

Abstract
The cardiovascular toxicity of older generation of tricyclic antidepressants (e.g. imipramine, desipramine, amitriptyline, clomipramine) and neuroleptics (e.g. haloperidol, droperidol, thioridazine, pimozide) is well established. These drugs inhibit cardiovascular Na+, Ca2+ and K+ channels often leading to life-threatening arrhythmia.
To overcome the toxicity of old generation of antidepressants and antipsychotics, selective serotonin reuptake inhibitor antidepressants (SSRIs: fluoxetine, fluvoxamine, paroxetine, sertraline, citalopram, venlafaxin) and several new antipsychotics (e.g. clozapine, olanzapine, risperidone, sertindole, aripiprazole, ziprasidone, quetiapine) were introduced during the past decade. Although these new compounds are not more effective in treating psychiatric disorders than older medications, they gained incredible popularity since they have been reported to have fewer and more benign side effect profile (including cardiovascular) than predecessors.
Surprisingly, an increasing number of case reports have demonstrated that the use of SSRIs and new antipsychotics (e.g. clozapine, olanzapine, risperidone, sertindole, aripiprazole, ziprasidone, quetiapine) is associated with cases of arrhythmias, prolonged QTc interval on electrocardiogram (ECG) and orthostatic hypotension in patients lacking cardiovascular disorders, raising new concerns about the putative cardiovascular safety of these compounds. In agreement with these clinical reports these new compounds indeed show marked cardiovascular depressant effects in different mammalian and human cardiovascular preparations by inhibiting cardiac and vascular Na+, Ca2+ and K+ channels. Taken together, these results suggest that the new generation of antidepressants and antipsychotics also have clinically important cardiac as well as vascular effects. Clinicians should be more vigilant about these potential adverse reactions and ECG control may be suggested during therapy, especially in patients with cardiovascular disorders.
The primary goal of this review is to shed light on the recently observed clinically important cardiovascular effects of new antidepressants and antipsychotics and discuss the mechanism beyond this phenomenon.
Keywords: antidepressants, neuroleptics, antipsychotics, QT prolongation, arrhythmia, cardiac ion channels, repolarization
INTRODUCTION
Cardiovascular mortality in psychiatric patients is high. Reports of sudden unexplained death in those taking psychotropic drugs, including neuroleptics and antidepressants, have raised the concern that part of this excess may be due to drug-induced arrhythmias, since many of these drugs have cardiac electrophysiological effects similar to those of quinidine. Indeed, it has recently been established that old generation of antidepressants (tricyclic antidepressants (TCAs) and antipsychotics (e.g. haloperidol, droperidol, thioridazine, pimozide) can be associated with increased risk of cardiac arrhythmias and sudden death [reviewed in 1–7].
In contrast, new generation of selective serotonin reuptake inhibitor antidepressants (SSRIs: fluoxetine, fluvoxamine, paroxetine, sertraline, citalopram, venlafaxin) and several new antipsychotics (e.g. clozapine, olanzapine, risperidone, sertindole, aripiprazole, ziprasidone, quetiapine risperidal) are considered to be free from the cardiotoxicity of their predecessors. However, there are increasing number of case reports on various arrhythmias and syncope associated with the use of these new compounds [reviewed in: 3, 5–9]. In addition recent studies have demonstrated that the new SSRIs and antipsychotics also exert potent cardiovascular depressant effects in various mammalian and human cardiovascular preparations by inhibiting cardiac and vascular Na+, Ca2+ and K+ channels. This review is concerned with the cardiovascular effects of new antidepressants and antipsychotics.
1. CARDIOVASCULAR EFFECTS OF ANTIDEPRES-SANTS
1.1. Clinical Evidence
1.1.1. Cardiovascular Effects of Tricyclic Antidepressants (TCAs)
The cardiovascular effects and toxicity of tricyclic antidepressants have been well documented in depressed patients without pre-existing cardiac disease [1, 3, 10–11]. The most common manifestation of such effect is the slowing of intraventricular conduction, manifested by prolonged PR, QRS and QT intervals on the standard ECG, and othostatic hypotension [12–15]. The prolonged conduction can be dangerous in overdose and depressed patients with preexisting conduction defect and in patients who have already been treated with a class I (Na+-channel blocking) antiarrhythmic agent [16–17]. In overdose, delayed conduction may result in a complete heart block or ventricular reentry arrhythmias. Any of these complications, or a combination of both, may lead to death [18–20]. Depressed patients with conduction disease, particularly bundle branch block, being treated with TCAs at therapeutic plasma levels, are at a higher risk of developing symptomatic AV block than those of free from conduction disorders [16–17]. Tricyclic antidepressants have also been found to exert I/A class antiarrhythmic effects [21–23]. Children seem to be especially vulnerable to cardiotoxic effects of high doses of tricyclic compounds. Death has occurred in children after accidental or deliberate overdose with only a few hundred milligrams of drug [24]. Since tricyclic antidepressants can cause orthostatic hypotension, induce arrhythmia at higher doses or tissue concentrations, and interact unpredictably with other drugs, as do the serotonin-reuptake inhibitors, they must be used with great caution in patients with cardiac disease [16–17].
1.1.2. Cardiovascular Effects of Selective Serotonin Reuptake Inhibitors (SSRIs)
The most human clinical studies with SSRIs like fluoxetine, fluvoxamin, paroxetine, sertaline and citalopram showed significant advantages over TCAs in producing fewer cardiotoxic, anticholinergic and antihistaminergic side effects in the treatment of major depressive disorders [reviewed in 3]. These newer compounds exhibited lower risk of inducing hypotension and a higher margin of safety in acute overdose than tricyclics [reviewed in 3]. However, it is interesting to note that the results of some animal studies were not always so clear cut. For example, early preclinical studies in cats with the highly selective serotonin reuptake inhibitor, citalopram, showed TCA-like cardiac effects at high doses [25], and the development of citalopram was delayed by reports of cardiotoxicity in dogs, eventually attributed to a species-specific metabolite not found in humans [26].
The SSRI drug, of that the most information is available, is fluoxetine [27] used for oral administration; it is chemically unrelated to tricyclic, tetracyclic antidepressant agents. Several clinical studies showed that compared to tricyclic antidepressants, fluoxetine causes significantly fewer anti-cholinergic, antihistaminergic and cardiovascular side effects [reviewed in: 3, 8–9]. However, even with fluoxetine one must be cautious in the conclusions drawn because the patients that have been carefully studied are, in general, depressed patients free of cardiovascular disease, and only very limited information is available in patients having cardiovascular disease as well [28–31]. The SSRIs do have cardiac effects, the best demonstrated of those being a mild bradycardia observed during chronic treatment with fluoxetine, fluvoxamin, paroxetine [reviewed in 3, 8]. This usually amounts to only a few beats per minute but it is the opposite of the tachycardia that has been associated with tricyclic drugs. Analysing large number of ECG recordings from citalopram-treated patients Enemark [32] reported that citalopram-treatment also reduced the heart rate. This reduction occurred within the first week of the treatment without further reduction later. In a small group of citalopram-treated patients (3–4%) with normal heart rate at baseline bradycardia was developed. Furthermore, citalopram treatment was associated with a non-specific, insignificant prolongation of QT interval irrespective of age. In younger group of the patients a statistically significant decrease in T-wave amplitude was also demonstrated [32]. Moreover, there are increasing number of case reports on dysrhythmia and syncope associated with fluoxetine and another SSRIs treatment and overdose [33–58]. A multicenter case-control study has shown that in the elderly the consumption of fluoxetine was significantly associated with an excess risk of syncope and orthostatic hypotension [59]. A significant blood pressure lowering effect of fluoxetine was reported in DOCA-hypertensive rats [60]. The authors suggested that a central action of fluoxetine on vasomotor center may be responsible for the reduction of blood pressure, but the possible direct cardiac and/or vascular effects of fluoxetine were not excluded or determined. Interestingly, several recent studies have provided evidence that fluoxetine and citalopram directly inhibit Ca2+ entry into vascular and intestinal smooth muscles resulting in vasodilation and intestinal relaxation, effects, which could be of significant therapeutic importance. [61–64]. Surprisingly results from recently published retrospective studies show that the use of new SSRIs, similarly to the old TCAs, increases the risk of falls and hip fracture among elderly people [reviewed in 9].
1.2. Cellular Electrophysiological Effects
Electrophysiological studies (using a broad range of in vitro models) demonstrated that both antidepressants and antipsychotics exerted their cardiac actions by modifying the different cardiac ionic currents during the action potential (Fig. 1).
Fig. 1.
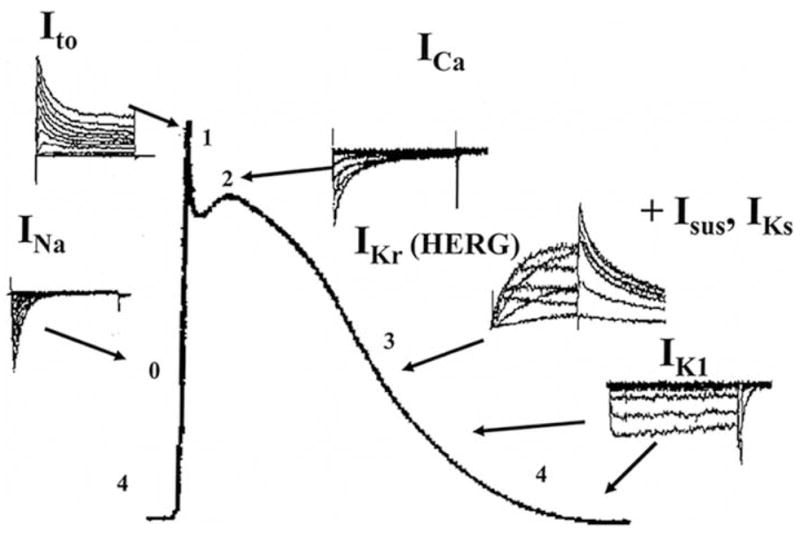
Ionic currents during cardiac action potential. Downward reperesents inward (depolarizing), upward shows outward (repolarizing) currents. INa= inward sodium current; ICa= inward calcium current; Ito= transient outward potassium current; IKr= rapid component of outward delayed rectifier potassium current; IKs= slow component of outward delayed rectifier potassium current; Isus= outward sustained potassium current; IK1= inward rectifier potassium current.
1.2.1. Cellular Electrophysiological Effects of TCAs
In electrophysiological studies on isolated mammalian multicellular cardiac preparations and single myocytes, TCAs, such as imipramine, chlorimipramine amitriptyline, desipramine, dibenzepin, lofepramine and amoxapine, were demonstrated to reduce the maximum velocity of depolarization (Vmax) of the action potential, an indirect index of the fast inward sodium current, INa. [65–69]. Furthermore, imipramine also blocks the outward delayed rectifier K+ current (IK) and the inward slow Ca2+ (ICa) currents in guinea-pig ventricular myocytes and transient outward K+ current (Ito) in rabbit atrial cells [70–72]. These direct membrane effects explain a variety of characteristic ECG abnormalities like prolongation of PQ, QRS, and QT, and cardiac adverse effects including tachyarrhythmias, heart block, congestive heart failure, observed during tricyclic antidepressants treatment and overdose [1, 3, 10–14, 19]. The effects of imipramine on action potential duration (APD) show important species dependence. In bovine ventricular [75] and Purkinje fibers [65], guinea-pig papillary muscles [76–77] and isolated ventricular myocytes [70] imipramine shortened the APD, whereas in rabbit and rat atrial fibers [78–79] it lengthened the APD. The different effects of imipramine on APD can be explained by the important differences in the ionic currents responsible for the repolarization among animal species. In guinea-pig ventricular myocytes where Ito is relatively little [74], the APD is controlled by the interaction between inward (INa and ICa) and outward currents (IK and IK1). Imipramine decreased the INa [81], IK and ICa but did not modify the IK1 [70, 75]. The reduction of the APD in bovine and guinea pig ventricular preparations could be explained mainly by inhibition of Ica [65, 70, 75, 77]. In contrast, in rat, rabbit and human atria [64, 80, 82] and rat ventricular myocytes the IK is negligible and Ito appears to be the most important outward K+ current responsible for action potential repolarization. Thus the reduction of Ito could explain the prolongation of the APD observed in above-mentioned species. More recently several antidepressants with different chemical structures (imipramine, amitriptyline, mianserine, maprotiline and trazodone) were reported to block transient outward K+ current (Ito) [73].
1.2.2. Cellular Electrophysiological Effects of SSRIs
1.2.2.1. Effects of SSRIs on Cardiac Action Potentials (APs) In vitro
We previously demonstrated that fluoxetine elicited a concentration dependent depression of the amplitude of action potential (APA), overshoot (OS) and the maximum rate of rise of depolarization phase (Vmax) in multicellular ventricular preparations of rats, rabbits and dogs without changing the resting membrane potential (RP) [84–85]. The significant threshold concentrations were more or less similar (3–10 μM) in various species (including the most sensitive isolated canine myocytes). Fluoxetine caused a nearly similar shortening of the duration of ventricular action potential (APD) in three species (guinea pig, rabbit, canine), but not in rats. Fluoxetine caused a concentration-dependent decrease in force of contraction in rat right ventricular papillary muscle with a calculated IC50 value of 9.86 μM. Citalopram similarly to fluoxetine elicited a concentration-dependent (10–100 μM) reduction of Vmax, decrease of APA, OS and shortening of APA in guinea-pig ventricular papillary muscle [86]. Fluoxetine and citalopram produced a dose-dependent decrease of V max (an indirect indicator of the fast Na+ channel activity), which suggests that they inhibited the activation of fast Na+ channels and exhibited class I anti-arrhythmic effects. A possible explanation of the decrease in APA and OS and shortening of the early part of repolarization (APD50) can be the inhibition of the calcium current (ICa). This latter mechanism may also be responsible for the negative inotropic effect of fluoxetine. The inhibitory effect of fluoxetine on peak Ca2+ current was proven in voltage clamped canine ventricular myocytes by IC50 value of 5.4 μM. This effect may cause lengthening of atrioventricular conduction. Considering its Na+ and Ca2+ currents inhibitory action, fluoxetine may have antiarrhythmic as well as pro-arrhythmic properties (due to impairement of atrioventricular or intraventricular conduction). As far as the different effects of fluoxetine on rat ventricular APD are concerned, these can be explained by the unique ion regulation characteristic to ventricular repolarization phase of rat markedly different from that of other mammalian species [87]. Similar cardiac electrophysiological effects with venlafaxine were observed in guinea-pig cardiac myocytes [88]. These direct cardiac effects of fluoxetine and citalopram are similar to those found by us for TCA clomipramine [86] and previously reported for the tri- and tetracyclic antidepressants [3].
1.2.2.2. Effects of SSRIs on Cardiac Ion Channels
Previous and recent studies demonstrated that fluoxetine and other SSRIs possess potent antagonistic properties on voltage-dependent ion channels in different tissues [84–85, 89–106]. The IC50 values of SSRIs for Na+, Ca2+ and K+ channels of mainly cardiac tissues are summarized in Table 1. Fluoxetine inhibited L-type of Ca2+(CaL2+) current in both rat and canine ventricular myocytes, but its potency was twice as high in rat (IC50 2.8 μM) than in canine myocytes (IC50 5.4 μM) [84, 95]. It is interesting to note that sertraline also inhibited the CaL2+ current in rat myocytes and its inhibitory activity (IC50= 2.3 μM) was similar to that of fluoxetine, while citalopram inhibited CaL2+ current of guinea-pig myocytes at much higher concentration (100 μM) [95, 105]. These data provide evidence that inhibition of cardiac CaL2+ current could play an important role in reducing cardiac contractility, heart rate and atrio-ventricular conduction. The proposed mechanism may explain the prolonged PR interval, AV block, hypotension, which are common cardiovascular complications of fluoxetine therapy.
Table 1.
IC50 Values of Antidepressants for Ionic Currents/Channels
| Compound | Model system | Potassium channels/currents | Calcium channels/current | Sodium channels/currents | References | ||
|---|---|---|---|---|---|---|---|
| KV/delayed rectifier (rapid) IKr | Transient outward Ito | HERG | CaL | ||||
| TRICYCLIC DRUGS | |||||||
| Imipramine | Guinea-pig ventricular myocytes | ≥ 1 μM | ≥ 1 μM | 77 | |||
| Rabbit atrial myocytes | ≥ 1 μM | ≥ 1 μM | 72 | ||||
| Guinea-pig ventricular myocytes | ≥ 1 μM | 70 | |||||
| Guinea-pig ventricular myocytes | 3μM | 166 | |||||
| Human cardiomyocyes | ≥ 1 μM | 167 | |||||
| Rat ventricular myocytes | 4 μM | 5 μM | 95 | ||||
| CHO | 3.4 μM | 168 | |||||
| Amitriptyline | CHO | ~4 μM
4.7 μM |
169
168 |
||||
| Rabbit atrial and ventricular myocytes | ≥ 1 μM | 170 | |||||
| Rat ventricular myocytes | 3.75 μM | 95 | |||||
| SELECTIVE SEROTONIN REUPTAKE INHIBITORS | |||||||
| Fluoxetine | Rat pheochromocytoma (PC12) cell | 16 μM | 13.4 μM | 25.6 μM | 171 | ||
| HEK - 293 | 1.5 μM
3.1 μM |
105
94 |
|||||
| Rat ventricular myocytes | 40 μM | 2.8 μM | 95 | ||||
| Canine ventricular myocytes | 5.4 μM | 84 | |||||
| Citalopram | HEK -293 | 3.97 μM | 105 | ||||
| Rat ventricular myocytes | ≥ 10 μM | 172 | |||||
| Sertaline | Rat ventricular myocytes | 38 μM | 2.3 μM | 95 | |||
| Venlafaxine | Guinea –pig ventricular myocytes | ~ 8μM | 88 | ||||
Fluoxetine and citalopram have a high inhibitory potency (IC50=3.1, 1.5 and 3,97 μM, respectively) on HERG potassium channel [94, 105]. The human ether-a-go-go-related gene, HERG, is believed to encode the protein, which underlies the rapid component of the delayed rectifier K+ current IKr. HERG encoded I Kr plays an important part in the repolarization of the cardiac action potential. Pharmacological inhibition of either heterologously expressed HERG or native IKr would thus be expected to correlate with ventricular action potential prolongation and associated prolongation of the QTc interval on ECG. Thus the HERG current inhibition by fluoxetine and citalopram may give an explanation for the arrhythmogenic side effects (ventricular tachycardias) of these drugs. It is very important to note that this current inhibition can occur at nearly therapeutic levels of these drugs, thus this effect should be considered during the therapy.
SSRIs also exhibit potent inhibitory effects on various voltage-dependent ion channels in non-cardiac tissues. Some of these effects are summarized in the Table 1, but the detailed description is beyond the scope of this review.
The inhibitory concentrations of SSRIs on cardiac APs and ion current were in the upper range of the therapeutic plasma levels [107]. However, it is difficult to relate in vivo plasma concentrations to in vitro concentrations as pharmacokinetic properties (tissue accumulation, metabolites) of the drug must also be considered. Under certain conditions (e.g. in case of drug interactions or reduced metabolism in elderly) the plasma concentration of SSRIs can reach even higher levels. Thus, a significant inhibition of various cardiovascular ion channels by SSRIs may occur in patients chronically treated with these compounds, resulting in certain pro- or arrhythmic effects. [reviewed in 3].
2. CARDIOVASCULAR EFFECTS OF NEUROLEPTICS
2.1. Clinical Evidence
The aim of this part of the review is to organise the available evidences on cardiac/cardiovascular side effects; proarrhythmic potential of antipsychotic drugs and to discuss their actions on cardiac ion currents as proposed explanation of their proarrhythmic effects.
Antipsychotic drugs represent a chemically various group of compounds. Antipsychotic drugs can be classified typical (older drugs acting on dopamine D1, D2, adrenergic α1, muscarinic cholinergic, 5-HT2 and histamine H1-receptors and associated with different side effects) and atypical (newer drugs inhibiting mainly both D2 and 5-HT2A receptors and have a higher efficacy and fewer side effects) groups. Among atypical antipsychotics clozapine shows marked difference from the others binding more to D4, 5-HT2 and α1 receptors than to D2 receptor. [108]. Aripiprazole is a first member of a new class of atypical antipsychotics have also unique properties showing a combined partial agonist activity at D2 and 5-HT1A receptors with an antagonism at 5-HT2A receptors [109–111].
Antipsychotic drugs have long been known to be associated with risk of cardiac arrhythmia and cardiac arrest. These arrhythmias are often reflected as changes in the electrocardiogram (ECG), prolongation of the QT interval, ventricular tachycardias, torsades de pointes (TdP). TdP is a potentially life-threatening ventricular tachyarrhythmia that is associated with syncope and sudden death. TdP is characterized by a twisting morphology of the QRS complex around the isoelectric baseline and can occur in congenital and acquired form induced by various cardiac and non-cardiac drugs. Among antipsychotics haloperidol, droperidol, pimozide, sertindole, thioridazine were found to cause definitively TdP [112–118]. Several other antipsychotics including typical (chlorpromazine, fluphenazine, mesoridazine, prochlorperazine, trifluperazine, sultopride) [119–121] (quetiapine, olanzapine, risperidone, ziprasidone) [122–127] have been reported to prolong the corrected QT interval (corrected for heart rate)(QTc). Both an Australian and a Finnish study of neuroleptic poisoning demonstrated that thioridazine caused the most frequently tachycardia, prolonged QTc, widened QRS, arrhythmias and sudden death [120, 128]. Thioridazine and droperidole were found to be associated with prolongation of QTc even at dosage used for therapy [129] and based on this study the indications of thioridazine were restricted and droperidol was voluntarily discontinued by the manufacturer in UK [114]. Pimozide, sultopiride and droperidole also prolong QTc interval and have been associated with TdP and sudden death, but far fewer data are available [121, 130]. The high-potency drug haloperidole can prolong QTc interval, causes TdP and sudden death at normal therapeutic doses [131], but the frequency by which these effects occur is less than with thioridazine [128]. Similar cardiovascular risks of traditional antipsychotics used at therapeutic dosage were published in the USA in a retrospective study investigating 481,744 persons (aged 15–84 years, from 1988 to 1993) [132].
The new atypical antipsychotics have greater efficacy and fewer side effects than older neuroleptics and with the exceptions of sertindole and ziprasidone they have not caused consistent statistically significant lengthening of QT or sudden cardiac death at therapeutic concentrations [118, 133]. Sertindole has been proven to be associated with a QT prolongation at therapeutic concentrations [117, 124], and both increasing evidence of unexplained sudden cardiac death and serious arrhythmias found by the Committee on Safety of Medicine in the United Kingdom resulted in a voluntary withdrawal of the drug by the manufacturer [134]. Albeit the known correlation between schizophrenia and increased cardiovascular mortality it may difficult to estimate the sudden death due to particular neuroleptics at therapeutic doses [135, 136]. Clozapine beyond the well-known agranulocytosis risk, is being associated with myocarditis, cardiomyopathy and arrhythmogenesis risk [137, 138]. It also reduced measures of heart rate variability associated with parasympathetic control [124]. In the study of overdoses, clozapine overdose was associated with sinus tachycardia (more than 66% of the patients) however, in the case of risperidone overdose more than 66% of the patients were asymptomatic [139]. However, there are data suggesting that risperidone could cause sudden death [125]. Neither olanzapine nor quetiapine had been implicated in cases of TdP or sudden death.
Ziprasidone is a new atypical drug with less side effects and in comparison with olanzapine and risperidone it does not appear to cause weight gain, hyperlipidemia and hyperglycaemia but prolongs the QT interval more than haloperidol, olanzapine, quetiapine and risperidone [127]. Although it was not associated with cardiac events during premarketing trials the appearance of unexpected life-threatening arrhythmias can not be excluded when the drug enters widespread use.
2.2. Mechanism of the Lengthening of QT Interval
The QT interval includes both depolarization and repolarization. Q wave represents the onset of ventricular depolarization, while T wave is the sign of the repolarization. Because the QT interval shortens with increasing heart rates, it is usually corrected for heart rate (QTc). Depolarization of ventricular cells is the result of a rapid influx of sodium ions through selective Na+channel and its duration measured by the QRS interval. Repolarization involves calcium, sodium, and different potassium channels. Whereas the participation of these ion channels in the repolarization is highly dependent on species, mainly potassium channels are responsible for this parameter. Concerning the specificity of QT prolongation as a marker of an effect on cardiac repolarisation, it should be kept in mind that the duration of the QT interval may be affected by both the velocity of repolarisation and ventricular conduction velocity. Class I antiarrhythmics as sodium channel blockers, decrease ventricular conduction velocity, cause widening of QRS complex and therefore lengthen the QT interval [140]. Similar action can be observed in the case of tricyclic antidepressants which by blocking Na+ as well K+ channels widen both the QRS and the QTc. The potassium channels (among them IKr) are most often involved in drug-induced QT prolongation and TdP. Drug blocking the IKr channel can induce QT prolongation and TdP and sometimes sudden death [141]. However, there is no close correlation between QTc interval prolongation and occurrence of TdP. Not all drugs that prolong the QTc interval produce TdP. Amiodarone, a class III antiarrhythmic drug, produces marked prolongation of QTc interval but does not evoke TdP. The calcium-channel blocker verapamil has been shown to prolong QT interval in a manner that is linearly correlated to its plasma concentration [142] but there are few described cases of verapamil-induced TdP [143]. No clear-cut dose-dependency can also be observed for QT prolongation or occurrence of TdP. In some cases the QT prolongation and occurrence of TdP is dose dependent but these parameters can also be observed at normal plasma levels of drugs, too [144]. In the latter several factors (hypokalaemia/magnesaemia, mutation of K+ channels) reducing the repolarization reserve of a given subject greatly increase the proarrhythmic potential of relatively low plasma level [5, 7].
The link between the lengthening of QT interval and TdP is seemingly very complex and affected by several factors including electrolyte imbalance, age, gender, disease (myocardial ischemia, infarction, hypertension, hypothyroidism, diabetes, renal or hepatic dysfunction) and concomitant medications.
2.3. Cellular Electrophysiological Effects of Neuroleptics
Most of antipsychotics are generally lengthen the action potential duration (APD) and inhibit the rapid component of the delayed rectifier current (IKr) but some of the typical antipsychotics including chlorpromazine and haloperidol, beyond their inhibitory effect on K+ current inhibit also Na+ and Ca2+ channels [81]. Such effects could be antiarrhythmic or cardiotoxic, depending on the health (e.g. post myocardial infarct) of the myocardium. The net effect on APD of antipsychotics depend on the overall balance between inward and outward currents during the plateau phase of AP and their relative sensitivity to the particular agent in question. Table 2. summarizes the inhibitory potency of antipsychotic drugs on K+(IKr, Ito, IK1, HERG) and other ion (Na+, Ca2+) currents. In human IKr is carried by the human ether-a-go-go (HERG) K+ channel, which can be expressed in homologous and heterologous cells in order to assess the potency (IC50) of a drug in inhibiting this channel. Haloperidol and droperidol prolong APD in guinea-pig ventricular myocytes and inhibit IKr and HERG with IC50 values of 20 nM-1.36 μM and 32.2 μM, respectively and the effects of haloperidol on HERG are over five or twenty times more potent than its effects on INa and ICaL, respectively [81, 145–146, 150, 163]. Thioridazine also lengthened APD in guinea-pig ventricular myocytes and potently inhibited IKr and HERG (IC50 values of 1.25 μM, 191 nM and 1 μM) [147, 149]. Comparative study showed that newer atypical antipsychotic ziprasidone, olanzapine, risperidone block HERG and IKr in a more or less similar concentration range [148].
Table 2.
IC50 Values of Antipsychotics for Ionic Currents/Channels
| Compound | Model system | Potassium channels/currents
|
Calcium channels/current
|
Sodium channels/currents INa | References | |||
|---|---|---|---|---|---|---|---|---|
| KV/delayed rectifier IKr | Transient outward Ito | Inward rectifier IK1 | HERG | ICaL | ||||
| Chlorpromazine | CHO | 1.47 μM | 157 | |||||
|
| ||||||||
| Clozapine | HEK-293 | >50 μM | 320 nM | 29 μM | 148 | |||
|
| ||||||||
| Droperidol | Ventricular myocytes HEK-293 | 28 nM | 32.2 μM | 145 | ||||
|
| ||||||||
| Fluspirilene | Xenopus oocytes | 2.34 μM | 150 | |||||
|
| ||||||||
| Haloperidol | PC12 cells | 20 μM | 164 | |||||
| Ventricular myocytes | 5 μM | 81 | ||||||
| HEK-293 | >10 μM | >100 μM | 26.8 nM | 14.4 μM | 148 | |||
| 20 nM | 165 | |||||||
| Xenopus oocytes | 1.36 μM | 150 | ||||||
| Xenopus oocytes | 1.0 μM | 146 | ||||||
|
| ||||||||
| Olanzapine | HEK-293 | >100 μM | >100 μM | 731 nM | >100 μM | 148 | ||
| CHO | 6013 nM | 147 | ||||||
|
| ||||||||
| Pimozide | HEK-293 | >100 μM | >100 μM | 54.6 nM | 21.9 μM | 148 | ||
| Xenopus oocytes | 1.74 μM | 150 | ||||||
| CHO | 18 nM | 154 | ||||||
|
| ||||||||
| Quetiapine | CHO | 5765 nM | 147 | |||||
|
| ||||||||
| Risperidone | canine ventricular myocytes | 0.92 μM | 151 | |||||
| HEK-293 | >100 μM | >100 μM | 148 nM | >50 μM | 148 | |||
| CHO | 167 nM | 147 | ||||||
| CHO | 261 nM | 155 | ||||||
|
| ||||||||
| Sertindole | HEK-293 | >10 μM | >10 μM | 14 nM | >10 μM | 148 | ||
| HEK-293 | 21 μM | 14.7 nM | 153 | |||||
| CHO | 3 nM | 147 | ||||||
|
| ||||||||
| Thioridazine | Ventricular myocytes | 1.25 μM | 149 | |||||
| tsA201 | ||||||||
| CHO | 1.25 μM | |||||||
| CHO | 1.07 μM | 158 | ||||||
| HEK-293 | 91 nM | 147 | ||||||
| 27 μM | 33.2 nM | 7 μM | 148 | |||||
|
| ||||||||
| Ziprasidone | HEK-293 | >10 μM | > 10 μM | 125 nM | >10 μM | 148 | ||
| CHO | 169 nM | 147 | ||||||
CHO =Chinese hamster ovary cells; PC12 cells=rat pheochromocytoma cells; HEK-293= Human embryonic kidney cells.
Figures (2 and 3) show that risperidone concentration-dependently increased APD in both guinea-pig ventricular muscle (Fig. 2A) and canine ventricular myocytes (Fig. 3B). This effect was most prominent on terminal phase of repolarization (APD90) (with EC50 values of 0.29 μM and 0.48 μM in guinea pig and canine myocytes, respectively) (Fig. 3C) and showed reverse rate dependence (Fig. 2B). Haloperidol had similar effect on APD (Fig. 2C) but reduced also the maximum velocity of depolarization (Vmax)(indirect indicator of Na+ channel activity)(Fig. 2D) while risperidone was ineffective on this parameter. We found that risperidone concentration-dependently inhibited IKr with an IC50 of 0.92 μM and practically had no effect on the other K+ currents (Ito with IC50 >10 μM, IK1 with IC50 >100 μM) [151]. Similar effects of risperidone on both APD and IKr in rabbit ventricular myocardium and myocytes were observed [152], while lower IC50 values (167 and 261 nM, respectively) were found in HERG channel by others [147, 155].
Fig. 2.

Cummulative concentration-dependent effects of risperidone(A, n=9) and haloperidole (C, n=5) on action potential duration measured at various levels of repolarization in guinea-pig ventricular papillary muscles. The preparations were treated with each concentration of drugs and paced at frequency of 1 Hz. B: Frequency-dependence of the lenghetening effect of 1 μM risperidone on APD90 studied in night guinea-pig ventricular preparations. D: Comparison of the effect of haloperidole (●) and risperidone (▲) on the maximal velocity of depolarization of AP (Vmax). The effect was demonstrated as the percent change of decrease of Vmax.
Fig. 3.
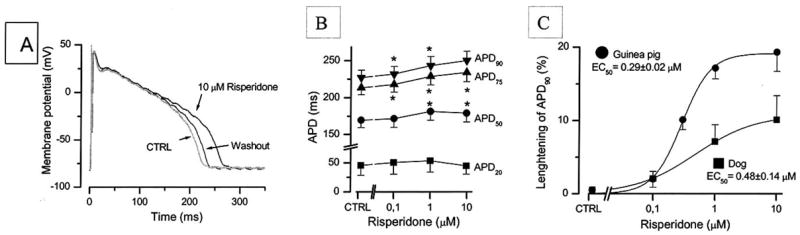
Effects of risperidone (10 μM) on action potential configuration in single canine ventricular myocytes paced a 1 Hz recorded 5 min after the drug superfusion (A). B: Cumulative concentration-dependent effects of risperidone on action potential duration measured at various level of repolarization in canine myocytes (n=5). C: Comparison of the APD prolongation effect of risperidone in canine ventricular myocytes (n=5) and guinea-pig papillary muscles (n=9). The risperidone induced prolongation of APD90 was normalized to control APD90 and expressed as its percent. Solid lines were obtained by fitting the data to the two-state Boltzmann model in order to determine EC50 values.
Sertindole was found to be a high affinity antagonist of the human cardiac K+ channel HERG (IC50= 3 and 14 nM) but was less active at blocking other K+ currents (Kv 1.5, Ito with IC50=2.1 and 10 μM, respectively) [147, 148, 153].
Pimozide potently inhibited cardiac HERG K+ channel (IC50 values of 18 and 174 nM) [147, 150], increased the risk of TdP [154] and also blocked ICaL in rat ventricular myocytes [156]. Beyond the inhibitory action on HERG channel sertindole and pimozide also blocked the human brain K+ channel erg3. Sertindole blocked erg3 channel currents with an IC50 of 43 nM, while pimozide had an IC50 value of 103 nM [157]. It was suggested that this inhibition of erg3 related K+ channels in the brain might contribute to their efficacy/side effect profiles.
Comparing the HERG channel inhibitory activity of seven antipsychotics drugs (olanzapine, pimozide, quetiapine, risperidone, sertindole, thioridazine, ziprasidone) to their binding affinities for D2 and 5-HT2A receptors the following selectivity rank was found: olanzepine > risperidone > ziprasidone > thioridazine > pimozide > sertindole. Sertindole and pimozide had the highest HERG channel inhibitory activity, while the lowest inhibitory activity can be observed in the case of olanzapine and quetiapine. These results also showed that sertindole, pimozide, thioridazine displayed little or no selectivity for dopamine D2 or 5-HT2A receptors relative to their HERG channel affinities, and olanzapine had the greatest selectivity for dopamine D2 and 5-HT2A receptor binding compared to the HERG channel. In the case of quetiapine the selectivity was not calculated due to its lack of affinity for the dopamine D2 receptor. Examining the relationship between plasma levels and QT prolongation for these drugs [127] the authors also found a good correlation between the ratio of total plasma drug concentration to HERG IC50 and their QTc prolongation effect [147]. Based upon this in vitro results drug’s selectivity (between their target receptor affinity and their HERG channel IC50 value) seems to be a predictive factor for appearance of QT prolongation in clinic. It would be expected that olanzepine and risperidone, displaying high selectivity, would have the least potential to produce QT prolongation in clinical settings.
However, it is widely accepted that most QT prolonging drugs inhibit IKr but the potency of drug-induced block of IKr does not show a clear correlation with the risk of QT prolongation and occurrence of TdP. In isolated feline hearts haloperidol prolongs QT interval more than sertindole [159], while sertindole is 10–300 times more potent as a blocker of HERG than haloperidole (Table 2). In addition, some authors found that thioridazine and chlorpromazine have similar potencies for inhibition of HERG [158] and in stepwise regression analysis of 495 psychiatric patients thioridazine was associated with QT prolongation, but chlorpromazine was not [128]. These data suggest that more than one drug-induced mechanisms exist that makes the heart vulnerable to (or protected from) QT prolongation by inhibition of the HERG K+ channel.
2.4. Perspectives
It is a widely held concept that most QT prolonging non cardiac drugs are potassium channel blockers, inhibit IKr and induce TdP. IC50 values for inhibiting IKr in human or other mammalian systems are important to gain insight into the mechanism of drug action, although extrapolation to the clinical setting must carefully consider concentration ranges and possible additional pharmacological effects. Antipsychotic drugs with complex pharmacological actions on different receptors (dopamine, serotonin, muscarinic) and moreover with additional inhibitory effects on Na+, Ca2+ channels may exert various antiarrhythmic/proarrhytmic actions and their effects on the QT interval in vivo may be quite variable depending on the animal species and experimental model. It is postulated that some antipsychotics (olanzapine, risperidone), having higher difference between concentration required for IKr inhibition and their therapeutic plasma levels will only cause cardiac complications in population of vulnerable patients [160] or patients suffering from other disorders (cardiac, or hepatic disease). Therefore, the inhibition of HERG/IKr at cellular level is an important, but not always a predictive arrhythmogenic property of a drug in vivo. The merits of various testing strategies in preclinical phases, including the advantages and limitations of preclinical assays and the current regulatory guidelines scrutinize these drugs, have recently been detailed [161–163]. Current strategies for the development of new psychotropic drugs should include voltage clamp experiments (using HERG) as well as in vivo QT animal models.
As far as the QT prolongation is concerned this sign per se does not necessary mean that the risk/benefit balance of an antipsychotic drug is negative. The final balance depends on other factors that must carefully be considered [5] and the various drugs do differ in their QT-prolonging potential. QTc interval is usually around 400 msec in duration. Greater duration than 440 msec can be considered abnormal. Considering a new drug showing a small or modest QT interval prolongation several questions arise. The first one is whether the prolongation is dose-dependent, because some patients will undoubtedly be either slow metabolizers or given unusually high doses (especially psychiatric patients). To predict the potential for drug-interactions it is also important to know the metabolic pathway of the drug.
The major problem that there is no consensus on the degree of QT prolongation which is clinically significant. It is very difficult to interpret the clinical data as case reports are sometimes questionable, inadequate, some of them involve extreme overdose and cannot be directly extrapolated to routine clinical practice. Some data derived from combination of different drugs, where the concomitant drug’s effect on QT prolongation or occurrence of TdP can not be excluded.
CONCLUSIONS
Collectively, results of numerous recent basic research and clinical studies suggest that the new generation of anti-depressants and antipsychotics also have clinically important cardiac as well as vascular effects. Clinicians should be more vigilant about these potential adverse reactions and ECG control may be suggested during therapy, especially in patients with cardiovascular disorders. The recent advances in our understanding of the cellular and molecular basis for the cardiac effects of antidepressants and antipsychotic drugs may help specialists in the better selection of the appropriate drugs for the given patient to avoid the unexpected, sometimes life-threatening cardiac arrhythmias.
Acknowledgments
This work was supported by the grant ETT 053/2003 of Hungarian Ministry of Health. The authors are indebted to Szilvia Bercsenyi, Mrs K. Barna, and Mr J. Balogh for their assistance during the preparation of the manuscript.
ABBREVIATIONS
- TCA(s)
Tricyclic antidepressant(s)
- SSRI(s)
Selective serotonin reuptake inhibitor antidepressant(s)
- ECG
Electrocardiogram
- 5-HT
5-hydroxytryptamine
- APD
Action potential duration
- APD50
Action potential duration measured at 50% and APD90 and 90% of repolarization
- OSP
Overshoot potential
- APA
Action potential amplitude
- Vmax
Maximum velocity of depolarization during the action potential upstroke
- RP
Resting membrane potential
- INa
Inward Na+ current
- ICa
Inward Ca2+ current
- Ito
Transient outward K+ current
- IKr
Rapid component of outward delayed rectifier K+ current
- IKs
Slow component of outward delayed rectifier K+ current
- Isus
Sustained outward K+ current
- IK1
Inward rectifier K+ current
- TdP
Torsade de pointes type ventricular tachycardia
- HERG
Human ether-a-go-go related gene
- CHO
Chinese hamster ovary cells
- HEK-293
Human embryonic kidney cells
References
References 173–175 are related articles recently published in Current Pharmaceutical Design.
- 1.Glassman AH. Cardiovascular effects of tricyclic antidepressants. Annu Rev Med. 1984;35:503–11. doi: 10.1146/annurev.me.35.020184.002443. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg RJ, Capone RJ, Hunt JD. Cardiac complications following tricyclic antidepressant overdose. Issues for monitoring policy. JAMA. 1985;254:1772–5. [PubMed] [Google Scholar]
- 3.Pacher P, Ungvari Z, Nanasi PP, Furst S, Kecskemeti V. Speculations on difference between tricyclic and selective serotonin reuptake inhibitor antidepressants on their cardiac effects. Is there any? Curr Med Chem. 1999;6:469–80. [PubMed] [Google Scholar]
- 4.De Ponti F, Poluzzi E, Montanaro N. QT-interval prolongation by non-cardiac drugs: lessons to be learned from recent experience. Eur J Clin Pharmacol. 2000;56:1–18. doi: 10.1007/s002280050714. [DOI] [PubMed] [Google Scholar]
- 5.De Ponti F, Poluzzi E, Cavalli A, Recanatini M, Montanaro N. Safety of non-antiarrhythmic drugs that prolong the QT interval or induce torsade de pointes: an overview. Drug Saf. 2002;25:263–86. doi: 10.2165/00002018-200225040-00004. [DOI] [PubMed] [Google Scholar]
- 6.Haddad PM, Anderson IM. Antipsychotic-related QTc prolongation, torsade de pointes and sudden death. Drugs. 2002;62:1649–71. doi: 10.2165/00003495-200262110-00006. [DOI] [PubMed] [Google Scholar]
- 7.Witchel HJ, Hancox JC, Nutt DJ. Psychotropic drugs, cardiac arrhythmia, and sudden death. J Clin Psychopharmacol. 2003;23:58–77. doi: 10.1097/00004714-200302000-00010. [DOI] [PubMed] [Google Scholar]
- 8.Pacher P, Ungvari Z, Kecskemeti V, Furst S. Review of cardiovascular effects of fluoxetine, a selective serotonin reuptake inhibitor, compared to tricyclic antidepressants. Curr Med Chem. 1998;5:381–90. [PubMed] [Google Scholar]
- 9.Pacher P, Ungvari Z. Selective serotonin-reuptake inhibitor antidepressants increase the risk of falls and hip fractures in elderly people by inhibiting cardiovascular ion channels. Med Hypotheses. 2001;57:469–71. doi: 10.1054/mehy.2001.1366. [DOI] [PubMed] [Google Scholar]
- 10.Burckhardt D, Raeder E, Muller V, Imhof P, Neubauer H. Cardiovascular effects of tricyclic and tetracyclic antidepressants. JAMA. 1978;239:213–6. [PubMed] [Google Scholar]
- 11.Glassman AH, Bigger JT., Jr Cardiovascular effects of therapeutic doses of tricyclic antidepressants. A review. Arch Gen Psychiatry. 1981;38:815–20. doi: 10.1001/archpsyc.1981.01780320095011. [DOI] [PubMed] [Google Scholar]
- 12.Kantor SJ, Bigger JT, Jr, Glassman AH, Macken DL, Perel JM. Imipramine-induced heart block. A longitudinal case study. JAMA. 1975;231:1364–6. [PubMed] [Google Scholar]
- 13.Vohra J, Burrows G, Hunt D, Sloman G. The effect of toxic and therapeutic doses of tricyclic antidepressant drugs on intracardiac conduction. Eur J Cardiol. 1975;3:219–27. [PubMed] [Google Scholar]
- 14.Giardina EG, Bigger JT, Jr, Glassman AH, Perel JM, Kantor SJ. The electrocardiographic and antiarrhythmic effects of imipramine hydrochloride at therapeutic plasma concentrations. Circulation. 1979;60:1045–52. doi: 10.1161/01.cir.60.5.1045. [DOI] [PubMed] [Google Scholar]
- 15.Glassman AH, Bigger JT, Jr, Giardina EV, Kantor SJ, Perel JM, Davies M. Clinical characteristics of imipramine-induced orthostatic hypotension. Lancet. 1979;1:468–72. doi: 10.1016/s0140-6736(79)90824-9. [DOI] [PubMed] [Google Scholar]
- 16.Roose SP, Glassman AH, Giardina EG, Walsh BT, Woodring S, Bigger JT. Tricyclic antidepressants in depressed patients with cardiac conduction disease. Arch Gen Psychiatry. 1987;44:273–5. doi: 10.1001/archpsyc.1987.01800150093011. [DOI] [PubMed] [Google Scholar]
- 17.Glassman AH, Roose SP, Bigger JT., Jr The safety of tricyclic antidepressants in cardiac patients. Risk-benefit reconsidered. JAMA. 1993;269:2673–5. [PubMed] [Google Scholar]
- 18.Crome P, Newman B. Fatal tricyclic antidepressant poisoning. J R Soc Med. 1979;72:649–53. doi: 10.1177/014107687907200905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Callaham M, Kassel D. Epidemiology of fatal tricyclic antidepressant ingestion: implications for management. Ann Emerg Med. 1985;14:1–9. doi: 10.1016/s0196-0644(85)80725-3. [DOI] [PubMed] [Google Scholar]
- 20.Hulten BA. TCA poisoning treated in the intensive care unit. Pharmacopsychiatry. 1990;23(Suppl 1):14–6. doi: 10.1055/s-2007-1014524. [DOI] [PubMed] [Google Scholar]
- 21.Bigger JT, Giardina EG, Perel JM, Kantor SJ, Glassman AH. Cardiac antiarrhythmic effect of imipramine hydrochloride. N Engl J Med. 1977;296:206–8. doi: 10.1056/NEJM197701272960407. [DOI] [PubMed] [Google Scholar]
- 22.Giardina EG, Bigger JT., Jr Antiarrhythmic effect of imipramine hydrochloride in patients with ventricular premature complexes without psychological depression. Am J Cardiol. 1982;50:172–9. doi: 10.1016/0002-9149(82)90025-x. [DOI] [PubMed] [Google Scholar]
- 23.Giardina EG, Barnard T, Johnson L, Saroff AL, Bigger JT, Jr, Louie M. The antiarrhythmic effect of nortriptyline in cardiac patients with ventricular premature depolarizations. J Am Coll Cardiol. 1986;7:1363–9. doi: 10.1016/s0735-1097(86)80158-9. [DOI] [PubMed] [Google Scholar]
- 24.Biederman J. Sudden death in children treated with a tricyclic antidepressant. J Am Acad Child Adolesc Psychiatry. 1991;30:495–8. doi: 10.1097/00004583-199105000-00023. [DOI] [PubMed] [Google Scholar]
- 25.Boeck V, Jorgensen A, Fredricson Overo K. Comparative animal studies on cardiovascular toxicity of tri- and tetracyclic antidepressants and citalopram; relation to drug plasma levels. Psychopharmacology (Berl) 1984;82:275–81. doi: 10.1007/BF00427669. [DOI] [PubMed] [Google Scholar]
- 26.Overo KF. In: Citalopram: A New Antidepressant from Lundbeck Research. Montgomery SA, editor. Amsterdam, The Netherlands: Excerta Medica; 1989. pp. 22–30. [Google Scholar]
- 27.Wong DT, Bymaster FP, Engleman EA. Prozac (fluoxetine, Lilly 110140), the first selective serotonin uptake inhibitor and an antidepressant drug: twenty years since its first publication. Life Sci. 1995;57:411–41. doi: 10.1016/0024-3205(95)00209-o. [DOI] [PubMed] [Google Scholar]
- 28.Zarzar MN, Kingsley RS. Use of fluoxetine in a person with cardiac arrhythmias. Psychosomatics. 1990;31:235–6. doi: 10.1016/S0033-3182(90)72207-4. [DOI] [PubMed] [Google Scholar]
- 29.Roose SP, Glassman AH, Attia E, Woodring S, Giardina EG, Bigger JT., Jr Cardiovascular effects of fluoxetine in depressed patients with heart disease. Am J Psychiatry. 1998;155:660–5. doi: 10.1176/ajp.155.5.660. [DOI] [PubMed] [Google Scholar]
- 30.Roose SP. Treatment of depression in patients with heart disease. Biol Psychiatry. 2003;54:262–8. doi: 10.1016/s0006-3223(03)00320-2. [DOI] [PubMed] [Google Scholar]
- 31.Pacher P, Ungvari Z. Use of selective serotonin reuptake inhibitors and myocardial infarction. Circulation. 2002;105:e84. doi: 10.1161/01.cir.0000012607.30379.f5. [DOI] [PubMed] [Google Scholar]
- 32.Enemark B. The importance of ECG monitoring in antidepressant treatment. Nord J Psychiatry. 1993;47(suppl 30):57–65. [Google Scholar]
- 33.Buff DD, Brenner R, Kirtane SS, Gilboa R. Dysrhythmia associated with fluoxetine treatment in elderly patients with cardiac disease. J Am Psychiatry. 1991;52:174–176. [PubMed] [Google Scholar]
- 34.Friedman EH. Fluoxetine-induced bradycardia. J Clin Psychiatry. 1991;52:174–176. [PubMed] [Google Scholar]
- 35.Friedman EH. The role of the right hemisphere in fluoxetine-induced bradycardia and syncope [letter] J Clin Psychiatry. 1991;52:138–139. [PubMed] [Google Scholar]
- 36.Gardner SF, Rutherford WF, Munger MA, Panacek EA. Drug-induced supraventricular tachycardia:a case repore of fluoxetine. Ann Emerg Med. 1991;20:194–7. doi: 10.1016/s0196-0644(05)81222-3. [DOI] [PubMed] [Google Scholar]
- 37.Spier SA, Frontera MA. Unexpected deaths in depressed medical patients treated with fluoxetine. J Clin Psychiatr. 1991;52:377–382. [PubMed] [Google Scholar]
- 38.Masquelier I, Saint JO, Bourdiol MC, Boiffin A, Bouchon JP. Bradycardia and hypothermia in an elderly patient receiving fluoxetine. Presse Med. 1993;22:553–58. [PubMed] [Google Scholar]
- 39.Drake WM, Gordon GD. Heart block in a patient on propranolol and fluoxetine. Lancet. 1994;343:425–426. doi: 10.1016/s0140-6736(94)91265-3. [DOI] [PubMed] [Google Scholar]
- 40.Hussein S, Kaufman BM. Bradycardia associated with fluoxetine in an elderly patient with sick sinus syndrome. Postgrad Med J. 1994;70:56–60. doi: 10.1136/pgmj.70.819.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberge RJ, Martin TG. Mixed fluoxetine loxapine overdose and atrial flutter. Ann Emerg Med. 1994;23:586–590. doi: 10.1016/s0196-0644(94)70083-4. [DOI] [PubMed] [Google Scholar]
- 42.Appleby M, Mbewu A, Clarke B. Fluoxetine and ventricular torsade-is there a link? Int J Cardiol. 1995;49:178–180. doi: 10.1016/0167-5273(94)02270-s. [DOI] [PubMed] [Google Scholar]
- 43.Graudins A, Vossler C, Wang R. Fluoxetine-induced cardiotoxicity with response to bicarbonate therapy. Am J Emerg Med. 1997;15:501–503. doi: 10.1016/s0735-6757(97)90194-7. [DOI] [PubMed] [Google Scholar]
- 44.Anderson J, Compton SA. Fluoxetine induced bradycardia in presenile dementia. Ulster Med J. 1997;66:144–145. [PMC free article] [PubMed] [Google Scholar]
- 45.Marchiando RJ, Cook MD. Probable terfenadine-fluoxetine-associated cardiac toxicity. Ann Pharmacotherapy. 1995;29:937–38. doi: 10.1177/106002809502900922. [DOI] [PubMed] [Google Scholar]
- 46.Ravina T, Suarez ML, Mendez-Castrillon J. Fluoxetine-induced QTU interval prolongation, T wave alternans and syncope. Int J Cardiol. 1998;65:311–3. doi: 10.1016/s0167-5273(98)00095-3. [DOI] [PubMed] [Google Scholar]
- 47.Neely JL. Tonic clonic seizures and tachycardia induced by fluoxetine (Prozac) overdose. WV Med J. 1998;94:283–5. [PubMed] [Google Scholar]
- 48.Ellison JM, Milofsky JE, Ely E. Fluoxetine- induced bradycardia and syncope in two patients. J Clin Psychiatry. 1990;51:385–386. [PubMed] [Google Scholar]
- 49.Cherin P, Colvez A, Deville de Periere G, Sereni D. Risk of syncope in the elderly and consumption of drugs: a case-control study. J Clin Epidemiol. 1997;50:313–320. doi: 10.1016/s0895-4356(96)00385-x. [DOI] [PubMed] [Google Scholar]
- 50.McAnally LE, Threlkeld KR, Dreyling CA. Case report of a syncopal episode associated with fluoxetine. Ann Pharmacother. 1992;26:1091–1091. doi: 10.1177/106002809202600909. [DOI] [PubMed] [Google Scholar]
- 51.Sternbach H. Fluoxetine-associated potentiation of calcium-channel blockers. J Clin Psychopharmacol. 1991;11:390–91. doi: 10.1097/00004714-199112000-00019. [DOI] [PubMed] [Google Scholar]
- 52.Livshits ATL, Danenberg HD. Tachycardia, orthostatic hypotension and profound weakness due to concomitant use of fluoxetine and nifedipine. Pharmacopsychiatry. 1997;30:274–275. doi: 10.1055/s-2007-979508. [DOI] [PubMed] [Google Scholar]
- 53.Rich JM, Njo L, Roberts KW, Smith KP. Unusual hypotension and bradycardia in a patient receiving fenfluramine, phentermine and fluoxetine. Anesthesiology. 1998;88:529–531. doi: 10.1097/00000542-199802000-00034. [DOI] [PubMed] [Google Scholar]
- 54.Langlois RP, Paquette D. Sustained bradycardia during fluvoxamine and buspirone intoxication. Can J Psychiatry. 1994;39:126–7. doi: 10.1177/070674379403900217. [DOI] [PubMed] [Google Scholar]
- 55.Konig F, Hafele M, Hauger B, Loble M, Wossner S, Wolfersdorf M. Bradycardia after beginnig therapy with metaprolol and paroxetine. Psychiart Prax. 1996;23:244–5. [PubMed] [Google Scholar]
- 56.Erfurth A, Loew M, Dobmeier P, Wendler G. ECG changes after paroxetine. 3 case reports. Nervenarzt. 1998;69:629–31. doi: 10.1007/s001150050321. [DOI] [PubMed] [Google Scholar]
- 57.Personne M, Persson H, Sjoberg G. Picture of poisoning in citalopram overdose. Convulsions and ECG Symptoms can occur. Lakartidningen. 1997;94:1603–4. [PubMed] [Google Scholar]
- 58.Rodriguez de la Torre B, Dreher J, Malevany I, Bagli M, Kolbinger M, Omran H, et al. Serum levels and cardiovascular effects of tricyclic antidepressants and selective serotonin reuptake inhibitors in depressed patients. Ther Drug Monit. 2001;23:435–40. doi: 10.1097/00007691-200108000-00019. [DOI] [PubMed] [Google Scholar]
- 59.Cherin P, Colvez A, Deville de Periere G, Sereni D. Risk of syncope in the elderly and consumption of drugs: a case-control study. Clin Epidemiol. 1997;50:313–20. doi: 10.1016/s0895-4356(96)00385-x. [DOI] [PubMed] [Google Scholar]
- 60.Fuller RW, Holland DR, Yen TT, Bemis KG, Stamm NB. Antihypertensive effects of fluoxetine and L-5-hydroxytryptophan in rats. Life Sci. 1979;25:1237–42. doi: 10.1016/0024-3205(79)90466-1. [DOI] [PubMed] [Google Scholar]
- 61.Pacher P, Ungvari Z, Kecskemeti V, Koller A. Serotonin reuptake inhibitor, fluoxetine, dilates isolated skeletal muscle arterioles. Possible role of altered Ca2+ sensitivity. Br J Pharmacol. 1999;127:740–6. doi: 10.1038/sj.bjp.0702571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ungvari Z, Pacher P, Koller A. Serotonin reuptake inhibitor fluoxetine decreases arteriolar myogenic tone by reducing smooth muscle [Ca2+]i. J Cardiovasc Pharmacol. 2000;35:849–54. doi: 10.1097/00005344-200006000-00004. [DOI] [PubMed] [Google Scholar]
- 63.Pacher P, Ungvari Z, Kecskemeti V, Friedmann T, Furst S. Serotonin reuptake inhibitors fluoxetine and citalopram relax intestinal smooth muscle. Can J Physiol Pharmacol. 2001;79:580–4. [PubMed] [Google Scholar]
- 64.Ungvari Z, Pacher P, Kecskemeti V, Koller A. Fluoxetine dilates isolated small cerebral arteries of rats and attenuates constrictions to serotonin, norepinephrine, and a voltage-dependent Ca(2+) channel opener. Stroke. 1999;30:949–54. doi: 10.1161/01.str.30.9.1949. [DOI] [PubMed] [Google Scholar]
- 65.Rawling D, Fozzard H. Effects of imipramine on cellular electrophisiological properties of cardiac Purkinje fibers. J Pharmacol Exp Ther. 1979;209:371–375. [PubMed] [Google Scholar]
- 66.Tamargo J, Rodriguez S. Electrophysiological actions of chlorimipramine on guinea-pig ventricular fibres. Experimentia. 1979;35:366–7. doi: 10.1007/BF01964356. [DOI] [PubMed] [Google Scholar]
- 67.Rodriguez S, Tamargo J. Elektrophysiological effects of imipramine on bovine ventricular muscle and Purkinje fibres. Br J Pharmacol. 1980;70:15–20. doi: 10.1111/j.1476-5381.1980.tb10899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang CM, Parker CH, Jr, Maxwell RA. Electrophysiological Effects of Antidepressants on Mammalian Hearts and Crayfish Giant Axon. J Cardiovasc Pharmacol. 1981;3:101–112. doi: 10.1097/00005344-198101000-00009. [DOI] [PubMed] [Google Scholar]
- 69.Muir WW, Strauch SM, Schaal SF. Effects of tricyclic antidepressant drugs on the electrophysiological properties of dog Purkinje fibers. J Cardiovasc Pharmacol. 1982;4:82–90. doi: 10.1097/00005344-198201000-00014. [DOI] [PubMed] [Google Scholar]
- 70.Delpon E, Tamargo J, Sanchez-Chapula J. Further characterization of the effects of imipramine on plateau currents in guinea-pig ventricular myocytes. N-S Arch Pharmacol. 1991;344:645–652. doi: 10.1007/BF00174748. [DOI] [PubMed] [Google Scholar]
- 71.Valenzuela C, Chapula SJ, Delpon E, Elizalde A, Perez O, Tamargo J. Imipramine Blocks Rapidly Activating and Delays Slowly Activating K+ Current Activation in Guinea Pig Ventricular Myocytes. Circ Res. 1994;74:687–699. doi: 10.1161/01.res.74.4.687. [DOI] [PubMed] [Google Scholar]
- 72.Delpon E, Tamargo J, Sanchez-Chapula J. Effects of imipramine on the transient outward current in rabbit atrial single cells. Br J Pharmacol. 1992;106:464–469. doi: 10.1111/j.1476-5381.1992.tb14357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Casis O, Sanchez-Chapula JA. Mechanism of block of cardiac transient outward K+ current (I(to)) by antidepressant drugs. J Cardiovasc Pharmacol. 1998;32:527–34. doi: 10.1097/00005344-199810000-00004. [DOI] [PubMed] [Google Scholar]
- 74.Giles WR, Imaizumi Y. Comparison of potassium currents in rabbit atrial and ventricular cells. J Physiol. 1988;405:123–145. doi: 10.1113/jphysiol.1988.sp017325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Isenberg G, Tamargo J. Effect of imipramine on calcium and potassium currents in isolated bovine ventricular myocytes. Eur J Pharmacol. 1985;108:121–131. doi: 10.1016/0014-2999(85)90716-2. [DOI] [PubMed] [Google Scholar]
- 76.Rouet RH, Tisne-Versalles J, Adamantidis MM, Vincent A, Dupuis BA. Fundam Comparative in vivo and in vitro study of the cardiac effects of midalcipran and imipramine. Clin Pharmacol. 1989;3:237–44. doi: 10.1111/j.1472-8206.1989.tb00454.x. [DOI] [PubMed] [Google Scholar]
- 77.Delpón E, Valenzua C, Tamargo J. Tonic and frequency dependent Vmax block induced by imipramine in guinea-pig ventricular muscle fibres. J Cardiovasc Pharmacol. 1990;15:414–420. doi: 10.1097/00005344-199003000-00011. [DOI] [PubMed] [Google Scholar]
- 78.Manzanares J, Tamargo J. Electrophysiological effects of imipramine in nontreated and in imipramine pretreated rat atrial fibres. Br J Pharmacol. 1983;79:167–175. doi: 10.1111/j.1476-5381.1983.tb10509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matsuo S. Comparative effects of imipramine and propranolol on the transmembrane potentials of the isolated rabbit atria. Jpn J Pharmacol. 1967;17:279–286. doi: 10.1254/jjp.17.279. [DOI] [PubMed] [Google Scholar]
- 80.Shibata E, Drury T, Refsum H, Aldrete V, Giles W. Contribution of a transient outward current to repolarization in human atrium. Am J Physiol. 1989;257:H1773–1781. doi: 10.1152/ajpheart.1989.257.6.H1773. [DOI] [PubMed] [Google Scholar]
- 81.Ogata N, Narahashi T. Block of sodium channels by psychotropic drugs in single guinea-pig cardiac myocytes. Br J Pharmacol. 1989;97:905–913. doi: 10.1111/j.1476-5381.1989.tb12031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Escande D, Coulombe A, Faivre J, Deroubaix E, Coraboeuf E. Two types of transient outward currents in adult human atrial cells. Am J Physiol. 1987;252:H142–H148. doi: 10.1152/ajpheart.1987.252.1.H142. [DOI] [PubMed] [Google Scholar]
- 83.Casis O, Sanchez-Chapula JA. Mechanism of block of cardiac transient outward K+ current (I(to)) by antidepressant drugs. J Cardiovasc Pharmacol. 1998;32:527–34. doi: 10.1097/00005344-199810000-00004. [DOI] [PubMed] [Google Scholar]
- 84.Pacher P, Magyar J, Szigligeti P, Banyasz T, Pankucsi C, Korom Z, et al. Electrophysiological effects of fluoxetine in mammalian cardiac tissues. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:67–73. doi: 10.1007/s002109900154. [DOI] [PubMed] [Google Scholar]
- 85.Magyar J, Rusznak Z, Harasztosi C, Kortvely A, Pacher P, Banyasz T, et al. Differential effects of fluoxetine enantiomers in mammalian neural and cardiac tissues. Int J Mol Med. 2003;11:535–42. [PubMed] [Google Scholar]
- 86.Pacher P, Bagi Z, Lako-Futo Z, Ungvari Z, Nanasi PP, Kecskemeti V. Cardiac electrophysiological effects of citalopram in guinea pig papillary muscle comparison with clomipramine. Gen Pharmacol. 2000;34:17–23. doi: 10.1016/s0306-3623(99)00048-8. [DOI] [PubMed] [Google Scholar]
- 87.Mitchell MR, Powell T, Terrar DA, Twist VW. The effects of ryanodin, EGTA and low-sodium on action potentials in rat and guinea-pig ventricular myocytes:evidence for two inward currents during the plateau. Br J Pharmacol. 1990;81:543–550. doi: 10.1111/j.1476-5381.1984.tb10107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Khalifa M, Daleau P, Drolet B, Turseon Block of sodium channels underlies unheraldded cardiac toxicity observed with the antidepressant agent venlafaxine. J Mol Cell Cardiol. 1998;30:A136. [Google Scholar]
- 89.Tytgat J, Maertens C, Daenens P. Effect of fluoxetine on a neuronal, voltage-dependent potassium channel (Kv1.1) Br J Pharmacol. 1997;122:1417–1424. doi: 10.1038/sj.bjp.0701545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pancrazio JJ, Kamatchi GL, Roscoe AK, Lynch C. Inhibition of neuronal Na+ channels by antidepressant drugs. J Pharmacol Exp Ther. 1998;284:208–214. [PubMed] [Google Scholar]
- 91.Farrugia G. Modulation of ionic currents in isolated canine and human jejunal circular smooth muscle cells by fluoxetine. Gastroenterology. 1996;110:1438–45. doi: 10.1053/gast.1996.v110.pm8613049. [DOI] [PubMed] [Google Scholar]
- 92.Reeve HL, Nelson DP, Archer SL, Weir EK. Effects of fluoxetine, phentermine and venlafaxine on pulmonary arterial pressure and electrophysiology. AJP. 1999;276:L213–219. doi: 10.1152/ajplung.1999.276.2.L213. [DOI] [PubMed] [Google Scholar]
- 93.Choi JS, Hahn SJ, Rhie DJ, Yoon SH, Jo VH, Kim MS. Mechanism of fluoxetine block of cloned voltage-activated potassium channel Kv1.3. J Pharmacol Exp Ther. 1999;291:1–6. [PubMed] [Google Scholar]
- 94.Thomas D, Gut B, Wndt-Nordahl G, Kiehn J. The antidepressant drug fluoxetine is an inhibitor of human either-a-go-go-related gene (HERG) potassium channels. J Pharmacol Exp Ther. 2002;300:543–8. doi: 10.1124/jpet.300.2.543. [DOI] [PubMed] [Google Scholar]
- 95.Park KS, Kong IK, Park KC, Lee JW. Fluoxetine inhibits L-type Ca2+ and transient outward K+ currents in rat ventricular myocytes. Yonsei Med J. 1999;40:144–151. doi: 10.3349/ymj.1999.40.2.144. [DOI] [PubMed] [Google Scholar]
- 96.Stauderman KA, Gandhi VC, Jones DJ. Fluoxetine-induced inhibition of synaptosomal (3H)5-HT release: possible Ca2+-channel inhibition. Life Sci. 1992;50:2125–2138. doi: 10.1016/0024-3205(92)90579-e. [DOI] [PubMed] [Google Scholar]
- 97.Deák F, Lasztóczi B, Pacher P, Petheö GL, Kecskeméti V, Spät A. Inhibition of voltage-gated calcium channels by fluoxetine in rat hippocampal pyramidal cells. Neuropharmacol. 2000;39:1029–1036. doi: 10.1016/s0028-3908(99)00206-3. [DOI] [PubMed] [Google Scholar]
- 98.Choi BH, Choi JS, Ahn HS, Kim MJ, Rhie DJ, Yoon SH, et al. Fluoxetine blocks cloned neuronal A-type K+ channels Kv1.4. Neuroreport. 2003;14:2451–5. doi: 10.1097/00001756-200312190-00032. [DOI] [PubMed] [Google Scholar]
- 99.Choi JS, Choi BH, Ahn HS, Kim MJ, Rhie DJ, Yoon SH, et al. Mechanism of block by fluoxetine of 5-hydroxytryptamine3 (5-HT3)-mediated currents in NCB-20 neuroblastoma cells. Biochem Pharmacol. 2003;66:2125–32. doi: 10.1016/j.bcp.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 100.Terstappen GC, Pellacani A, Aldegheri L, Graziani F, Carignani C, Pula G, et al. The antidepressant fluoxetine blocks the human small conductance calcium-activated potassium channels SK1, SK2 and SK3. Neurosci Lett. 2003;346:85–8. doi: 10.1016/s0304-3940(03)00574-3. [DOI] [PubMed] [Google Scholar]
- 101.Wang SJ, Su CF, Kuo YH. Fluoxetine depresses glutamate exocytosis in the rat cerebrocortical nerve terminals (synaptosomes) via inhibition of P/Q-type Ca2+ channels. Synapse. 2003;48:170–7. doi: 10.1002/syn.10200. [DOI] [PubMed] [Google Scholar]
- 102.Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by fluoxetine (Prozac) Br J Pharmacol. 2003;138:1119–28. doi: 10.1038/sj.bjp.0705172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hajdu P, Ulens C, Panyi G, Tytgat J. Drug- and mutagenesis-induced changes in the selectivity filter of a cardiac two-pore background K+ channel. Cardiovasc Res. 2003;58:46–54. doi: 10.1016/s0008-6363(02)00831-3. [DOI] [PubMed] [Google Scholar]
- 104.Bian JT, Yeh JZ, Aistrup GL, Narahashi T, Moore EJ. Inhibition of K+ currents of outer hair cells in guinea pig cochlea by fluoxetine. Eur J Pharmacol. 2002;453:159–66. doi: 10.1016/s0014-2999(02)02421-4. [DOI] [PubMed] [Google Scholar]
- 105.Witchel HJ, Pabbathi VK, Hofmann G, Paul AA, Hancox JC. Inhibitory actions of the selective serotonin re-uptake inhibitor citalopram on HERG and ventricular L-type calcium currents. FEBS Lett. 2002;512:59–66. doi: 10.1016/s0014-5793(01)03320-8. [DOI] [PubMed] [Google Scholar]
- 106.Maertens C, Droogmans G, Verbesselt R, Nilius B. Block of volume-regulated anion channels by selective serotonin reuptake inhibitors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:158–65. doi: 10.1007/s00210-002-0567-5. [DOI] [PubMed] [Google Scholar]
- 107.Pato MT, Murphy DL, Devane CL. Sustained plasma concentrations of fluoxetine/or norfluoxetine four or eight weeks after fluoxetine discontinuation. J Clin Psychopharmacol. 1991;11:224–225. doi: 10.1097/00004714-199106000-00024. [DOI] [PubMed] [Google Scholar]
- 108.Potter WZ, Hollister L. In: Basic and Clinical Pharmacology Antipsychotic agents and lithium. Katzung B, editor. New York: McGraw-Hill Company; 2001. pp. 478–97. [Google Scholar]
- 109.Burris KD, Molski TF, Xu C, Ryan E, Tottori K, Kikuchi T, et al. Aripiprazole, a novel antipsychotic is a high-affinity partial agonist at human D2 receptors. J Pharmacol Exp Ther. 2002;302:381–89. doi: 10.1124/jpet.102.033175. [DOI] [PubMed] [Google Scholar]
- 110.Jordan S, Koprivica V, Chen R, Tottori K, Kikuchi T, Altar CA. The antipsychotic aripiprazole as a potent partial agonist at the human 5-HT1A receptor. Eur J Pharmacol. 2002;441:137–40. doi: 10.1016/s0014-2999(02)01532-7. [DOI] [PubMed] [Google Scholar]
- 111.Taylor DM. Aripiprazole: a review of its pharmacology and clinical use. Int J Clin Pract. 2003;63:49–54. [PubMed] [Google Scholar]
- 112.Henderson RA, Lane S, Henry JA. Life-threatening ventricular arrhythmia (torsades de pointes) after haloperidol overdose. Hum Exp Toxicol. 1991;10:59–62. doi: 10.1177/096032719101000110. [DOI] [PubMed] [Google Scholar]
- 113.Metzger E, Friedman R. Prolongation of the corrected QT and torsades de pointes cardiac arrhythmia associated with intravenous haloperidol in the medically ill. J Clin Psychopharmacol. 1993;13:128–32. [PubMed] [Google Scholar]
- 114.Committee on Safety of Medicines. QT interval prolongation with antipsychotics. Curr Problems in Pharmacovigilance. 2001;27:4. [Google Scholar]
- 115.Krahenbuhl S, Sauter B, Kupferschmiedt H, Krause M, Wyss PA, Meier PJ. Case report: reversible QT prolongation with torsades de pointes in a patient with pimozide interaction. Am J Med Sci. 1995;309:315–6. doi: 10.1097/00000441-199506000-00003. [DOI] [PubMed] [Google Scholar]
- 116.Barnett AA. Safety concerens over antipsychotic drug, sertindole. Lancet. 1996;348:256–57. [Google Scholar]
- 117.Pezawas I, Quinner S, Moertl D, Tauscher J, Barnes C, Kufferle B, et al. Efficacy, cardiac safety and tolerability of sertindole: a drug surveillance. In Clin Psychopharmacol. 2000;15:207–14. doi: 10.1097/00004850-200015040-00003. [DOI] [PubMed] [Google Scholar]
- 118.Glassman AH, Bigger JT., Jr Antipsychotic drugs: prolonged QTc interval, torsades de pointes, suddend death. Am J Psychiatry. 2001;158:1774–82. doi: 10.1176/appi.ajp.158.11.1774. [DOI] [PubMed] [Google Scholar]
- 119.Warner JP, Barnes TR, Henry JA. Electrocardiographic changes in patients receiving neuroleptic medication. Acta Psychiatr Scand. 1996;93:311–3. doi: 10.1111/j.1600-0447.1996.tb10653.x. [DOI] [PubMed] [Google Scholar]
- 120.Bucley NA, Whyte JM, Dawson AH. Cardiotoxicity more common in thioridazine overdose than with other neuroleptics. J Toxicol Clin Toxicol. 1995;33:199–204. doi: 10.3109/15563659509017984. [DOI] [PubMed] [Google Scholar]
- 121.Lande G, Drouin E, Gauthier C, Chevallier JC, Godin JF, Chiffoleau A, et al. Arrhythmogenic effects of sultopride chlorhydrate clinical and cerllular electrophysiological correlation. Ann Fr Anaesth Reanim. 1992;11:629–35. doi: 10.1016/s0750-7658(05)80782-8. [DOI] [PubMed] [Google Scholar]
- 122.Gajwani P, Pozuelo L, Tesar GE. QT interval prolongation associated with quietapine (Seroquel) overdose. Psychosomatica. 2000;31:63–5. doi: 10.1016/S0033-3182(00)71175-3. [DOI] [PubMed] [Google Scholar]
- 123.Beelen AP, Yeo KT, Lewis LD. Asymptomatic QTc prolongation associated with quetiapine fumarate overdose in a patient being treated with risperidone. Hum Exp Toxicol. 2001;20:215–219. doi: 10.1191/096032701678766778. [DOI] [PubMed] [Google Scholar]
- 124.Angelink MW, Majewski T, Wurthmann C, Lukas K, Ullrich H, Linka T, et al. Effects of newer atypical antipsychotics on autonomic neurocardiac function: a comparison between amisupride, olanzapine, sertindole and clozapine. J Clin Psychopharmacol. 200;21:8–13. doi: 10.1097/00004714-200102000-00003. [DOI] [PubMed] [Google Scholar]
- 125.Ravin DS, Levenson JW. Fatal cardiac event following initiation of risperidone therapy. Ann Pharmacother. 1997;31:867–70. doi: 10.1177/106002809703100712. [DOI] [PubMed] [Google Scholar]
- 126.Zarate CA, Baldessarini RJ, Siegel AJ, Nakamura A, McDonald J, Muir-Hutchinson LA, et al. Risperidone in elderly: a pharmacoepidemiologic study. J Clin Psychiatry. 1997;58:311–7. doi: 10.4088/jcp.v58n0705. [DOI] [PubMed] [Google Scholar]
- 127.Psychopharmacological Drugs Advisory Committee: Briefing document for Zeldox capsules(Ziprasidone HCl) The Food and Drug Administration (FDA) of the U.S. Department of Health and Human Services; Rockville, MD; 18 July 2000; NDA-825: 1–173.
- 128.Mehtonen OP, Aranko K, Malkonen L, Vapaatalo H. A survey of sudden death associated with the use of antipsychotic or antidepressant drugs: 49 cases in Finland. Acta Pschaitr Scand. 1991;84:58–64. doi: 10.1111/j.1600-0447.1991.tb01421.x. [DOI] [PubMed] [Google Scholar]
- 129.Reilly JG, Ayis SA, Ferrier IN, Jones SJ, Thomas SH. QTc-interval abnormalities and psychotropic drug therapy in psychiatric patients. Lancet. 2000;355:1048–52. doi: 10.1016/s0140-6736(00)02035-3. [DOI] [PubMed] [Google Scholar]
- 130.Flockhart DA, Drici MD, Kerbusch T, Soukhova N, Richard E, Pearle PL, et al. Studies on the mechanism of fatal clarithromycinpimozide interaction in a patient with Tourette syndrome. J Clin Psychopharmacol. 2000;20:317–24. doi: 10.1097/00004714-200006000-00005. [DOI] [PubMed] [Google Scholar]
- 131.Hunt N, Stern TA. The association between intravenous haloperidol and torsade de pointes three cases and a literature review. Psychosomatics. 1995;36:541–49. doi: 10.1016/S0033-3182(95)71609-7. [DOI] [PubMed] [Google Scholar]
- 132.Ray WA, Meredith S, Thapa PB, Meador KG, Hall K, Murray KT. Antipsychotics and the risk of sudden cardiac death. Arch Gen Psychiatry. 2001;58:1161–1167. doi: 10.1001/archpsyc.58.12.1161. [DOI] [PubMed] [Google Scholar]
- 133.Czekalla J, Beasley CM, Jr, Delva MA, Berg PH, Grundy S. Analysis of the QTc interval during olanzapine treatment of patients with schizophrenia and related psychosis. J Clin Psychiatry. 2001;62:191–98. doi: 10.4088/jcp.v62n0310. [DOI] [PubMed] [Google Scholar]
- 134.Suspension of Avaialbility of Serdolect (Sertindole). London, UK Department of Health. Committee on Safety of Medicine, Dec2, 1998
- 135.Ruschena D, Mullen PE, Burgess P, Cordner SM, Barry-Walsh J, Drummer OH, et al. Sudden death in psychiatric patients. Br J Psychiatry. 1998;172:331–336. doi: 10.1192/bjp.172.4.331. [DOI] [PubMed] [Google Scholar]
- 136.Brown S, Inskip H, Barraclough B. Causes of the excess mortality of schizophrenia. Br J Psychiatry. 2000;177:212–21. doi: 10.1192/bjp.177.3.212. [DOI] [PubMed] [Google Scholar]
- 137.Kilian JG, Kerr K, Lawrence C, Celermajer DS. Myocarditis and cardiomyopathy associated with clozapine. Lancet. 1999;354:1841–1845. doi: 10.1016/s0140-6736(99)10385-4. [DOI] [PubMed] [Google Scholar]
- 138.Hagg S, Spigset O, Bate A, Sonderstorm DG. Myocarditis releted to clozapine treatment. J Clin Psychopharmacol. 2001;21:382–388. doi: 10.1097/00004714-200108000-00005. [DOI] [PubMed] [Google Scholar]
- 139.Capel MM, Colbridge MG, Henry JA. Overdose profiles of new antipsychotic agents. Int J Neuropsychopharmacol. 2000;3:51–4. doi: 10.1017/S1461145700001760. [DOI] [PubMed] [Google Scholar]
- 140.Sheridan DJ. Drug-onduced proarrhythmic effects: assessment of changes in QT interval. Br J Clin Pharmacol. 2000;50:297–302. doi: 10.1046/j.1365-2125.2000.00274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Witchel HJ, Hancox JC. Familial and acqired long QT syndrome and the cardiac rapid delayed rectifier potassium current. Clin Exp Pharmacol Physiol. 2000;27:753–66. doi: 10.1046/j.1440-1681.2000.03337.x. [DOI] [PubMed] [Google Scholar]
- 142.DeCicco M, et al. Pharmacokinetic and pharmcodynamic effects of high doses continous intravenous verapamil infusion. Crtl Care Med. 1999;27:332–339. doi: 10.1097/00003246-199902000-00040. [DOI] [PubMed] [Google Scholar]
- 143.Winters SL, Schetzer P, Kupersmith J, Gomes JA. Verpamil-induced polyporphous ventricular tachycardia. J Am Coll Cardiol. 1985;6:257–259. doi: 10.1016/s0735-1097(85)80287-4. [DOI] [PubMed] [Google Scholar]
- 144.Roden DM. Acquired long QT syndromes and the risk of proarrhythmia. J Cardiovasc Electrophysiol. 2000;11:938–940. doi: 10.1111/j.1540-8167.2000.tb00077.x. [DOI] [PubMed] [Google Scholar]
- 145.Drolet B, Zhang S, Deschenes D, Rail J, Nadeau S, Zhou Z, et al. Droperidol lengthens cardiac repolarization due to block of the rapid component of the delayed rectifier potassium current. J Cardiovasc Electrophysiol. 1999a;10:1597–1604. doi: 10.1111/j.1540-8167.1999.tb00224.x. [DOI] [PubMed] [Google Scholar]
- 146.Suessbrich H, Schönherr R, Heinemann SH, Attali B, Lang F, Busch AE. The inhibitory effect of the antipsychotic drug haloperidol on HERG potassium channels expressed in Xenopus oocytes. Br J Pharmacol. 1997;120:968–74. doi: 10.1038/sj.bjp.0700989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kongsamut S, Kang J, Chen XL, Roehr J, Rampe D. A comparison of the receptor binding and HERG channel affinities for a series of antipsychotic drugs. Europ J Pharmacol. 2002;450:37–41. doi: 10.1016/s0014-2999(02)02074-5. [DOI] [PubMed] [Google Scholar]
- 148.Crumb WJ, Beasley CM, Jr, Thornton A, Breier A. Cardiac ion channel blocking profile of olanzapine and other antipsychotics. 38th Annual Meeting American College of Neuropsychopharmacology; Acapulco, Mexico. 1999. [Google Scholar]
- 149.Drolet B, Vincent F, Rail J, Chahine M, Deschenes D, Nadeau S, et al. Thioridazine lengthens repolarization of cardiac ventricular myocytes by blocking the delayed rectifier potassium current. J Pharmacol Exp Ther. 1999b;288:1261–68. [PubMed] [Google Scholar]
- 150.Shuba YM, Degtiar VE, Osipenko VN, Naidenov VG, Woosley RL. Testosterone-mediated modulation of HERG blockade by proarrhythmic agents. Biochem Pharmacol. 2001;62:41–9. doi: 10.1016/s0006-2952(01)00611-6. [DOI] [PubMed] [Google Scholar]
- 151.Magyar J, Bányász T, Bagi Zs, Pacher P, Szentandrassy N, et al. Electrophyssiological effects of risperidone inmammalian cardiac cells. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:350–6. doi: 10.1007/s00210-002-0595-1. [DOI] [PubMed] [Google Scholar]
- 152.Gluais P, Bastide M, Caron J, Adamantidis M. Risperidone prolongs cardiac action potential through reduction of K+ currents in rabbit myocytes. Eur J Pharmacol. 2002;444:123–32. doi: 10.1016/s0014-2999(02)01626-6. [DOI] [PubMed] [Google Scholar]
- 153.Rampe D, Murawsky MK, Grau J, Lewis EW. The antipsychotic agent sertindole is a high affinity antagonist of the human cardiac potassium channel HERG. J Pharmacol Exp Ther. 1998;286:788–93. [PubMed] [Google Scholar]
- 154.Kang UG, Kwon JS, Ahn YM, Chung SJ, Ha JH, Koo ZJ, et al. Electrocardiographic abnormabilities in patients treated with clozapine. J Clin Psychiatry. 2000;61:441–46. doi: 10.4088/jcp.v61n0609. [DOI] [PubMed] [Google Scholar]
- 155.Drolet B, Yang T, Daleau P, Roden DM, Turgeon J. Risperidone prolong cardiac repolariyation by blocking the rapid component of the delayed rectifier potassium current. J Cardiovasc Pharmacol. 2003;41:934–37. doi: 10.1097/00005344-200306000-00016. [DOI] [PubMed] [Google Scholar]
- 156.Enyeart JJ, Dirksen RT, Sharma VK, Williford DJ, Sheu SS. Antipsychotic pimozide is a potent Ca2+ channel blocker in heart. Mol Pharmacol. 1990;37:752–757. [PubMed] [Google Scholar]
- 157.Kang J, Chen XL, Rampe D. The antipsychotic drugs sertindole and pimozide block erg3, a human brain K+ channel. Biochem Biophys Res Commun. 2001;286:499–504. doi: 10.1006/bbrc.2001.5434. [DOI] [PubMed] [Google Scholar]
- 158.Tie H, Walker BD, Valenzuela SM, Breit SN, Campbell TJ. The heart of psychotropic drug therapy. Lancet. 2000;355:1825. doi: 10.1016/s0140-6736(05)73083-x. [DOI] [PubMed] [Google Scholar]
- 159.Drici MD, Wang WX, Liu XK, Woosley RL, Flockhart DA. Prolongation of QT interval in isolated feline hearts by antipsychotic drugs. J Clin Psychopharmacol. 1998;18:477–81. doi: 10.1097/00004714-199812000-00011. [DOI] [PubMed] [Google Scholar]
- 160.Abbott GW, Sesti F, Splawski I, Buck M, Lehmann MH, Timothy KW, et al. MiRpP1 forms IKr potassium with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–87. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 161.vonMoltke LL, Greenblatt DJ, Schmide J, Wright CE, Harmatz JS, Shader RJ. In vitro approaches to predicting drug interactions in vivo. Biochem. 1998;55:113–22. doi: 10.1016/s0006-2952(97)00239-6. [DOI] [PubMed] [Google Scholar]
- 162.Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Anzelevitch C, et al. The potential for QT prolongation and pro-arrhythmia by non- antiarrhythmic drugs: Clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Cardiovasc Res. 2000;47:219–33. doi: 10.1016/s0008-6363(00)00119-x. [DOI] [PubMed] [Google Scholar]
- 163.Fermini B, Fossa AA. The impact of drug-induced QT interval prolongation on drug discovery and development. Nature Review. 2003;2:439–47. doi: 10.1038/nrd1108. [DOI] [PubMed] [Google Scholar]
- 164.Ito K, Nakayawa K, Koiyumi S, Liu M, Takeuchi K, Hashimoto T, et al. Inhibition of antipszchotic drugs of L/type Ca2+ channel current in PC12 cells. Eur J Pharmacol. 1996;314:143–50. doi: 10.1016/s0014-2999(96)00500-6. [DOI] [PubMed] [Google Scholar]
- 165.Gessner G, Heinemann SH. Inhibition of hEAG1 and HERG1 potassium channels by clofilium and its tertiary analogue LY97241. Br J Pharmacol. 2003;138:161–71. doi: 10.1038/sj.bjp.0705025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Habuchi Y, Furukawa T, Tanaka H, Tsujimura Y, Yoshimura M. Block of Na+ channels by imipramine in guinea-pig cardiac ventricular cells. J Pharmacol Exp Ther. 1991;256:1072–1081. [PubMed] [Google Scholar]
- 167.Bou-Abboud E, Nattel S. Molecular mechanisms of the reversal of imipramine-induced sodium channel blockade by alkalinization in human cardiac myocytes. Cardiovasc Res. 1998;38:395–404. doi: 10.1016/s0008-6363(98)00011-x. [DOI] [PubMed] [Google Scholar]
- 168.Teschemacher AG, Seward EP, Hancox JC, Witchel HJ. Inhibition of the current of the heterologously expressed HERG potassium channels by imipramine and amitriptyline. Brit J Pharmacol. 1999;128:479–485. doi: 10.1038/sj.bjp.0702800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jo SH, Youm JB, Lee CO, Earm YE, Ho WK. Blockade of the HERG human cardiac K + channel by the antidepressant drug amitriptyline. Brit J Pharmacol. 2000;129:1474–1480. doi: 10.1038/sj.bjp.0703222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Barber M, Starmer CF, Grant AO. Blockade of cardiac sodium channels by amitriptyline and diphenylhidantoin. Evidence for two use-dependent binding sites. Circ Res. 1991;69:677–696. doi: 10.1161/01.res.69.3.677. [DOI] [PubMed] [Google Scholar]
- 171.Hahn SJ, Cho JS, Rhie DJ, Oh CS, Jo YH, Kim MS. Inhibition by fluoxetine of voltage-activated ion channels in rat PC12 cells. Eur J Pharmacol. 1999;367:113–118. doi: 10.1016/s0014-2999(98)00955-8. [DOI] [PubMed] [Google Scholar]
- 172.Hamplova-Peichlova J, Krusek J, Pacit I, Slavicek L, Lisa V, Vyskocil F. Citalopram inhibits L-type calcium channel current in rat cardiomycytes in culture. Physiol Res. 2002;51:317–321. [PubMed] [Google Scholar]
- 173.de Paulis T. The discovery of epidepride and its analogs as high-affinity radioligands for imaging extrastriatal dopamine D(2) receptors in human brain. Curr Pharm Design. 2003;9(8):673–96. doi: 10.2174/1381612033391135. [DOI] [PubMed] [Google Scholar]
- 174.Ito C. Analysis of overall gene expression induced by amphetamine and phencyclidine: novel targets for the treatment of drug psychosis and schizophrenia. Curr Pharm Design. 2002;8(2):147–53. doi: 10.2174/1381612023396528. [DOI] [PubMed] [Google Scholar]
- 175.Takano H, Ohtsuka M, Akazawa H, Toko H, Harada M, Hasegawa H, Nagai T, Komuro I. Pleiotropic effects of cytokines on acute myocardial infarction: G-CSF as a novel therapy for acute myocardial infarction. Curr Pharm Design. 2003;9(14):1121–7. doi: 10.2174/1381612033455008. [DOI] [PubMed] [Google Scholar]