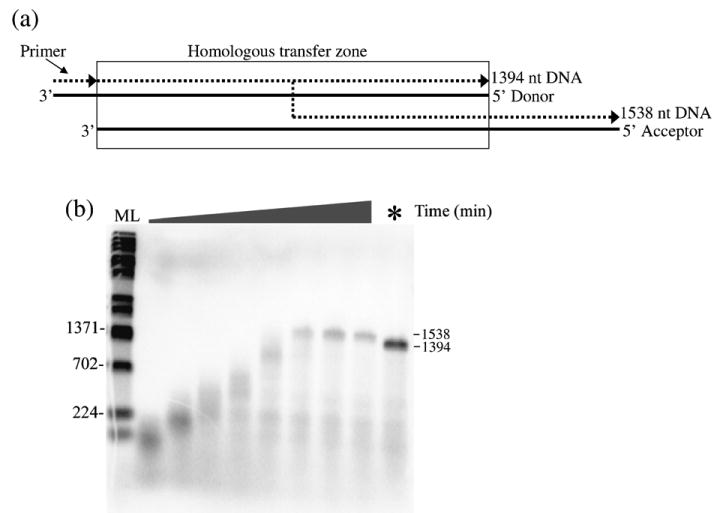
(a) and (b). Time-course strand transfer assay shows that strand transfer is involved in the synthesis of long DNA products. (a) A schematic diagram of the strand transfer assay is shown. The RNA (continuous lines) without the dimer signal was used as the donor and was hybridized to a 20 nt 5′ 32P-labeled DNA (broken lines) primer. A longer RNA without the binding site for the donor primer was used as acceptor. Full extension on the donor produced a 1394 nt DNA while transfer and subsequent extension on the acceptor produced a 1538 nt product. The region of homology (homologous transfer zone) between the donor and acceptor is boxed. (b) An autoradiogram of a strand transfer experiment. The reactions were carried out in the presence of 4 μM NC for time points 2, 5, 10, 15, 30, 45, 60 and 75 min as shown from left to right. In the last lane (*), the reaction was carried out by adding excess donor RNA to the reaction instead of acceptor RNA and this reaction was carried out for 75 min. The positions of full-length DNA products from the donor (1394) or transfer products (1538) are indicated. Other markings are as in Figure 1(b). A repeat experiment yielded similar results.
Determination of the rate of strand transfer
To determine the rate of strand transfer during the synthesis of long DNA products, an acceptor template with several mutations was made. The acceptor in this case lacked the 20 base primer binding site present on the 3′ end of the donor RNA and contained 5 nt changes, the first being 268 nt from the 3′ end and the other four spaced at 250 nt intervals (see Figure 5). Donor RNA was prehybridized to the primer at a 1:1 ratio. Fourfold excess acceptor RNA (16 nM acceptor, 4 nM donor) was added in reactions with 4 μM NC and 80 nM RT. Long DNA products were isolated, amplified by PCR, and cloned and sequenced as described in Materials and Methods. Transfer between each of the mutations could be easily monitored because the products would contain the bases from the donor until transfer to the acceptor took place, at which point a base change to one of the mutated bases would occur in the sequence. Therefore, there were five regions within the donor where transfers could be detected (Figure 5). The actual cross-over could have occurred anywhere in the 250 base region (268 for the first region) that preceded the mutation. Of the eight total clones sequenced, six transferred before the first mutation (numbers 1–4 and 7 and 8), while one transferred between the first and second (number 6) and the other between the fourth and fifth (number 5). Several clones also showed jumps back to a donor after jumping to an acceptor. Jumps to the donor should be limited because of the four-fold excess of acceptor in the reactions; however, the effective concentration of the acceptor and donor may be slightly different than the actual concentration ratio of 4:1. It is also reasonable to assume if jumping back to the donor occurred that jumps to other acceptors were also frequent, though these cannot be detected. Since the initial jump in six of the eight clones occurred before the first mutation, which was 268 nucleotides from the 3′ end of the acceptor, the transfer rate was clearly high with an average rate of at least one jump per 300 nucleotides synthesized. It is not possible to estimate the exact rate from this experiment because the jumps could have occurred anywhere within the 268 nt region and more mutations would be required for an accurate determination. The possibility of the results being skewed by a transfer “hotspot” in the first region seem unlikely, since the tendency to jump back to the donor suggests that the rate of transfer was high throughout the template. Overall the experiment showed that most long products are synthesized as a result of several strand transfer events.
Figure 5.
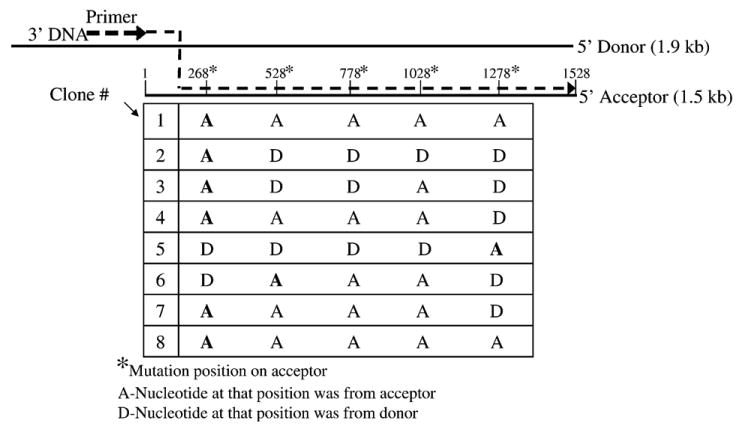
Analysis of transfer products in reactions with mutated acceptor template indicates that transfer occurs frequently during synthesis of long products. The Figure denotes the composition of the DNA products in reactions with the 1.9 kb donor and an acceptor template that was mutated at five positions (as indicated by asterisks) so that cross-over regions could be determined. The acceptor did not contain the primer binding site so all reactions initiated on the donor. Eight full-length clones were sequenced and are numbered 1–8 at the left (see Materials and Methods). Labels A or D below each mutated base position indicate whether that base was derived from the donor (D) or acceptor (A) for a given clone. Since all DNA extension started on the donor, the first A that appears on the clone indicates that a cross-over occurred somewhere in the preceding region. These positions are in bold type for each clone. In some cases products switched back to the donor after transferring to the acceptor. The depiction of strand transfer (dotted line) in the schematic at the top of the Figure illustrates a jump before the first mutation which occurred in six of the eight clones.
Long DNA products in vitro are synthesized in a high molecular mass aggregate that forms rapidly in the presence of NC and contains NC, RT, primer, and RNA
Stable HIV-1 nucleocapsid complexes can be generated in vitro when NC protein is added to RNA.40,42,43 The complexes were large and could be pelleted by slow speed centrifugation and were found to be competent for DNA synthesis.40 To test for the formation of aggregates in the reactions that produce long DNAs, the assay was performed using the 1.9 kb RNA as template in the presence or absence of NC. After 1 h the material was centrifuged for 1 min at 12,000g in a microfuge. The pellet and supernatant fractions were then run on a 1% alkaline agarose gel. The pellet fraction of the reactions with NC had the full-length DNA while the supernatant fraction showed no products (see Figure 6, 1 h reactions). All products in reactions without NC were shorter and contained mostly in the supernatant. The results suggest that high molecular mass aggregates are formed during synthesis of long DNA products.
Figure 6.
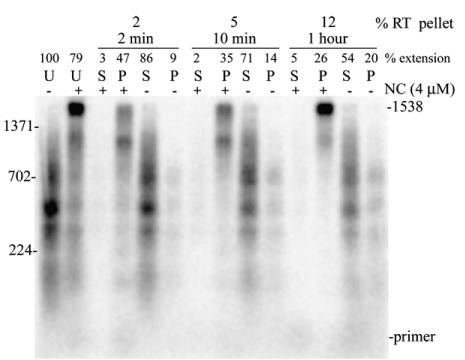
Reverse transcription assay demonstrating that high molecular mass aggregates promote synthesis of long DNA in vitro. (a) The 1.9 kb RNA was used as template. Lanes with reactions with NC (4 μM) are denoted as + and those without NC as −. U denotes lanes with uncentrifuged control reactions. 2 min, 10 min and 1 h denote the time at which reactions were centrifuged after the start of reverse transcription. Pellet (P) and supernatant (S) fractions were obtained after centrifuging reactions for 1 min at 12,000 centrifuge. The pellet and supernatant fractions obtained after centrifugation at 2 and 10 min were further subjected to reverse transcription as described in Materials and Methods. The percentage of RT in the pellet at 2 min, 10 min and 1 h of reverse transcription are indicated. Other markings were as in Figure 1(b). The experiment was repeated twice and yielded similar results.
NC is known to form precipitates at high concentrations so the high molecular mass pellet fractions could be a result of NC precipitation rather than a discrete high molecular mass functional aggregate. In order to determine whether the aggregates were functional and promoted synthesis of long DNA, the reactions were centrifuged early (2 and 10 min) after the start of reverse transcription and pellet and supernatant fractions were incubated for 58 and 50 min, respectively, at 37 °C. In these experiments the pellet was resuspended in the original reaction buffer containing divalent cation and dNTPs but no additional RT. The addition of dNTPs and divalent cation was required for efficient synthesis in the pellet fractions, indicating that substantial quantities of these components do not pellet with the aggregates. In each case, the pellet fractions of the reactions with NC had the larger products and the supernatant fractions showed just a small amount of DNA products. Addition of RT, dNTPs, or divalent cation to the supernatants did not improve synthesis. The low DNA synthesis in the supernatant can be explained by the low level of template in this fraction (see below). The results show that aggregates are formed early in the reactions and that long DNA products are formed in the aggregate fraction after extended incubation with RT. This suggests that aggregates play an important role in long DNA synthesis in vitro. Activity analysis indicated that most of the RT remained in the supernatant even after 1 h of reverse transcription (Figure 6(a)). This could explain the smaller amount of full-length products in the 2 and 10 versus 1 h pellet fractions. To determine if functional aggregates could form in the absence of RT and if exogenously added RT could access the aggregate, reactions were pre-incubated with 4 μM NC as described in Materials and Methods but for 10 min instead of 5. The material was then centrifuged for 1 min at 12,000g. The pellet was resuspended in reaction buffer containing dNTPs and divalent cation then RT was added and incubation was continued for 1 h. Full-length products similar to those observed in the 1 h pellet in Figure 6 were observed (data not shown). This indicated that RT was able to access preformed aggregates but was not required to form functional aggregates.
The constituents of the pellet fraction for the 1.9 kb template were analyzed using different levels of template RNA. The amount of fully extended products and total primer extension were increased in the presence of excess template (Figure 7). In the experiment shown, at 1:1 template:primer (4 nM each) only a small amount of full-length product was observed and only 20% of the primer extension level of reaction without NC was observed. The amount of primer extension and full-length products both increased with 34, 41, 51 and 70% of −NC reactions observed with 2:1, 3:1, 4:1 and 8:1 template:primer, respectively. This showed that the apparent inhibition of primer extension in the presence of NC can be partially overcome by adding more template. At a 1:1 concentration of primer: template (4 nM each), most of the primer and RT was in the supernatant (Figure 7) and increasing the template concentration pulled down more primer and slightly more RT into the pellet. In contrast to RT and primer, most of the template was in the pellet fraction at all template concentrations. Analysis of the pellet constituents in the NC titration assay shown in Figure 1(b) (see indicated numbers above lanes) showed that long product synthesis correlated with the presence of primer and template in the pellet fraction, which occurred at NC concentrations ≥ 2 μM.
Processivity of RT has no role in the synthesis of long DNA products in vitro
NC has been shown to have a modest effect on the processivity of RT.18,19 This is generally attributed to melting by NC of some secondary structures in the template. Above we showed that strand transfer plays a major role in producing long products, however, an increase in processivity could also have contributed. To test this, the 1.9 kb RNA template was used and the reverse transcription assay was carried out in the absence or presence of poly(rA)-oligo(dT) trap. The trap sequesters enzyme molecules that dissociate from the substrate thereby limiting synthesis to a single binding event between the enzyme and the substrate.45 To test the effectiveness of the trap, it was added to the reaction before the enzyme and incubation was continued for 1 h. A low level of small extension products were observed in reactions without NC while no products were observed with NC, indicating that the trap sequestered RT over the entire reaction (Figure 8, lanes marked as C). Assays performed in the absence of NC with trap showed products up to about 800 nt in length. Several dilutions of these reactions (lanes shown as −NC, +trap) are shown to make it easier to compare them to the reactions with NC (lane+NC, +trap). Due to the inhibition by NC of primer extension (described above), several reactions were combined for the sample shown in lane +NC, +trap (see Materials and Methods). No notable increase in the average length of products was evident in the reactions with NC. This indicates that there was no significant increase in processivity in the presence of NC and this was not a factor in production of the long DNA synthesis products.
NC finger mutants lacking either finger 1 or 2 or switching their positions also stimulate the synthesis of long DNA products in vitro
HIV NC has two non-identical zinc fingers, an N and a C-terminal finger, denoted 1 and 2, respectively. Three NC mutants, 1.1 NC, 2.2 NC and 2.1 NC were used. In mutant 1.1, finger 1 replaces finger 2 giving the protein two copies of finger 1. In 2.2, finger 2 replaces finger 1 giving this protein two copies of finger 2 and 2.1 NC is a finger switch mutant in which the positions of the zinc fingers are switched. It was previously reported that the N and C-terminal zinc fingers of NC are not biologically equivalent.46–48 Previous work from several laboratories has shown that the two fingers possess different functional activities with finger 1 being more important for helix-destabilizing activity than finger 2.47,48 Results showed that 1.1 and 2.1 retained helix-destabilizing activity while this activity was significantly reduced in 2.2. In contrast, all the mutants were able to stimulate the annealing of non-structured complements, suggesting that they retained “aggregating/condensing” activity that is required to bring nucleic acids into close proximity.47,49 To determine if the finger mutants could stimulate the production of long DNAs, an NC titration was performed as described for wild-type NC using the 1.9 kb RNA as template, and the DNA products were run on a 1% alkaline agarose gel (Figure 9). All three NC finger mutants increased the proportion of full-length DNA products similar to wild-type NC. These results are consistent with data from the previous report using the 874 nt template.39 Since results with 2.2 were similar to those with wild-type, this suggests that the full helix destabilizing activity of NC is not required for the synthesis of long DNAs in vitro.
Figure 9.
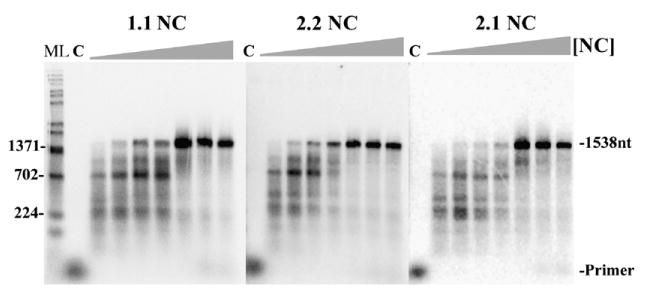
Reverse transcription assay with increasing concentrations of NC finger mutants showing that mutants with low helix-destabilizing activity can still promote long product formation. Shown are autoradiograms of assays with 1.9 kb RNA template with increasing concentrations (left to right, 0, 0.5, 1, 2, 4, 6, 8 μM) of NC finger mutants 2.2, 1.1 and 2.1 (as indicated). Other markings are as in Figure 1(b). See Results for description of NC mutants. A repeat experiment yielded similar results.
Discussion
Here in vitro reverse transcription reactions capable of efficiently producing single-stranded DNA up to 4 kb from genomic RNA of HIV were characterized. The reactions required large amounts of RT (at least 80 nM) and NC (Figures 1(b) and 2). Long products were synthesized in large aggregates that could be pelleted by slow speed centrifugation (Figure 6). The aggregates were functional and synthesized long DNAs when supplemented with dNTPs and Mg2+. They were consistent with previously reported NC aggregates that were able to carry out reverse transcription (see Introduction), but were not examined for the ability to produce long DNAs.
NC clearly increased the proportion of full-length DNA products (Figures 1(b) and 2). This effect of NC is consistent with data from a previous report using a smaller (874 nt) template,39 and was observed only at high NC concentrations, i.e. 2–8 μM for the 1.9 kb RNA. At these concentrations there is enough NC to completely coat all the nucleic acids in the reaction at approximately one NC per 7 nt. This is typically observed in the HIV virion with the ribonucleoprotein complex consisting of the dimeric RNA genome in association with 2000 to 3000 molecules of NC protein.50 However, it was not clear from our results that templates were completely coated or that this was necessarily required for long products to form. In fact the high MgCl2 concentration (6 mM) used would tend to decrease NC binding and it is unlikely that full saturation was achieved under standard reaction conditions.51 Lowering the concentration to levels presumably more consistent with the free Mg2+ in cells (1 mM or less52) resulted in less long products, probably due to slowing the polymerization or strand transfer rate, and did not improve the amount of total primer extended even though NC binding would be greater under these conditions (data not shown). Even with 6 mM Mg2+, with 4 μM NC nearly all the template was found in aggregates whether 4 nM or 32 nM template was used (Figure 7, 1:1 and 8:1 template:primer). At the 32 nM concentration the nucleotide:NC ratio is about 15:1, so even if 100% of the NC was bound to RNA only about 50% coating would have been achieved. Therefore complete coating of the RNA is not required for aggregate formation. The results also suggest that long product formation is more dependent on the amount of template and primer in aggregates than the extent of RNA coating. This was evident from Figure 7, where increasing the [RNA] template at a fixed [NC], which would decrease coating under the conditions used, increased the proportion of primers in the aggregate and substantially increased full-length products.
Inhibition of total DNA synthesis was also observed with high NC concentrations, again consistent with the previous report.39 In our experiments this inhibition was less dramatic and could be partly overcome by the addition of excess template when the 1.9 kb template was used. Part of the inhibition clearly resulted from a partitioning of the primer and template (Figure 7). At low template concentrations (1:1 and 2:1 template:primer) about half of the primer fractionated with the supernatant while over 80% of the template was in the pellet. This is consistent with longer nucleic acids being aggregated more efficiently by NC than very shorter oligonucleotides.42 Since essentially all the primer is extended in reactions without NC (Figures 1(b), 7 and 8)), then presumably it is all bound to the template after hybridization. Therefore the partitioning observed in the presence of NC must involve a portion of the primer dissociating from the template. This could be caused by NC’s helix-destabilizing activity, however, NC mutant 2.2 in which this activity is strongly reduced behaved essentially like wild-type in the reactions (Figure 9). Another possibility is that displacement from the template is proportional to the level of NC coating, with less primer displacement occurring as coating decreases. This would be consistent with more primer partitioning with the aggregate fraction when the level of template was increased in the reactions (Figure 7), and also with too much NC inhibiting extension (Figure 1(b), 6 and 8 μM NC). Other experiments also showed that using a 50 rather than 20 nt primer did modestly improve primer utilization (data not shown). This could result from the longer primer being more difficult to displace and more easily aggregated by NC. The interplay between the amount of NC, RT, and primer-template appears quite complex, with too little NC likely resulting in poor aggregate formation and too much decreasing primer extension. We are currently continuing work to understand how aggregate formation leads to primer segregation and inhibition of extension.
Although NC clearly enhanced the production of longer products with the 4 kb template, a lower proportion of fully extended products was observed (Figure 2 and data not shown), while attempts to synthesize even longer products (7 kb) failed. The 4 kb template is essentially a longer version of the 1.9 kb template that extends further toward the 3′ end of the HIV genome. The reason for the apparent decrease in efficiency with the longer templates was not clear but could have resulted from several factors. First, the efficiency of synthesizing full-length RNA transcripts in vitro decreases with increasing size.53 Therefore for the 4 and 7 kb transcripts a larger proportion of the RNA likely consists of incompletely synthesized strands lacking the primer binding site. Although formaldehyde gel analysis indicated that much of the 4 kb preparations consisted of long RNAs, this technique cannot resolve small differences in RNA length so it was unclear if the large RNAs were all completely synthesized. How this would affect the reaction is unclear. Also, these reactions required long incubation times (2–3 h to observed fully extended products) and the activities of RT and NC may have been decreasing, although adding additional RT to the reactions after 1.5 h did not affect product production. Finally, there may simply be a limit to the size of the products that can be efficiently produced in the aggregates.
Synthesis of long DNA products in vitro did not seem to require NC’s full helix destabilizing activity as a mutant NC with very low activity (NC 2.2, Figure 9) was as effective as wild-type in the assays. This suggests that the aggregation/condensation activity of NC may be more pivotal for synthesis of long DNA products in vitro. This activity is likely responsible for the large aggregates observed in reactions with NC. Although we do not have definitive evidence showing that long products can only be synthesized in aggregates, that conclusion would be consistent with the results. Aggregated complexes isolated just 2 min into the reactions were capable of long DNA synthesis and long products were only associated with material in the reactions that pelleted with slow speed centrifugation (Figure 6).
One possible explanation for the large products made in aggregates is that RNA molecules would be in close proximity to each other, thereby allowing strand transfers to occur more easily. This could further be facilitated by the high local concentrations of NC and RT in the complexes. Tanchou et al.40 have shown that strand transfer occurs within HIV-1 nucleoprotein complexes in vitro. Further, time-course experiments using donor and acceptor RNAs showed that the long DNA products resulted from internal strand transfer events (Figure 4), and other experiments showed that a change in processivity was not a factor (Figure 8). With respect to the latter, it is important to note that the assays were conducted with poly(rA)-oligo(dT) trap and it is not clear how this may have affected synthesis of long DNAs in aggregates. It is possible that some factor required for long DNA production was altered by the trap and therefore enhanced processivity cannot be completely ruled out. However, overall the results are more supportive of a strand transfer mechanism.
This is further supported by the high rate of strand transfer determined using the assay with mutated acceptor template (Figure 5). A rate of at least one transfer per 300 nt was calculated. This rate was a rough approximation for reasons stated in Results. With respect to the recombination rates estimated in cell infections the rates in the in vitro system were comparable to rates calculated in macrophages (about 30 recombinations over the approximately 9000 base genome or one per 300 bases) but considerably higher than those in T cells (about 1 in 1000). Rates in vitro may be greater because each aggregate probably contains several hundred or more templates (based on the aggregates centrifuging in the microfuge in less than 1 min) where viruses only have two genome copies.
The finding that the 2.2, 1.1 and 2.1 NC finger mutants were effective in the assays is not inconsistent with strand transfer, as all the mutants were able to stimulate transfer in assays with different regions of the HIV genome.54 In those experiments stimulation was dependent on the particular genome region used and 2.2 was somewhat less effective with highly structured regions, a finding consistent with this mutant being highly defective in catalyzing transfer on substrates with the highly structured TAR genome region.48
In vitro reverse transcription reactions have been critical in understanding the process of reverse transcription. Despite this reactions that authentically mimic the cellular process with respect to producing long DNA products have not been described. Here we show that long products can be efficiently produced under the right conditions in NC-induced aggregates. In the virion, NC and RTare condensed within the virus core and present at millimolar concentrations.55 Limitations in protein and nucleic acid solubility make it impossible to reproduce these concentrations in the test tube. However, aggregates with high concentrations of NC, RT and RNA can clearly form in vitro. The fact that they can efficiently produce long DNA products suggests that the condensing effect of the virion environment may be a key for efficient reverse transcription, and cellular factors may not be required. Still, the system described here had limitations and we were unable to efficiently produce genome-length products. In this regard the system resembled ERT and NERT reactions, which, although synthesizing some full-length minus sense DNAs, produce mostly uncompleted products (see Introduction). It remains to be determined whether the inability to produce genome length products in vitro was due to technical limitations associated with making and maintaining long RNAs in vitro, structural differences between the aggregates and cellular complexes, or the absence in the system of cellular or other viral components. Still, the aggregate systems appear to more closely represent what is presumably occurring during cellular reverse transcription, and future analysis should lead to a better understanding of how replication occurs during viral infection.
As a final note, it may also be possible to use these types of systems to improve the synthesis of cDNA from RNA. This would depend on the source of the RNA. For example it probably would not be particularly useful for producing cDNAs from pooled cellular mRNA. Because such a pool would have several different mRNAs with unique sequences, a mechanism dependent on homologous strand transfer would be unlikely to improve synthesis, especially for those mRNAs not present at high concentrations. However, for those cases where a single RNA species is being used (for example the RNA genome from a particular virus), a strand transfer mechanism could allow efficient synthesis of longer RNA segments. This could abrogate the need for cloning long genomes by combining smaller segments.
Materials and Methods
Materials
Plasmid pBKBH10S was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, from Dr John Rossi. This plasmid contains an 8.9 kb SstI fragment (nt 222–9154 of the RNA genome) from HIV-1 BH10 inserted into the SstI site. The fragment has all HIV-1 gene coding regions but does not contain the HIV-1 LTR.56 PCR primers and primers used to prime templates in reverse transcription assays were obtained from Integrated DNA Technologies, Inc. The HIV-RT clone was a generous gift from Dr Samuel H. Wilson (National Institute of Environmental Health Sciences, Research Triangle Park, NC). HIV-RT was purified as described.57 The protein was purified to homogeneity and the purity of the protein was evaluated using Coomassie Blue staining of 10% (w/v) SDS-PAGE gels.58 The subunits p51 and p66 of RT were in a 1:1 ratio (data not shown). Aliquots of HIV RT were stored frozen at −80 °C and fresh aliquots were used for each experiment. The HIV NC clone was a generous gift from Dr Charles McHenry (University of Colorado). NC was purified to apparent homogeneity (as judged from Coomassie Blue staining of 17.5% SDS-PAGE gels58 according to the protocol described).36 Quantification was by absorbance at 280 nm using a molar extinction coefficient of 8350 cm−1 M−1.36 Aliquots of NC were stored frozen at −80 °C, and fresh aliquots were used for each experiment. NC finger mutants 1.1, 2.2 and 2.1 were a gift from Dr Robert Gorelick (SAIC, Frederick, MD). These proteins were expressed and purified as described,59 and quantified by amino acid analysis on a Beckman Systems 6300 amino acid analyzer (Beckman Coulter, Inc., Full-erton, CA). Taq polymerase was from Eppendorf. T7 RNA polymerase, SP6 RNA polymerase, DNase I-RNase-free, and RNase-DNase-free were from Roche Diagnostics. RNase inhibitor was from Promega. T4 polynucleotide kinase and restriction enzymes EcoRI and HincII were obtained from New England Biolabs. Proteinase K was obtained from Eastman Kodak Co. Radiolabeled compounds were obtained from Amersham. Sephadex G-25 spin columns were from Amika Corp. RNA cleanup kit was from Qiagen and Mini-prep kits were from Qiagen. GeneTailor site-directed mutagenesis kit, Topo TA cloning kit and high fidelity Platinum Taq DNA polymerase were obtained from Invitrogen. All other chemicals were from Sigma or Fisher Scientific.
Site-directed mutagenesis of acceptor RNA templates for strand transfer rate determination experiments (see Figure 5)
The mutant primers listed in Table 1 were used to introduce approximately equally spaced mutations into the region from position 1 to 1518 (genome bases 222–1739 based on HXB-2 numbering of genomic RNA) of the HIV insert in pBKBH10S plasmid. The mutations correspond to positions 250, 500, 750, 1000 and 1250 from the 5′ end of the HIV insert (see Figure 5). The primers carrying each mutation were extended during temperature cycling by high fidelity Platimum Taq DNA polymerase obtained from Invitrogen (as per manufacturer’s protocol). After temperature cycling, the product with the desired mutation was transformed into MAX Efficiency® DH5™-T1R One Shot® chemically competent cells. Mini preps were obtained using Qiagen mini-prep kits. Sequencing was done using primer 5′-GTTCTAGGTGATATGGCCTGATG-3′ to check for mutation incorporation at positions 250 and 500, primer 5′-GACCAACAAGGTTTCTGTCATC-3′ to check for mutation incorporation at positions 750 and 1000 and primer 5′-TCTGGCTGTGTGCCCTTCTTTG-3′ to check for mutation incorporation at position 1250.
Table 1.
Primers for synthesis of mutations in acceptor
| Basea | position on HIV genomeb | Sequence of primers in the 5′ and 3′ orientationsc |
|---|---|---|
| 250 | 471 | 5′-gagctagaacgattcgcagtttatcctggcctgttag-3′
5′-aactgcgaatcgttctagctccctgcttgcccatac-3′ |
| 500 | 721 | 5′-gacacagcagtcaggtcagcctaaattaccctatag-3′
3′-ggctgacctgactgctgtgtcctgtgtcagc-3′ |
| 750 | 971 | 5′-gaggaagctgcagaatgggaaagagtacatccagtg-3′
5′-tcccattctgcagcttcctcattgatggtctc-3′ |
| 1000 | 1221 | 5′-aagaaccttttagagactatctagaccggttctataaaac-3′
5′-atagtctctaaaaggttcttttggtccttgtc-3′ |
| 1250 | 1471 | 5′-cagctaccataatgatgcagacaggcaattttaggaac-3′
5′-tctgcatcattatggtagctgtatttgttacttg-3′ |
The base number refers to the position of the nt from the 5′ end of the RNA transcript.
This number refers to the position of the nt from the 5′ end of the HIV genome (strain HXB2).
Primer pairs that are complementary to opposite strands of pBKBH10S plasmid. One of the primers in each primer pair has the desired mutation indicated by underlined bold letters.
PCR amplification of DNA substrates for donor RNA (for time-course reactions (Figure 4)) and for acceptor RNA (for strand transfer rate determination (mutated acceptor) and time-course (non-mutated version))
Two PCR primers, 5′-GATTTAGGTGACACTATAGGAATTAGATCGATGGGAAAA-3′ and 5′-CTGAAGCTCTCTTCTGGTGG-3′ were designed to yield donor RNA to be used in strand transfer time-course reactions (see below) and amplified DNA from position 145 to 1538 on the HIV insert of pBKBH10S (genome bases 366–1759 based on HXB-2 numbering of genomic RNA). Also, two PCR primers, 5′-GATTTAGGTGACACTATAGAGCTCTCTCGACGCAGGACT-3′ and 5′-GGCTGTTGGCTCTGGTCTGC-3′ were designed to yield RNA templates to be used as acceptor RNA for the strand transfer reactions (strand transfer time-course and determining rate of transfer) and amplified DNA from position 1 to 1518 (genome bases 222–1739) of pBKBH10S. Two versions of the acceptor were used, with one having the five mutations introduced by site-directed mutagenesis (see above). The acceptor RNA lacks the primer binding site present on the above donor. An SP6 promoter sequence was included on one primer in each primer pair to allow transcription of the DNA by SP6 RNA polymerase. PCR reactions were performed with Taq polymerase according to the enzyme manufacturer’s protocol using the provided buffer. One hundred pmol of each primer was used. Reactions included 30 cycles of denaturation, annealing and extension at temperatures of 94 °C for 1 min, 50 °C for 1 min and 72 °C for 2 min, respectively, followed by one cycle of extension at 72 °C for 5 min. The PCR products were run on a 1% (w/v) agarose gel, extracted into a dialysis tube and purified as described,58 and used to prepare RNA as described below.
Preparation of RNA substrates
Five different RNAs were used in the experiments, with two being derived directly from plasmid DNA and three from PCR products. RNAs of approximately 1.9 and 4 kb were made by first digesting pBKBH10S with restriction enzymes HincII and EcoRI, respectively. The digests were then extracted with phenol:chloroform:isoamyl alcohol (25:24:1, by vol.) and precipitated with ethanol. Run-off transcription (performed according to the enzyme manufacturer’s protocol) was then conducted using 5 μg of the digest plasmid and T7 RNA polymerase enzyme to generate 1.9 and 4 kb RNAs. Run-off transcription was also performed using ~5 μg of purified PCR DNAs described above and SP6 RNA polymerase to generate a 1.4 (donor RNA for time-course experiments (Figure 4)) and 1.5 kb acceptor RNA with (for strand transfer rate determinations (Figure 5)) or without (for time-course (Figure 4)) mutations. All transcription reactions were treated with 2 μl of ten units/μl of DNase I-RNase-free enzyme for 15 min to digest the template DNA. The RNA was purified using a Qiagen RNA cleanup kit. The amount of recovered RNA was determined spectrophotometrically from optical density. The RNA preparations were checked by formaldehyde-agarose gel electrophoresis. Most of the observed products ran at a position consistent with the expected size of full-length RNA transcript although some faster migrating small products were also observed. These may have resulted from incomplete transcription or a low level of breakdown.
Preparation of 1.5 kb single-stranded DNA by asymmetric PCR
Two PCR primers, 5′-GATTTAGGTGACACTATAGAGCTCTCTCGACGCAGGACT-3′ and 5′-CTGAAGCTCTCTTCTGGTGG-3′ were used at 100 pmol and 1 pmol, respectively, in PCR reactions performed with Taq polymerase and 0.1 μg of plasmid pBKBH10S according to the enzyme manufacturer’s protocol using the provided buffer. Reactions included 50 cycles of denaturation, annealing and extension at temperatures of 94 °C for 1 min, 50 °C for 1 min and 72 °C for 2 min, respectively followed by one cycle of extension at 72 °C for 5 min. The PCR product, which is single-stranded plus-strand DNA, was run on a 1% agarose gel, extracted by dialysis and purified as described.58 The amount of recovered DNA was determined spectrophotometrically from optical density.
RNA-DNA hybridization
DNA primers that bound specifically to the RNA templates: 5′-CTGAAGCTCTCTTCTGGTGG-3′ to the 1.9 kb and 1.4 kb templates and 5′-GCTTGATTCCCGCCCACCAA-3′ to the 4 kb template were 32P-labeled at the 5′-end with T4 polynucleotide kinase according to the manufacturer’s protocol. Each of the RNA templates was hybridized to the complementary labeled primer by mixing primer:transcript at a ~1:1 ratio in 50 mM Tris-HCl (pH 8.0), 1 mM dithiothreitol, 80 mM KCl. The mixture was heated to 70 °C for 5 min and then slowly cooled to room temperature.
Reverse transcription reactions with or without NC
RNA template-DNA primer hybrids (4 nM final concentration of RNA) were pre-incubated for 5 min along with additional template RNA (12 nM) in the presence or absence of NC (4 μM) in 21 μl of buffer (see below) at 37 °C. The reactions were initiated by addition of 4 μl of HIV-RT (80 nM final in reactions). The following reagents at the indicated final concentrations were also included in the reaction mixtures: 50 mM Tris-HCl (pH 8.0), 1 mM dithiothreitol, 80 mM KCl, 6 mM MgCl2, 100 μM dNTPs, 5 mM AMP (pH 7.0), 25 μM ZnCl2 and 0.2 units/μl RNase inhibitor. Reactions were allowed to incubate for 75 min. In some reactions the amounts of excess template, RT, or NC were varied as indicated. The reactions were stopped by adding 2 μl of a solution containing 200 mM EDTA (pH 8.0) and 5 ng of RNase-DNase-free enzyme and allowed to digest for 20 min at 37 °C. Nine μl of proteinase K at 2 mg/ml in 1.25% (w/v) SDS, 15 mM EDTA (pH 8.0), 10 mM Tris-HCl (pH 8.0) was then added to the above mixture, which was placed at 65 °C for 1 h. Finally 7 μl of 6× alkaline dye (300 mM NaOH, 6 mM EDTA, 15% (v/v) glycerol, 0.15% (w/v) bromophenol blue) was added to the mixture and the samples were resolved on 1% alkaline agarose gel containing 50 mM NaOH and 1 mM EDTA (pH 8). Similar reactions were conducted during the analysis of mutant NC proteins (1.1, 2.2 and 2.1). Time-course experiments with the 4 kb RNA template were done in the presence of 8 μM NC and reactions were stopped at time points −0.5, 1, 2 and 3 h. For all experiments extended DNA products were observed using a Bio-Rad Molecular Imager FX. The amount of product formed was quantified by phosphor-imager analysis. The amount of total primer extension without NC was taken as 100% extension and the percentage of extension at different NC concentrations was calculated as percentage of the primer extension without NC.
Strand transfer time-course reaction
The 1.4 kb RNA template, hybridized to DNA primer as described above (4 nM final concentration of RNA in reaction) were pre-incubated for 5 min along with non-mutated 1.5 kb acceptor RNA (12 nM) in the presence of NC (4 μM) in 21 μl of buffer (as in reverse transcription experiment) at 37 °C. The reactions were initiated by addition of 4 μl of HIV-RT (80 nM final in reactions) and stopped as described above at time points 2, 5, 10, 15, 30, 45, 60 and 75 min and samples were resolved on 1% alkaline agarose gel containing 50 mM NaOH and 1 mM EDTA (pH 8).
Determining the rate of strand transfer during synthesis of long DNA products in vitro
The 1.9 kb RNA template (donor) was hybridized to DNA primer as described above (4 nM final concentration of RNA in reaction) and pre-incubated for 5 min along with 1.5 kb mutated acceptor RNA (16 nM) in the presence of NC (4 μM) in 21 μl of buffer (as in reverse transcription experiment) at 37 °C. Assay conditions were as described above except that 50 μl reactions were performed. The reactions were initiated by addition of 8 μl of HIV-RT (80 nM final in reactions) and stopped as described above after 75 min. Reactions were processed, and then electrophoresed on 5% polyacrylamide denaturing gels. Full-length DNA products were located by autoradiography, excised, and eluted overnight in a TE buffer (10 mM Tris-HCL (pH 8.0), 1 mM EDTA). The eluate was separated from the gel by centrifugation and subsequent filtration through a 0.45 micron disposable syringe filter. The DNAs were recovered by precipitation in ethanol with 300 mM sodium acetate. The recovered DNA was amplified by PCR. Approximately 0.2 fmol (as judged by counts per minute (cpm) of the samples detected using a scintillation counter) of full-length DNA were amplified by PCR. Primers, 5′-GATTTAGGTGACACTATAGAGCTCTCTCGACGCAGGACT-3′ and 5′-CTGAAGCTCTCTTCTGGTGG-3′ were used to PCR amplify the strand transfer samples. PCR reactions were performed with Taq polymerase according to the enzyme manufacturer’s protocol using the provided buffer. Thirty-two pmol of each primer was used. Reactions included 25 cycles of denaturation, annealing and extension at temperatures of 94 °C for 1 min, 50 °C for 1 min and 72 °C for 3 min, respectively, followed by one cycle of extension at 72 °C for 5 min. The PCR products were run on a 1% agarose gel, extracted by dialysis and purified as described.58 The purified PCR products were ligated into Topo vector (Invitrogen), which was used to transform Top10_ Escherichia coli competent cells (as per manufacturer’s protocol). Only white colonies were picked. Minipreps were prepared using a Qiagen miniprep kit, and each clone was sequenced using three primers: M13 reverse primer, T7 promoter primer and 5′-GACCAACAAGGTTTCTGTCATC-3′. Controls conducted to test for recombination resulting from the PCR reactions showed a low level of PCR-derived recombination (approximately one transfer per 1700 nt synthesized) that was insignificant in comparison to the rate for strand transfer reactions (approximately one transfer per 300 nt synthesized (see Results)) (data not shown).
Experiments testing aggregate formation
Reactions were set up and initiated similar to the reverse transcription experiment described above except that the final reaction volume was 50 μl. As indicated in Figure 6, the reactions were centrifuged for 1 min at 12,000g at the end of 2, 10 or 60 min after initiating with RT. The pellet and the supernatant fractions from the 2 and 10 min samples were then subjected to reverse transcription (58 and 50 min, respectively) after adding back the reverse transcription buffer (minus RT) described above to the pellet fractions only. Pellet and supernatant fractions from all reactions were stopped and treated with proteinase K and samples were resolved on alkaline agarose gels as described above.
Experiments testing proportion of primer, template and RT in the aggregate
To test the proportion of primer in pellet, reactions were set up similar to the reverse transcription experiment described above, i.e. RNA template was hybridized to DNA primer (4 nM final concentration of RNA in reaction) pre-incubated for 5 min along with excess RNA (8, 16 and 32 nM) in the presence of NC (4 μM) in 50 μl of buffer (as in reverse transcription experiment) at 37 °C. The final reaction volume was 50 μl and RT was not added to initiate the reaction. The reactions were centrifuged for 1 min at 12,000 g at the end of 10 min of pre-incubation with NC. Pellet and supernatant fractions from all reactions were treated with proteinase K and samples were resolved on 8% polyacrylamide gels. The radiolabeled primers in each fraction were observed and quantified using a Bio-Rad Molecular Imager FX. To test the proportion of RNA template in pellet, the RNA was labeled with [32P] CTP during transcription and hybridized to DNA primer (4 nM final concentration of RNA in reaction) and was pre-incubated for 5 min along with excess labeled RNA (8, 16 and 32 nM) in the presence of NC (4 μM) in 50 μl of buffer (as in reverse transcription experiment) at 37 °C. As in the above experiment, the final reaction volume was 50 μl and RT was not added to initiate the reaction. The reactions were centrifuged for 1 min at 12,000g at the end of 10 min of pre-incubation with NC. The amount of radiolabeled template in the pellet and supernatant fractions from all reactions was measured using a scintillation counter. To test the proportion of RT in pellet, reactions were set up and initiated similar to the experiment testing aggregate formation described above except that the DNA was not radiolabeled. The reactions were centrifuged for 1 min at 12,000 g at the end of 2, 10 or 60 min after initiating with RT. The pellet and the supernatant fractions were separated and the reverse transcription buffer was added back to the pellet fractions only. The amount of RT present in the pellet and supernatant fractions of each reaction was determined by measuring incorporation of [3H]TTP on poly(rA)-oligo (dT). Reactions (30 μl) included 33 mM Tris-HCl (pH 8), 50 mM KCl, 1.3 mM DTT, 3.3 mM MgCl2, 0.11 mg/ml poly(rA)-oligo(dT20) (8:1 w:w), and 5 μM TTP (spec. act. approximately 10 Ci/mmol). The amount of RT added from samples was adjusted to keep the reactions in the linear range. Incubations were for 20 min at 37 °C and samples were stopped with 1 μl of 250 mM EDTA (pH 8) then spotted on DE-81 filter disks (Whatman). Dried disks were washed three times for approximately 10 min each with 0.5 M NaPO4 (pH 7) and once with 80%(v/v) ethanol, then dried and counted in a scintillation counter.
Experiments testing processivity of HIV-RT
RNA template (1.9 kb)-DNA primer hybrids (4 nM final concentration of RNA) were pre-incubated for 5 min at 37 °C with threefold excess (12 nM final concentration) RNA template and 4 μM final concentration of NC in 17 μl of buffer containing 50 mM Tris-HCl (pH 8.0), 1 mM dithiothreitol, 80 mM KCl, 5 mM AMP (pH 7.0), 25 μM ZnCl2 and 0.2 units/μl RNase inhibitor. Four μl of HIV-RT (80 nM final concentration in reactions) was then added and further incubated for 3 min at 37 °C. Reactions were initiated by adding 4 μl of a supplement containing MgCl2 and dNTPs in the above buffer such that the final concentrations were 6 mM and 100 μM, respectively. In the reactions with trap, 5 μg of poly(rA)-oligo(dT)20 (8:1, w/w) was included in the supplement to sequester RT molecules that dissociated from the substrate. In control reactions to test the effectiveness of the trap (see Results), the trap mix was added before adding the enzyme, incubated for 3 min at 37 °C and then the reactions were initiated by adding enzyme. Reactions were allowed to incubate at 37 °C for 1 h. The reactions were stopped and treated with proteinase K as described above. The samples were extracted with phenol:chloroform:isoamyl alcohol (25:24:1, by vol.) and precipitated with ethanol. Due to the inhibition by NC of primer extension (described above), ten reactions with NC in the presence of trap were combined. The samples were resuspended in 5 μl of water and 5 μl of 2× formamide dye (90% formamide, 10 mM EDTA (pH 8.0), 0.1% xylene cyanol, 0.1% bromphenol blue) was added and the samples were resolved on a 5% denaturing polyacrylamide gel containing 7 M urea.
Gel electrophoresis
Alkaline agarose gels (1%) containing 50 mM NaOH and 1 mM EDTA (pH 8), 1% native agarose gels in Tris-Borate-EDTA buffer and denaturing 5% polyacrylamide gels (19:1) (acrylamide:bisacrylamide), containing 7 M urea were prepared and subjected to electrophoresis as described.58
Acknowledgments
We thank the AIDS Research and Reference Reagent Program for plasmid pBKBH10S and Dr Charles McHenry from the University of Colorado for the plasmid clone for wild-type NC. We thank Dr Robert Gorelick from Science Applications International Corp., Frederick, MD for NC mutant proteins. This work was supported by National Institute of General Medicine grant number GM051140.
Abbreviations used
- HIV
human immunodeficiency virus
- RNase H
ribonuclease H
- RT
reverse transcriptase
- NC
nucleocapsid protein
- ERT
endogenous reverse transcription
- NERT
natural endogenous reverse transcription
- kb
kilobase
- nt
nucleotide
Footnotes
Edited by J. Karn
References
- 1.Lori F, di Marzo Veronese F, de Vico AL, Lusso P, Reitz MS, Jr, Gallo RC. Viral DNA carried by human immunodeficiency virus type 1 virions. J Virol. 1992;66:5067–5074. doi: 10.1128/jvi.66.8.5067-5074.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trono D. Partial reverse transcripts in virions from human immunodeficiency and murine leukemia viruses. J Virol. 1992;66:4893–4900. doi: 10.1128/jvi.66.8.4893-4900.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zack JA, Haislip AM, Krogstad P, Chen IS. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J Virol. 1992;66:1717–1725. doi: 10.1128/jvi.66.3.1717-1725.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu J, Cunningham JM. Minus-strand DNA is present within murine type C ecotropic retroviruses prior to infection. J Virol. 1993;67:2385–2388. doi: 10.1128/jvi.67.4.2385-2388.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Bagasra O, Niikura M, Poiesz BJ, Pomerantz RJ. Intravirion reverse transcripts in the peripheral blood plasma on human immunodeficiency virus type 1-infected individuals. J Virol. 1994;68:7591–7597. doi: 10.1128/jvi.68.11.7591-7597.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grewe C, Beck A, Gelderblom HR. HIV: early virus-cell interactions. J Acquir Immune Defic Syndr. 1990;3:965–974. [PubMed] [Google Scholar]
- 7.Negroni M, Buc H. Mechanisms of retroviral recombination. Annu Rev Genet. 2001;35:275–302. doi: 10.1146/annurev.genet.35.102401.090551. [DOI] [PubMed] [Google Scholar]
- 8.Nermut MV, Fassati A. Structural analyses of purified human immunodeficiency virus type 1 intracellular reverse transcription complexes. J Virol. 2003;77:8196–8206. doi: 10.1128/JVI.77.15.8196-8206.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haseltine WA, Kleid DG, Panet A, Rothenberg E, Baltimore D. Ordered transcription of RNA tumor virus genomes. J Mol Biol. 1976;106:109–131. doi: 10.1016/0022-2836(76)90303-x. [DOI] [PubMed] [Google Scholar]
- 10.Rothenberg E, Baltimore D. Synthesis of long, representative DNA copies of the murine RNA tumor virus genome. J Virol. 1975;17:168–174. doi: 10.1128/jvi.17.1.168-174.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothenberg E, Smotkin D, Baltimore D, Weinberg RA. In vitro synthesis of infectious DNA of murine leukaemia virus. Nature. 1977;269:122–126. doi: 10.1038/269122a0. [DOI] [PubMed] [Google Scholar]
- 12.Gilboa E, Goff S, Shields A, Yoshimura F, Mitra S, Baltimore D. In vitro synthesis of a 9 kbp terminally redundant DNA carrying the infectivity of Moloney murine leukemia virus. Cell. 1979;16:863–874. doi: 10.1016/0092-8674(79)90101-6. [DOI] [PubMed] [Google Scholar]
- 13.Mitra SW, Goff S, Gilboa E, Baltimore D. Synthesis of a 600-nucleotide-long plus-strand DNA by virions of Moloney murine leukemia virus. Proc Natl Acad Sci USA. 1979;76:4355–4359. doi: 10.1073/pnas.76.9.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boone LR, Skalka AM. Viral DNA synthesized in vitro by avian retrovirus particles permeabilized with melittin. II Evidence for a strand displacement mechanism in plus-strand synthesis. J Virol. 1981;37:117–126. doi: 10.1128/jvi.37.1.117-126.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Zhang Y, Spicer TP, Abbott LZ, Abbott M, Poiesz BJ. Reverse transcription takes place within extracellular HIV-1 virions: potential biological significance. AIDS Res Hum Retroviruses. 1993;9:1287–1296. doi: 10.1089/aid.1993.9.1287. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Dornadula G, Pomerantz RJ. Endogenous reverse transcription of human immunodeficiency virus type 1 in physiological microenviroments: an important stage for viral infection of nondividing cells. J Virol. 1996;70:2809–2824. doi: 10.1128/jvi.70.5.2809-2824.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H, Dornadula G, Alur P, Laughlin MA, Pomerantz RJ. Amphipathic domains in the C terminus of the transmembrane protein (gp41) permeabilize HIV-1 virions: a molecular mechanism underlying natural endogenous reverse transcription. Proc Natl Acad Sci USA. 1996;93:12519–12524. doi: 10.1073/pnas.93.22.12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji X, Klarmann GJ, Preston BD. Effect of human immunodeficiency virus type 1 (HIV-1) nucleocapsid protein on HIV-1 reverse transcriptase activity in vitro. Biochemistry. 1996;35:132–143. doi: 10.1021/bi951707e. [DOI] [PubMed] [Google Scholar]
- 19.Druillennec S, Caneparo A, de Rocquigny H, Roques BP. Evidence of interactions between the nucleocapsid protein NCp7 and the reverse transcriptase of HIV-1. J Biol Chem. 1999;274:11283–11288. doi: 10.1074/jbc.274.16.11283. [DOI] [PubMed] [Google Scholar]
- 20.Rong L, Liang C, Hsu M, Kleiman L, Petitjean P, de Rocquigny H, et al. Roles of the human immunodeficiency virus type 1 nucleocapsid protein in annealing and initiation versus elongation in reverse transcription of viral negative-strand strong-stop DNA. J Virol. 1998;72:9353–9358. doi: 10.1128/jvi.72.11.9353-9358.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan R, Giedroc DP. Recombinant human immunodeficiency virus type 1 nucleocapsid (NCp7) protein unwinds tRNA. J Biol Chem. 1992;267:6689–6695. [PubMed] [Google Scholar]
- 22.Feng YX, Campbell S, Harvin D, Ehresmann B, Ehresmann C, Rein A. The human immunodeficiency virus type 1 Gag polyprotein has nucleic acid chaperone activity: possible role in dimerization of genomic RNA and placement of tRNA on the primer binding site. J Virol. 1999;73:4251–4256. doi: 10.1128/jvi.73.5.4251-4256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brule F, Marquet R, Rong L, Wainberg MA, Roques BP, Le Grice SF, et al. Structural and functional properties of the HIV-1 RNA-tRNA(Lys)3 primer complex annealed by the nucleocapsid protein: comparison with the heat-annealed complex. Rna. 2002;8:8–15. doi: 10.1017/s1355838202010981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shehu-Xhilaga M, Kraeusslich HG, Pettit S, Swanstrom R, Lee JY, Marshall JA, et al. Proteolytic processing of the p2/nucleocapsid cleavage site is critical for human immunodeficiency virus type 1 RNA dimer maturation. J Virol. 2001;75:9156–9164. doi: 10.1128/JVI.75.19.9156-9164.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez-Rodriguez L, Tsuchihashi Z, Fuentes GM, Bambara RA, Fay PJ. Influence of human immunodeficiency virus nucleocapsid protein on synthesis and strand transfer by the reverse transcriptase in vitro. J Biol Chem. 1995;270:15005–15011. doi: 10.1074/jbc.270.25.15005. [DOI] [PubMed] [Google Scholar]
- 26.Johnson PE, Turner RB, Wu ZR, Hairston L, Guo J, Levin JG, Summers MF. A mechanism for plus-strand transfer enhancement by the HIV-1 nucleocapsid protein during reverse transcription. Biochemistry. 2000;39:9084–9091. doi: 10.1021/bi000841i. [DOI] [PubMed] [Google Scholar]
- 27.You JC, McHenry CS. Human immunodeficiency virus nucleocapsid protein accelerates strand transfer of the terminally redundant sequences involved in reverse transcription. J Biol Chem. 1994;269:31491–31495. [PubMed] [Google Scholar]
- 28.Allain B, Lapadat-Tapolsky M, Berlioz C, Darlix JL. Transactivation of the minus-strand DNA transfer by nucleocapsid protein during reverse transcription of the retroviral genome. EMBO J. 1994;13:973–981. doi: 10.1002/j.1460-2075.1994.tb06342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo J, Henderson LE, Bess J, Kane B, Levin JG. Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J Virol. 1997;71:5178–5188. doi: 10.1128/jvi.71.7.5178-5188.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeStefano JJ. Interaction of human immunodeficiency virus nucleocapsid protein with a structure mimicking a replication intermediate. Effects on stability, reverse transcriptase binding, and strand transfer. J Biol Chem. 1996;271:16350–16356. [PubMed] [Google Scholar]
- 31.DeStefano JJ. Human immunodeficiency virus nucleocapsid protein stimulates strand transfer from internal regions of heteropolymeric RNA templates. Arch Virol. 1995;140:1775–1789. doi: 10.1007/BF01384341. [DOI] [PubMed] [Google Scholar]
- 32.Raja A, DeStefano JJ. Kinetic analysis of the effect of HIV nucleocapsid protein (NCp) on internal strand transfer reactions. Biochemistry. 1999;38:5178–5184. doi: 10.1021/bi9828019. [DOI] [PubMed] [Google Scholar]
- 33.Guo J, Wu T, Anderson J, Kane BF, Johnson DG, Gorelick RJ, et al. Zinc finger structures in the human immunodeficiency virus type 1 nucleocapsid protein facilitate efficient minus- and plus-strand transfer. J Virol. 2000;74:8980–8988. doi: 10.1128/jvi.74.19.8980-8988.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hooker CW, Scott J, Apolloni A, Parry E, Harrich D. Human immunodeficiency virus type 1 reverse transcription is stimulated by tat from other lentiviruses. Virology. 2002;300:226–235. doi: 10.1006/viro.2002.1554. [DOI] [PubMed] [Google Scholar]
- 35.Klarmann GJ, Schauber CA, Preston BD. Template-directed pausing of DNA synthesis by HIV-1 reverse transcriptase during polymerization of HIV-1 sequences in vitro. J Biol Chem. 1993;268:9793–9802. [PubMed] [Google Scholar]
- 36.You JC, McHenry CS. HIV nucleocapsid protein. Expression in Escherichia coli, purification, and characterization. J Biol Chem. 1993;268:16519–16527. [PubMed] [Google Scholar]
- 37.Khan R, Giedroc DP. Nucleic acid binding properties of recombinant Zn2 HIV-1 nucleocapsid protein are modulated by COOH-terminal processing. J Biol Chem. 1994;269:22538–22546. [PubMed] [Google Scholar]
- 38.Karpel RL, Henderson LE, Oroszlan S. Interactions of retroviral structural proteins with single-stranded nucleic acids. J Biol Chem. 1987;262:4961–4967. [PubMed] [Google Scholar]
- 39.Drummond JE, Mounts P, Gorelick RJ, Casas-Finet JR, Bosche WJ, Henderson LE, et al. Wild-type and mutant HIV type 1 nucleocapsid proteins increase the proportion of long cDNA transcripts by viral reverse transcriptase. AIDS Res Hum Retroviruses. 1997;13:533–543. doi: 10.1089/aid.1997.13.533. [DOI] [PubMed] [Google Scholar]
- 40.Tanchou V, Gabus C, Rogemond V, Darlix JL. Formation of stable and functional HIV-1 nucleoprotein complexes in vitro. J Mol Biol. 1995;252:563–571. doi: 10.1006/jmbi.1995.0520. [DOI] [PubMed] [Google Scholar]
- 41.Lener D, Tanchou V, Roques BP, Le Grice SF, Darlix JL. Involvement of HIV-I nucleocapsid protein in the recruitment of reverse transcriptase into nucleoprotein complexes formed in vitro. J Biol Chem. 1998;273:33781–33786. doi: 10.1074/jbc.273.50.33781. [DOI] [PubMed] [Google Scholar]
- 42.Stoylov SP, Vuilleumier C, Stoylova E, De Rocquigny H, Roques BP, Gerard D, Mely Y. Ordered aggregation of ribonucleic acids by the human immunodeficiency virus type 1 nucleocapsid protein. Biopolymers. 1997;41:301–312. doi: 10.1002/(SICI)1097-0282(199703)41:3<301::AID-BIP5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 43.Le Cam E, Coulaud D, Delain E, Petitjean P, Roques BP, Gerard D, et al. Properties and growth mechanism of the ordered aggregation of a model RNA by the HIV-1 nucleocapsid protein: an electron microscopy investigation. Biopolymers. 1998;45:217–229. doi: 10.1002/(SICI)1097-0282(199803)45:3<217::AID-BIP4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 44.DeStefano JJ, Mallaber LM, Rodriguez-Rodriguez L, Fay PJ, Bambara RA. Requirements for strand transfer between internal regions of hetero-polymer templates by human immunodeficiency virus reverse transcriptase. J Virol. 1992;66:6370–6378. doi: 10.1128/jvi.66.11.6370-6378.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeStefano JJ, Buiser RG, Mallaber LM, Bambara RA, Fay PJ. Human immunodeficiency virus reverse transcriptase displays a partially processive 3′ to 5′ endonuclease activity. J Biol Chem. 1991;266:24295–24301. [PubMed] [Google Scholar]
- 46.Gorelick RJ, Chabot DJ, Rein A, Henderson LE, Arthur LO. The two zinc fingers in the human immunodeficiency virus type 1 nucleocapsid protein are not functionally equivalent. J Virol. 1993;67:4027–4036. doi: 10.1128/jvi.67.7.4027-4036.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heath MJ, Derebail SS, Gorelick RJ, DeStefano JJ. Differing roles of the N- and C-terminal zinc fingers in human immunodeficiency virus nucleocapsid protein-enhanced nucleic acid annealing. J Biol Chem. 2003;278:30755–30763. doi: 10.1074/jbc.M303819200. [DOI] [PubMed] [Google Scholar]
- 48.Guo J, Wu T, Kane BF, Johnson DG, Henderson LE, Gorelick RJ, Levin JG. Subtle alterations of the native zinc finger structures have dramatic effects on the nucleic acid chaperone activity of human immunodeficiency virus type 1 nucleocapsid protein. J Virol. 2002;76:4370–4378. doi: 10.1128/JVI.76.9.4370-4378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levin JG, Guo J, Rouzina I, Musier-Forsyth K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog Nucl Acid Res Mol Biol. 2005;80:217–286. doi: 10.1016/S0079-6603(05)80006-6. [DOI] [PubMed] [Google Scholar]
- 50.Henderson LE, Bowers MA, Sowder RC, 2nd, Serabyn SA, Johnson DG, Bess JW, Jr, et al. Gag proteins of the highly replicative MN strain of human immunodeficiency virus type 1: post-translational modifications, proteolytic processings, and complete amino acid sequences. J Virol. 1992;66:1856–1865. doi: 10.1128/jvi.66.4.1856-1865.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vo MN, Barany G, Rouzina I, Musier-Forsyth K. Mechanistic studies of mini-TAR RNA/DNA annealing in the absence and presence of HIV-1 nucleocapsid protein. J Mol Biol. 2006;363:244–261. doi: 10.1016/j.jmb.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 52.Goldschmidt V, Didierjean J, Ehresmann B, Ehresmann C, Isel C, Marquet R. Mg2+ dependency of HIV-1 reverse transcription, inhibition by nucleoside analogues and resistance. Nucl Acids Res. 2006;34:42–52. doi: 10.1093/nar/gkj411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ikeda RA, Richardson CC. Enzymatic properties of a proteolytically nicked RNA polymerase of bacteriophage T7. J Biol Chem. 1987;262:3790–3799. [PubMed] [Google Scholar]
- 54.Derebail SS, Heath MJ, DeStefano JJ. Evidence for the differential effects of nucleocapsid protein on strand transfer in various regions of the HIV genome. J Biol Chem. 2003;278:15702–15712. doi: 10.1074/jbc.M211701200. [DOI] [PubMed] [Google Scholar]
- 55.Benjamin J, Ganser-Pornillos BK, Tivol WF, Sundquist WI, Jensen GJ. Three-dimensional structure of HIV-1 virus-like particles by electron cryotomography. J Mol Biol. 2005;346:577–588. doi: 10.1016/j.jmb.2004.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang PS, Cantin EM, Zaia JA, Ladne PA, Stephens DA, Sarver N, Rossi JJ. Ribozyme-mediated site-specific cleavage of the HIV-1 genome. Clin Biotechnol. 1990;2:23–31. [Google Scholar]
- 57.Hou EW, Prasad R, Beard WA, Wilson SH. High-level expression and purification of untagged and histidine-tagged HIV-1 reverse transcriptase. Protein Expr Purif. 2004;34:75–86. doi: 10.1016/j.pep.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 58.Sambrook JR, Russell DW. Molecular Cloning: A Laboratory Manual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 59.Carteau S, Gorelick RJ, Bushman FD. Coupled integration of human immunodeficiency virus type 1 cDNA ends by purified integrase in vitro: stimulation by the viral nucleocapsid protein. J Virol. 1999;73:6670–6679. doi: 10.1128/jvi.73.8.6670-6679.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laughrea M, Shen N, Jette L, Darlix JL, Kleiman L, Wainberg MA. Role of distal zinc finger of nucleocapsid protein in genomic RNA dimerization of human immunodeficiency virus type 1; no role for the palindrome crowning the R-U5 hairpin. Virology. 2001;281:109–116. doi: 10.1006/viro.2000.0778. [DOI] [PubMed] [Google Scholar]