

Abstract
A mutation within subdomain 1 of the hepatitis C virus (HCV) NS3 helicase (NS3-Q221L) (M. Yi, Y. Ma, J. Yates, and S. M. Lemon, J. Virol. 81:629-638, 2007) rescues a defect in production of infectious virus by an intergenotypic chimeric RNA (HJ3). Although NS3-Gln-221 is highly conserved across HCV genotypes, the Leu-221 substitution had no effect on RNA replication or NS3-associated enzymatic activities. However, while transfection of unmodified HJ3 RNA failed to produce either extracellular or intracellular infectious virus, transfection of HJ3 RNA containing the Q221L substitution (HJ3/QL) resulted in rapid accumulation of intracellular infectious particles with release into extracellular fluids. In the absence of the Q221L mutation, both NS5A and NS3 were recruited to core protein on the surface of lipid droplets, but there was no assembly of core into high-density, rapidly sedimenting particles. Further analysis demonstrated that a Q221N mutation minimally rescued virus production and led to a second-site I399V mutation in subdomain 2 of the helicase. Similarly, I399V alone allowed only low-level virus production and led to selection of an I286V mutation in subdomain 1 of the helicase which fully restored virus production, confirming the involvement of both major helicase subdomains in the assembly process. Thus, multiple mutations in the helicase rescue a defect in an early-intermediate step in virus assembly that follows the recruitment of NS5A to lipid droplets and precedes the formation of dense intracellular viral particles. These data reveal a previously unsuspected role for the NS3 helicase in early virion morphogenesis and provide a new perspective on HCV assembly.
Hepatitis C virus (HCV) is a small, enveloped virus that is classified within the genus Hepacivirus of the family Flaviviridae. Over 2% of the world's population is chronically infected with this virus and is thus at risk for progressive hepatic fibrosis, cirrhosis, and hepatocellular carcinoma (2, 37). The HCV genome is a positive-sense RNA about 9,600 nucleotides in length, encoding a single polyprotein that is processed by host and viral proteases into at least 10 distinct structural and nonstructural proteins (3, 36, 39). The structural proteins core, E1, and E2, are processed from the N-terminal third of the polyprotein, while the nonstructural proteins NS3, NS4A, NS4B, NS5A, and NS5B are derived from the C-terminal two-thirds of the polyprotein and are sufficient to support viral RNA replication (28). p7 and NS2 are located within the polyprotein between these structural and nonstructural proteins. p7 is a small membrane protein with ion channel activity (9). NS2 contains a cysteine-like protease domain within its C-terminal residues and, together with the N-terminal region of NS3, constitutes an autoprotease that cleaves the junction between NS2 and NS3 (29, 36, 43). The functional role of these two proteins in the life cycle of HCV has been uncertain since they are not required for viral RNA replication, but increasing data support their involvement in the assembly and release of infectious virus (12, 40, 51).
New opportunities to study the entire life cycle of HCV, especially the processes involved in virus entry, assembly, and release, have been provided by the development of cell culture-based systems for production of infectious HCV from genotype 2a JFH1 or genotype 1a H77S cDNAs (26, 45, 52, 53). While it has been clear that the core protein and the two envelope proteins, E1 and E2, must be involved in assembly, little was known of the late events in the virus life cycle before the advent of these cell culture-infecting viruses. Using JFH1 virus, Gastaminza et al. (7) found that infectious HCV particles are assembled within an intracellular compartment (presumably the endoplasmic reticulum) and then undergo further maturation to a less-dense particle before or during their release into the extracellular milieu. Intra- and extracellular viral particles are similar in size but differ in their buoyant density, indicating that their physical properties are different (7). Other recent data suggest that hepatocellular processes that are normally involved in the assembly and secretion of very low density lipoproteins may contribute at multiple steps in the assembly and release of infectious particles, including an early step in the assembly of infectious, high-density particles within the endoplasmic reticulum and their subsequent transformation to lower-density particles that are exported from the cell (6, 11). Other work indicates that the lipid droplet, a cellular organelle involved in the storage of neutral lipids, plays a central role in the assembly of infectious intracellular particles (32). Both core and NS5A proteins associate with the surface of lipid droplets (30, 38), and recruitment of NS5A to the lipid droplet appears to be essential for virus assembly (32). As indicated above, there has also been growing recognition that p7 and NS2 also have functions related to the assembly and release of infectious virus (12, 40, 51). It is not yet clear how these various sets of observations relate to each other, however, and much remains to be learned about the processes of virus assembly and release.
We have previously reported the construction of intergenotypic, chimeric HCV RNAs that retain the robust RNA replication properties of the JFH1 virus from which their nonstructural proteins (NS3 to NS5B) are derived (51). We have shown that a number of mutations selected during the replication of these chimeric RNAs in transfected cells dramatically promote the release of infectious virus into cell culture supernatants (51). These mutations appear to compensate for incompatibilities in genome segments derived from different genotypes of HCV in these chimeras and thus may provide useful insights into the mechanisms involved in assembly and release of infectious virus. The genome of the H-NS2/NS3-J chimera (referred to here as HJ3) is comprised of genotype 1a H77c sequence spanning the core to the NS2 region, placed within the corresponding region of the genotype 2a JFH1 genome (51). Unlike both parental viral genomes (45, 52, 53), which release infectious virus into supernatant fluids within 48 h of transfection, this chimeric RNA does not produce any infectious virus despite evidence for robust RNA replication in the transfected cells. However, approximately 9 days after electroporation, large amounts of infectious virus begin to be released from the cells (51). Sequencing results from multiple transfection experiments with HJ3 suggested that a single amino acid change (NS3-Q221L) within the helicase domain of NS3 was responsible for this change in the virus production phenotype. This was confirmed when the mutation was engineered back into the HJ3 chimera (HJ3/QL) (51). This was an intriguing finding, since the HCV NS3 protein had not been implicated previously in either virus assembly or release.
NS3 is a multifunctional protein with several associated enzymatic activities. The N-terminal residues of NS3 contribute to the NS2/NS3 autoprotease which is responsible for cis-cleavage between the NS2 and NS3 sequences in the polyprotein (47). The N-terminal third of the protein also possesses serine protease activity, which is dependent upon an interaction with NS4A cofactor sequence and is responsible for processing the nonstructural proteins at the NS3-NS4A, NS4A-NS4B, NS4B-NS5A, and NS5A-NS5B junctions (3). The NS3/4A protease has also been shown to modulate innate cellular antiviral responses by cleaving two cellular adaptor proteins, TRIF and MAVS (also known as IPS-1, Cardif, or VISA), that play important roles in signal transduction leading to beta interferon synthesis (22, 24, 31). NS3 also possesses NTPase and helicase activities residing within the C-terminal two-thirds of the protein and forms a domain that is structurally distinct from the protease (16, 33). The mutation we identified as responsible for the production of infectious virus by the HJ3 chimeric RNA, NS3-Q221L, is located near the N terminus of the helicase/NTPase domain.
Interestingly, the NS3 protein of Kunjin virus (KUN), a “classical” flavivirus assigned to the genus Flavivirus of the Flaviviridae family, has also been implicated in virus assembly (27). Although an exact role has not been determined for the KUN NS3 protein, there is genetic evidence that the NS3 protein must be expressed in cis to generate infectious virus. Recent data indicate that it is in fact the Kunjin NS3 protein, and not the nucleotide sequence encoding it, that is involved in assembly (35). In addition, a viral assembly defect caused by a mutation within the NS2A sequence of yellow fever virus (YFV), another flavivirus, can be rescued by a second-site mutation within the NS3 helicase domain, suggesting that NS3 is involved in production of infectious YFV (20). Thus, although not recognized previously for HCV, there are strong precedents for the involvement of NS3 in assembly of other members of the Flaviviridae.
Here, we describe experiments aimed at elucidating how the NS3-Q221L mutation promotes the production of infectious HJ3 virus. Our results indicate the involvement of multiple subdomains within the HCV NS3 helicase at an early step in viral assembly that is required for the formation of infectious, high-density intracellular particles containing core protein in association with viral RNA.
MATERIALS AND METHODS
Plasmids.
The construction of plasmids containing the chimeric cDNAs pHJ3 (pH-NS2/NS3-J), pHJ3/QL (pH-NS2/NS3-J/QL), and pHJ2 (pH-(NS2)-J) has been described previously (51). pHJ2/QL and pJFH1/QL were constructed by replacing the small AvrII/NsiI fragment in each with the corresponding fragment containing the Q221L substitution in pHJ3/QL. Additional substitutions of the NS3 Gln-221 residue to Leu (TTA), Ile (ATT), Val (GTT), and Glu (GAA) were introduced into HJ3 by QuikChange mutagenesis (Stratagene) followed by exchange of the AvrII/NsiI fragment. pHJ3/ΔE1p7 contains an in-frame deletion spanning the E1p7 coding region, with the sequence encoding the C terminus of the core fused directly to the N terminus of NS2. It was constructed by QuikChange mutagenesis (Stratagene) using primers sets described previously (52). pHJ3/QL/ΔE1p7 was constructed by replacing the large EcoRI/AvrII fragment of pHJ3/ΔE1p7 with that of pHJ3/QL. pHJ3/GND was constructed by replacing the large EcoRI/AvrII fragment of pHJ3 with that of pJFH1/GND. pJFH1/QL/ΔE1E2 was constructed by replacing the large EcoRI/AvrII fragment of pJFH1/ΔE1E2 with that of pJFH1/QL (45). Polyprotein expression vectors were constructed by amplifying sequences encoding the NS2-NS4A segment from pHJ3 and pHJ3/QL by PCR, and subcloning these into the vector pcDNA6/V5-His, modified to contain the encephalomyocarditis virus internal ribosome entry site sequence upstream of the inserted sequence and under the control of the cytomegalovirus and T7 promoters. To construct pJtat2Aneo, the sequence spanning the encephalomyocarditis virus internal ribosome entry site and JFH1 NS3-3′NTR region of psgJFH1 (kindly provided by Takaji Wakita) was first inserted into pHtat2ANeo (50) at KpnI/XbaI restriction sites. The EcoRI/ApaLI fragment of sgJFH1 containing the JFH1 5′NTR sequence was subsequently spliced into the resulting plasmid to make pJtat2ANeo. The Q221L mutation was engineered into pJtat2ANeo by replacement of the AvrII/NsiI fragment of pJtat2ANeo with the corresponding fragment from pHJ3/QL. The sequences of each of the introduced base changes and the modified DNA segments were verified by DNA sequence analysis.
For construction of vectors expressing the glutathione S-transferase (GST)-fused JFH1 NS3 protein with or without the Q221L mutation, the JFH1 NS3 sequence was amplified from pJFH1 or pJFH1/QL by PCR using the primer set JNS3-s (5′-CGGGATCCGGTTCAGCTCCCATCACTGCTTATG-3′), which contains a BamHI site followed by codons for a Gly-Ser linker preceding NS3, and JNS3-as (5′-CGGAATTCTCAGGTCATGACCTCAAGGTC-3′), which contains an EcoRI site following the termination codon. PCR fragments were digested with BamHI/EcoRI and cloned into related sites in pGEX-4T-3 (Amersham Biosciences) to make pGST-JFH1/NS3 and pGST-JFH1/NS3/QL. To express the NS2-NS4A segments of the polyprotein in order to determine cis-processing between NS2 and NS3, cDNAs encoding the NS2-NS3-NS4A segments of HJ3 and HJ3/QL were PCR amplified and cloned into the pcDNA6/V5-His vector (Invitrogen), yielding pHJ3/NS2-4A and pHJ3/NS2-4A/QL, respectively.
Cells.
FT3-7, Huh-7.5, and En5-3 cells are all clonal derivatives of Huh7 human hepatoma cell lines (49, 51). FT3-7 and Huh7.5 cells were grown in Dulbecco's modified Eagle's medium (GIBCO) containing 10% fetal bovine serum and 1× penicillin-streptomycin at 37°C in a 5% CO2 environment. En5-3 cells were cultured under identical conditions except for the addition to the medium of 2 μg/ml of blasticidin (Invitrogen, Carlsbad, CA). Huh7/191-20 cells, which conditionally express the full-length HCV core protein (residues 1 to 191) under control of the Tet-Off promoter, were maintained as described by Li et al. (23).
HCV RNA transfection and virus production.
HCV RNAs were transcribed in vitro and electroporated into cells as described previously (50). In brief, for electroporation, 10 μg of in vitro-synthesized HCV RNA was mixed with 5 × 106 FT3-7 cells in a 2-mm cuvette and pulsed twice at 1.4 kV and 25 μF in a Gene Pulser II apparatus (Bio-Rad). Cells were subsequently seeded into 12-well plates for analysis of HCV RNA or into six-well plates for immunoblot detection of HCV proteins. For virus production, transfected cells were seeded into 25-cm2 flasks and fed with medium containing 10% fetal bovine serum. Cells were split every 3 to 4 days.
Quantitation of HCV RNA.
Total RNA was isolated from cell lysates using an RNeasy kit (Qiagen) in accordance with the manufacturer's instructions. RNA was isolated from cell culture supernatants and gradient fractions (see below) using a QIAamp viral RNA kit (Qiagen). For monitoring RNA replication in transfected cells, we assayed viral RNA abundance in a quantitative real-time reverse transcription-PCR (RT-PCR) carried out in a BioRad iQ5 real-time PCR detection system using TaqMan chemistry and the forward primer HCV84FP (5′-GCCATGGCGTTAGTATGAGTGT-3′), reverse primer HC300R (5′-CCCTATCAGGCAGTACCACAA-3′), and detection probe 6-carboxyfluorescein (FAM)-TCTGCGGAACCGGTGAGTACACC-dual-labeled probe black hole quencher 1. We also used a semiquantitative, moderately long-range RT-PCR assay to determine the relative quantity of HCV RNA present in density gradient fractions. It was carried out using reagents provided with the One-Step RT-PCR kit (Qiagen) (52). One to 3 μl of RNA was reverse transcribed in a 50-μl reaction mixture at 45°C for 30 min, followed by inactivation of the reverse transcriptase at 95°C for 15 min. Products were then amplified by PCR for 30 cycles, each comprising 94°C for 30 s and 68°C for 2.5 min. Primer sets targeting the JFH1 NS3 coding regions (nucleotides 3460 to 5322) were described previously (52).
Immunoblotting of viral proteins.
Immunoblots of cell lysates were probed with antibody to core (C7-50; 1:30,000 dilution; BioReagents) or anti-glyceraldehyde 3-phosphate dehydrogenase (anti-GAPDH; catalog no. 4300, 1:20,000 dilution; Ambion) followed by horseradish peroxidase-conjugated anti-mouse immunoglobulin G (catalog no. 1030-05, 1:30,000; Southern Biotechnology). Proteins were visualized by chemiluminescence using reagents provided with the ECL Advance kit (Amersham Biosciences).
FACS detection of core protein expression.
FT3-7 cells were trypsinized 2 and 4 days following transfection, resuspended at 2 × 106 cells/ml, and fixed with a 1:1 mixture of methanol and acetone for 10 min. The cells were blocked with 5% milk solution for 5 min, followed by incubation with mouse monoclonal anti-core antibody (C7-50; Affinity BioReagents) at a 1:300 dilution for 2.5 h at room temperature. Cells were then washed twice with phosphate-buffered saline (PBS) before incubation with allophycocyanin-conjugated anti-mouse antibody (A865; Invitrogen) at a dilution of 1:100 for 1 h at room temperature. Subsequently, the cells were washed twice with PBS, resuspended in 1 ml of fluorescence-activated cell sorter (FACS) sheath fluid (BD Biosciences) prior to analysis on a Becton-Dickinson FACS scanner.
Secreted alkaline phosphatase assay.
Secreted alkaline phosphatase (SEAP) activity was measured in 10-μl aliquots of transfected cell supernatant culture fluids using the Phospha-Light chemiluminescent reporter assay (Tropix) with the manufacturer's suggested protocol reduced in scale as described previously (49, 50). The luminescent signal was read using a TD-20/20 luminometer (Turner Designs).
HCV infectivity assays.
Cell lysates were prepared for intracellular virus infectivity assays as described by Gastaminza et al. (7). Briefly, cell pellets harvested after trypsinization were resuspended in complete medium, washed twice with PBS, and lysed by four cycles of freezing and thawing. Lysates were clarified by centrifugation at 4,000 rpm for 5 min prior to inoculation onto naïve Huh-7.5 cells. For virus titration, 100-μl aliquots of serial 10-fold dilutions of supernatant cell culture fluids (clarified by low-speed centrifugation), clarified freeze-thaw cell lysates, or iodixanol or sucrose gradient fractions (see below) were inoculated onto naïve Huh-7.5 cells seeded 24 h previously into eight-well chamber slides (Nalge Nunc) at 3 × 104 cells/well. Cells were maintained at 37°C in a 5% CO2 environment and fed with 200 μl of medium 24 h later. Following 48 h of additional incubation, cells were fixed in 1:1 methanol-acetone at room temperature for 9 min and then stained with monoclonal antibody C7-50 to core protein (1:300; Affinity BioReagents) for 2 h at 37°C, washed with PBS twice, and incubated with fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin G (1:100; Southern Biotechnology) for 30 min at 37°C. Clusters of infected cells staining for core antigen were considered to constitute a single infectious focus-forming unit (FFU), as described previously (51-53). Infectivity titers (FFU/ml) were calculated from the results obtained with sample dilutions yielding 5 to 100 FFU.
Laser-scanning confocal microscopy.
Transfected cells were seeded onto eight-well chamber slides and 2 to 3 days later washed three times with PBS, fixed with 4% paraformaldehyde, and permeabilized with 50 mg/ml digitonin for 10 min (30, 32). Cells were labeled with monoclonal antibody C7-50 to core protein (1:400; Affinity BioReagents) and rabbit polyclonal antibody to NS5A (1:300; a generous gift of Craig Cameron) or NS3 (1:250; kindly provided by Michinori Kohara), followed by goat anti-mouse immunoglobulin G conjugated to Alexa 488 and goat anti-rabbit immunoglobulin G conjugated to Alexa 594 (1:200; Invitrogen). Neutral lipids present in lipid droplets were visualized by staining with LipidTOX Deep Red (Invitrogen). Nuclei were visualized by counterstaining with 4′,6′-diamidino-2-phenylindole (1:1,000). Slides were examined with a Zeiss LSM 510 Meta laser-scanning confocal microscope.
Isopycnic gradient centrifugation of HCV particles.
Two days after electroporation of cells with HCV RNA, the cell culture medium was changed to serum-free medium (complete medium minus serum; see above). Twenty-four hours later, supernatant fluids were collected, clarified by low-speed centrifugation, concentrated ∼10-fold using a Centricon PBHK Centrifugal Plus-20 filter unit with an Ultracel PL membrane (100-kDa exclusion; Millipore), and layered on top of a preformed, continuous 10 to 40% iodixanol gradient (OptiPrep; Sigma-Aldrich) prepared in Hanks balanced salt solution (Invitrogen). Clarified cell lysates (prepared after multiple freeze-thaw cycles as described in the preceding paragraph) were similarly layered on top of iodixanol gradients. Gradients were centrifuged in an SW60 rotor (Beckman Coulter) at 45,000 rpm for 16 h at 4°C, and nine fractions (500 μl each) were collected from the top of the tube. The densities of gradient fractions were estimated from the refractive index using a Milton-Roy refractometer.
Rate zonal centrifugation of HCV particles.
HCV RNA transfected cell lysates prepared by multiple freeze-thaw cycles as described above were subjected to rate zonal centrifugation as described by Gastaminza et al. (7). Briefly, 250 μl of lysate was loaded on a preformed continuous 10 to 50% sucrose gradient prepared in TNE (10 mM Tris, pH 8.0, 150 mM NaCl, 2 mM EDTA) and centrifuged for 1 h at 40,000 rpm (∼200,000 × g) in an SW41 Ti rotor at 4°C. A total of 40 fractions (300 μl each) were collected from the top of the gradient and subjected to immunoblot analysis to determine the distribution of core protein and virus titration to determine the location of infectious virus within the gradient. The densities of fractions were estimated as described above.
Quantitative core protein ELISA.
Core protein was quantified in gradient fractions using the Ortho Trak-C enzyme-linked immunoassay (ELISA) kit (Ortho-Clinical Diagnostics) with minor modifications to the manufacturer's recommended procedures. Gradient samples were prediluted 10- to 200-fold in the dilution buffer supplied with the kit (so that values for the optical density at 490 nm [OD490] values were within range of the standard solutions) prior to incubation in the ELISA plates at room temperature for 1 h. The quantity of core protein present in fractions was estimated from the OD490 by reference to a standard curve.
Expression of GST-NS3 fusion proteins.
GST-fused NS3 proteins were expressed using ArcticExpress competent cells (Stratagene). Protein expression was induced by bacterial growth in 1 mM isopropyl-β-d-thiogalactopyranoside at 10 to 13°C for 24 h. Bacteria were lysed by sonication after resuspension in a lysis buffer comprised of PBS containing 1% Triton X-100 and protease inhibitor cocktail (Roche Applied Science). After affinity capture of GST-NS3 with glutathione-Sepharose 4B beads (GE Healthcare) in lysis buffer, the beads were washed five times with 50 mM Tris buffer (pH 8.0) before elution of GST-fused proteins with 20 mM glutathione.
NS3/4A protease assay.
NS3 protease activity was quantified by fluorescent resonance energy transfer (FRET) assay using reagents provided with the SensoLyte 520 HCV protease assay Kit (AnaSpec). Briefly, serial twofold dilutions of GST-NS3 protein (starting at 4 pmol), with or without a 10-fold excess of NS4A cofactor peptide (Pep4AK; AnaSpec), were prepared in 1× assay buffer provided with the kit. After addition of the 5-FAM/QXL 520 FRET substrate, the fluorescence signal was measured kinetically at 5-min intervals for 30 min in a SpectraMax M2 microplate reader (Molecular Devices). Vmax values for each reaction were calculated using the SpectraMax M2 software package.
The ability of mutated NS3 sequences to direct the NS2/NS3 cis-cleavage was assessed by gel analysis of the products of in vitro translation reactions. To express the NS2-NS4A segment of the polyprotein, rabbit reticulocyte lysates (Promega) were programmed with RNA transcribed in vitro from the polyprotein expression vectors pHJ3/NS2-4A and pHJ3/NS2-4A/QL (see above). Translation reactions were carried out in a 50-μl reaction volume containing 1 μg RNA, 2 μl [35S]methionine (1,000 Ci/mmol at 10 mCi/ml; ICN), 2.5 μl canine pancreatic microsomal membranes (Promega), and 1 μl of amino acid mixture lacking methionine (Promega) at 30°C for 15 to 180 min. Reactions were stopped by the addition of sodium dodecyl sulfate (SDS) sample buffer followed by boiling for 10 min. Translation products were separated by electrophoresis on a 12% SDS-polyacrylamide gel, followed by autoradiography.
NS3 helicase and NTPase assays.
The helicase and NTPase activities of GST-NS3 proteins were assayed as described by Welbourn et al. (47). Briefly, for the helicase assay, an 18-mer DNA oligonucleotide with the sequence 5′-ACCGCTGCCGTCGCTCCG-3′ was end labeled with T4 polynucleotide kinase and [γ-32P]ATP (Amersham) and purified by passage through a Sephadex G-25 column. The 32P-labeled 18-mer release strand was annealed to an unlabeled 36-mer DNA oligonucleotide with the sequence 5′-CGAGGGAGACGAGGAGACGGAGCGACGGCAGCGGT-3′ by mixing the oligonucleotides at a 1:3 ratio in water and heating the mixture to 95°C for 2 min followed by slow cooling to room temperature. Threefold dilutions of the GST-NS3 protein (starting at 40 pmol) were added to 100 fmol of the annealed oligonucleotides and preincubated for 10 min at room temperature in 30 μl helicase buffer (25 mM morpholinepropanesulfonic acid-NaOH, pH 7, 3 mM MgCl2, 2 mM dithiothreitol, and 0.1 μg/μl bovine serum albumin) before adding 4 mM ATP and shifting to 30°C to initiate the unwinding reaction. After 30 min, the reaction was stopped by the addition of 30 μl of 2× stop buffer (50 mM EDTA, pH 8, 0.8% SDS, 0.04% Nonidet P-40, 20% glycerol, 0.4 mg/ml bromphenol blue, and a 20-fold excess of unlabeled release strand), and the reaction products were separated by electrophoresis on an 8% polyacrylamide gel followed by exposure to X-ray film or PhosphorImager analysis (Storm 860; Molecular Dynamics) to quantify the products. NTPase reactions were carried out for 30 min at room temperature in 100-μl reaction mixtures containing 0.125 to 4.0 pmol of GST-NS3 and 2 mM ATP in 50 mM HEPES (pH 7.5), 2.5 mM MgCl2, and 0.04 μg/μl poly(U). Free phosphate released by the NTPase reaction was quantified using a malachite green phosphate assay kit (BioAssay System) according to the manufacturer's recommended procedures.
RESULTS
A mutation in the NS3 helicase rescues assembly of infectious intracellular virus particles by the chimeric HJ3 RNA.
Infectious virus particles are released from FT3-7 (Huh7) cells within 48 h of transfection with either genome-length wild-type JFH1 (genotype 2a HCV) RNA or H77S (genotype 1a) RNA, which contains five cell culture-adaptive mutations (52). In contrast, the chimeric HJ3 RNA (previously “H-NS2/NS3-J”) (51), which contains the core-NS2 sequence of H77S virus within the background of JFH1 (Fig. 1A), does not produce infectious virus (<10 FFU/ml) until approximately 9 days after transfection, when cells transfected with this RNA begin to release 103 to 105 FFU/ml. Semiquantitative RT-PCR suggested that the absence of infectious virus production was not due to a failure of the HJ3 RNA to replicate (51). In four replicate transfection experiments (Table 1), the late release of infectious virus was associated with the appearance of a novel amino acid substitution at a conserved residue within the helicase domain of NS3 (Q221L). Confirming its importance, introduction of this mutation into the HJ3 chimeric RNA (HJ3/QL) resulted in the immediate (48 h posttransfection) release of infectious virus from transfected cells (51) (Fig. 1B). Remarkably, transfection of the modified HJ3/QL RNA resulted in the production of approximately 30-fold more infectious virus particles than its JFH1 parent (Fig. 1B). Thus, this NS3 mutation appears to compensate for an unknown incompatibility within the intergenotypic chimeric HJ3 sequence that results in a profound defect in the release of infectious virus by RNA-transfected cells.
FIG. 1.

(A) Organization of the chimeric HCV RNAs included in this study. Three genome-length HCV RNAs are shown, JFH1, HJ3, and HJ2, while Jtat2ANeo is a dicistronic subgenomic replicon containing nonstructural sequence derived from JFH1 virus. The JFH1 polyprotein coding sequence is shown as open boxes, and the H77 sequence is shaded. The base positions of the H77 and JFH1 nucleotides flanking the site of the fusion in the HJ2 chimera are shown, as is the location of the Q221L mutation within the helicase domain of NS3. (B) Virus yield assays showing the effect of the Q221L mutation on the efficiency of infectious virus assembly and release by various HCV RNAs. FT3-7 cells were transfected with HJ3, HJ2, and JFH1 RNAs with or without the Q221L mutation as indicated. At day 3 posttransfection, supernatant culture fluids were collected and assayed for released virus, while cell lysates were prepared by multiple freeze-thaw cycles and assayed for cell-associated virus. The data shown represent the means of the three replicate experiments, ± standard deviations. The titer (FFU/ml) of intracellular (shaded bars) and extracellular (open bars) virus was determined as described previously (51).
TABLE 1.
Mutations identified in the core-NS3 polyprotein sequence of vHJ3
| Residue | Mutation found in RNA transfection expt no.
|
|||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| E1 170 | Y170H | |||
| NS3 132 | I132Va | |||
| NS3 221 | Q221L | Q221L | Q221L | Q221L |
Mixed sequence with wild type.
Gastaminza et al. (7) recently reported that infectious intracellular JFH1 particles can be released from infected cells by repetitive freeze-thawing preceded by extensive washing of the cells. This intracellular virus has a greater buoyant density than the extracellular virus and appears to be a late assembly intermediate that is a precursor of mature extracellular virus (7). To determine whether the NS3 mutation is required by the chimeric HJ3 RNA for the intracellular assembly of this precursor particle, or only its release from the cell, we determined the abundance of intracellular infectious virus in HJ3 RNA-transfected cells, using the methods described by Gastaminza et al. (7). Lysates prepared from cells transfected with the modified HJ3/QL RNA contained approximately 104 FFU/ml infectious virus (Fig. 1B). In contrast, no infectious virus was present in lysates of cells transfected with HJ3 RNA lacking the Q221L mutation. These results thus indicate that the HJ3 chimera is defective in a step in the assembly process that is required for the production of infectious intracellular virus particles. The Q221L substitution within the NS3 helicase domain is capable of specifically correcting that defect, confirming a previously unsuspected role for NS3 in infectious particle assembly.
Since the NS3-NS5B sequence of the HJ3 chimera is identical to that of JFH1 virus (Fig. 1A), it was of interest to determine whether the Q221L substitution would have any effect on production of infectious virus by cells transfected with JFH1 RNA. Remarkably, JFH1 RNA containing this substitution (JFH1/QL) produced about 100-fold more intracellular as well as extracellular infectious virus than the wild-type JFH1 RNA (Fig. 1B). However, the relative proportions of intracellular versus extracellular virus were not altered by the mutation, suggesting that the substitution affects assembly and not release of infectious JFH1 virus. Equally remarkable, the Q221L mutation had a much lesser effect on virus production by a second chimera, H-(NS2)-J (referred to here as “HJ2”), in which the H77-JFH1 fusion is located within the center of the NS2 coding region (Fig. 1A) (51). This chimera is capable of producing infectious virus particles immediately (48 h) after transfection, although the amino acid sequence of its polyprotein differs from that of HJ3 at only 19 positions, all within the C-terminal half of NS2. Interestingly, insertion of the Q221L mutation into HJ2 led to only minimal increases in intracellular infectious virus titers and had no effect on the titer of infectious HJ2 virus released from the cells (Fig. 1B). Since the nucleotide sequences of HJ3 and HJ2 differ in only in the C-terminal segment of NS2 (Fig. 1A), these data suggest the possibility of a functional interaction between NS2 and the helicase domain of NS3 (see Discussion).
Impact of the Q221L mutation on the kinetics of viral RNA accumulation.
Although previous semiquantitative assays suggested that enhanced RNA replication is unlikely to explain the dramatic difference in virus production by the HJ3 and HJ3/QL RNAs (51), it was important to exclude this possibility. Quantitative RT-PCR assays demonstrated that the abundance of HJ3 RNA encoding the wild-type Gln-221 residue increased substantially during the first 48 h after transfection and then declined (Fig. 2A). In contrast, there was a sustained increase (over 96 h) in HJ3/QL RNA abundance following transfection (Fig. 2A). Similarly, immunoblot assays showed that core protein expression was maximal 48 h after transfection of HJ3 RNA, followed by a decline at later time points, while cells transfected with HJ3/QL demonstrated sustained expression of core, with increasing abundance at each time point up to 96 h (Fig. 2C). These results suggest that the Q221L mutation alters the kinetics of RNA and core protein accumulation in cells transfected with HJ3 RNA, resulting in a late, sustained increase in viral RNA and core protein expression not seen in the absence of the NS3 mutation. Interestingly, E1-p7 coding region deletion mutants derived from HJ3 and HJ3/QL (HJ3/ΔE1p7 and HJ3/QL/ΔE1p7, respectively), which are incapable of producing infectious virus, demonstrated increases in RNA abundance similar to HJ3 (Fig. 2A). Importantly, HJ3/ΔE1p7 and HJ3/QL/ΔE1p7 were indistinguishable in these assays, suggesting that the Q221L mutation does not significantly influence RNA replication.
FIG. 2.

Replication of intergenotypic chimeric HJ3 (A) and JFH1 (B) RNAs with or without the NS3-Q221L mutation. Viral RNA abundance was measured by quantitative RT-PCR assay of cell lysates harvested at 4.5, 24, 48, 72, and 96 h after transfection of the indicated RNA into FT3-7 cells. The data represent a triplicate analysis, ± standard deviations. (C) Immunoblots showing HCV core protein abundance at 48, 72, and 96 h after transfection of the indicated RNAs; GAPDH was included as a loading control. (D) Subgenomic JFH1 replicon RNA replication assay. Relative SEAP activity induced by replication of Jtat2ANeo (lightly shaded bars) and Jtat2ANeo/QL (heavily shaded bars) at 24, 48, 72, and 96 h after transfection of the RNAs is shown. Results are normalized to the SEAP activity present at the same time point in media from cultures of cells transfected in parallel with the replication-defective Ntat2ANeo/ΔGDD RNA. The data shown are means ± ranges from two independent experiments.
We next determined whether the Q221L mutation has a similar effect on the kinetics of wild-type JFH1 RNA accumulation in transfected cells. Both JFH1 and JFH1/QL RNA accumulated continuously up to 96 h posttransfection in a similar fashion (Fig. 2B). However, the mutated JFH1/QL RNA produced a modest increase in core protein expression between 48 and 96 h posttransfection, while core protein expression was stable over this interval in cells transfected with the wild-type RNA (Fig. 2C). On the other hand, E1/E2 deletion mutants derived from JFH1 and JFH1/QL (JFH1/ΔE1E2 and JFH1/QL/ΔE1E2, respectively) showed a pattern of RNA accumulation similar to HJ3, in that RNA abundance was decreased after 48 h (compare Fig. 2A and B). This can be explained by the inability of these RNAs to produce infectious virus and the lack of spread of this RNA in the cell culture. Taken together, these data provide strong evidence that the Q221L mutation does not enhance the replication efficiency of either the HJ3 or JFH1 RNA.
We also determined whether the Q221L mutation altered the replication of a subgenomic JFH1 RNA that lacked any possibility for infectious virus production. We inserted the Q221L mutation into a dicistronic, subgenomic JFH1 replicon RNA, Jtat2ANeo, in which the NS3-NS5B sequence is derived from the JFH1 sequence (Fig. 1A). The Jtat2ANeo expresses the human immunodeficiency virus tat protein from its upstream cistron and induces SEAP by transfected En5-3 (Huh7) cells in an amount proportional to the abundance of replicon RNA (49). Consistent with previous results, the level of SEAP activity induced by a control, genotype 1b replicon RNA containing a lethal mutation in NS5B (Ntat2ANeo/ΔGDD) (49) was maximal 24 h after transfection and declined to near background levels by 96 h (data not shown). Cells transfected with the Jtat2ANeo RNA showed increasing SEAP expression relative to that expressed by cells transfected with this replication-incompetent control RNA throughout this interval (Fig. 2D). Importantly, there was no difference in the quantity of SEAP produced by cells transfected with Jtat2ANeo replicon RNAs with and without the Q221L mutation (Fig. 2D). Thus, the Q221L mutation does not directly affect viral RNA replication, and it is likely that increased packaging and export of viral RNAs containing the Q221L mutation accounts for the altered kinetics of viral RNA and core protein accumulation shown in Fig. 2A to C.
The Q221L mutation reduces the accumulation of core protein and results in spread of virus in RNA-transfected cells.
The abundance of HCV RNA and viral proteins could be affected not only by RNA replication but also by spread of virus within a culture. To assess the proportion of cells supporting RNA replication following transfection, we examined cell cultures for core protein expression by indirect immunofluorescence microscopy. These results revealed equal numbers of cells labeled with antibody to core, but a generally lower intensity of cell labeling 2 days after transfection with HJ3/QL RNA, compared to cells transfected with HJ3 RNA (Fig. 3A). At 4 days, the proportion of HJ3-transfected cells that labeled with anti-core was markedly reduced, while that of HJ3/QL-transfected cells was substantially increased. We quantified these differences by FACS analysis (Fig. 3B). These results confirmed that there was a significantly greater abundance of core protein in HJ3-transfected versus HJ3/QL-transfected cells 2 days posttransfection, although the proportion of cells expressing core was similar in the two cultures (∼17%). By day 4 posttransfection, the fraction of HJ3-transfected cells expressing detectable core protein was markedly decreased (1.8%), while the percent of HJ3/QL-transfected cells expressing core had approximately doubled (39.9%).
FIG. 3.

Expression of core antigen at 2 and 4 days after transfection of cell cultures with the HJ3 and HJ3/QL RNAs. (A) Core antigen detected by indirect immunofluorescence microscopy. (B) The percentage of HCV RNA-transfected cells expressing HCV core antigen was quantified by FACS analysis. The length of the open doubleheaded arrow is proportional to the intensity of core staining and is greater in the graph depicting HJ3-transfected rather than HJ3/QL-transfected cells.
These results are consistent with the immunoblots shown in Fig. 2C and indicate several important differences between HJ3 and HJ3/QL RNA. HJ3 RNA accumulates rapidly over the first 48 h following transfection (Fig. 2A) but is unable to produce virus that can spread within the culture, in contrast to HJ3/QL, which spreads efficiently (Fig. 3A and B). We speculate that cells transfected with HJ3 accumulate more core protein, compared to HJ3/QL, due to the lack of virus release (Fig. 1). The decrease observed in the size of the core antigen-positive cell population between 2 and 4 days following transfection of HJ3 RNA suggests that the efficient replication of this chimera coupled with the accumulation of viral RNA and proteins is toxic to the cell, as reported for JFH1 RNA (54), likely resulting in cell death and overgrowth of nontransfected cells. Timpe et al. (44) recently demonstrated that HCV spreads between cultured cells by two distinct mechanisms and that it is capable of direct cell-to-cell spread in a fashion that is resistant to neutralizing antibodies as well as spread through the release of infectious extracellular virus. Importantly, the data presented in Fig. 3A and B indicate that the HJ3 chimeric RNA, while replicating efficiently, is unable to spread by either mechanism.
Q221L is not required for recruitment of NS5A or NS3 to lipid droplets.
To further characterize the influence of the Q221L mutation on core protein expression, we examined cells by laser-scanning confocal microscopy. Miyanari et al. (32) showed recently that cellular lipid droplets play an important role in infectious HCV production and that an essential early step in this process is the recruitment of NS5A by core protein, which localizes to the surface of lipid droplets (30). This was readily demonstrable by confocal imaging of cells transfected with the HJ3 RNA lacking the Q221L mutation (Fig. 4A, middle panels). Core protein was abundant in these cells and decorated the surface of cytoplasmic lipid droplets identified with a neutral lipid stain. NS5A had a similar pattern of expression and generally colocalized with core on the surface of lipid droplets. Interestingly, the distribution of core and NS5A in HJ3 RNA-transfected cells closely resembled that observed in JFH1 RNA-transfected cells (Fig. 4A, compare middle and top panels). Colocalization of both core and NS5A on lipid droplets was also evident in cells transfected with HJ3/QL, although overall viral protein expression was somewhat reduced (Fig. 4A, bottom panels). These results thus demonstrate that the NS3-Q221L mutation is not required for recruitment of NS5A to core present on the surface of lipid droplets and that the failure of virus production by HJ3-transfected cells is not due to a block in this step of the assembly process.
FIG. 4.

Laser-scanning confocal microscopic imaging. FT3-7 cells were transfected with JFH1, HJ3, or HJ3/QL RNA and 2 days later washed and fixed, as described in Materials and Methods, and labeled with probes specific for core protein (green), NS5A (red), and neutral lipids (white) (A), or core protein (green), NS3 (red), and neutral lipids (white) (B), prior to examination in a Zeiss LSM 510 Meta instrument. At the right is an enlarged area from the merged image showing a single lipid droplet (see box).
NS3 also localized to the surface of lipid droplets in HJ3-transfected cells, in some cases colocalizing strongly with core protein (Fig. 4B, middle panels). This was also observed in JFH1-transfected cells (Fig. 4B, upper panels). As with core and NS5A, the overall intensity of staining for NS3 was reduced and assumed a somewhat more punctuate pattern within the cytoplasm of cells transfected with HJ3/QL (Fig. 4B, bottom panels). However, there was clear colocalization of NS3 and core, predominantly on the surface of lipid droplets. Thus, the Q221L mutation appeared to have no effect on the subcellular distribution of NS3 or its ability to colocalize with core (and presumably NS5A) on the surface of lipid droplets.
Q221L is required for the assembly of high-density intracellular HJ3 particles.
The absence of infectious intracellular virus in cells transfected with HJ3 RNA without the Q221L mutation (Fig. 1) could reflect either a lack of infectivity associated with viral particles assembling within the cells or the complete absence of particle assembly. To distinguish between these possibilities, we collected supernatant culture fluids and cell lysates 3 days after transfection with either HJ3 or HJ3/QL RNA and determined the distribution of HCV core protein and HCV infectivity in fractions of iodixanol gradients that had been centrifuged to equilibrium. The results obtained with such isopycnic gradients are shown in Fig. 5. As expected, no infectious virus was detected in any fraction from gradients loaded with cell lysates or culture supernatants from HJ3-transfected cells (Fig. 5A, left panel). In contrast, infectious virus was readily detectable in fractions from gradients loaded with either cell lysates or culture supernatants from cells transfected with HJ3/QL RNA (Fig. 5A, right panel). Similar to the density profiles of intracellular and extracellular JFH1 virus reported previously by Gastaminza et al. (7), extracellular HJ3/QL virus was distributed widely across the gradient, with a broad peak between ∼1.03 and 1.12 g/cm3 (Fig. 5A, right panel). Intracellular virus was distributed more narrowly, with peak infectivity between ∼1.09 and 1.12 g/cm3 and a greater-than-100-fold difference in the infectious titers of fractions 2 (1.03 g/cm3) and 6 (1.12 g/cm3) (Fig. 5A, right panel).
FIG. 5.

Equilibrium gradient ultracentrifugation of viral particles present in cell culture supernatant and cell lysates collected 3 days after transfection of FT3-7 cells with RNA encoding HJ3 (left panels) and HJ3/QL (right panels). (A) Distribution of infectious virus present in gradient fractions as determined by FFU assay. Only HJ3/QL produced infectious virus, and this was present in both lysates and supernatant fluids. HJ3 did not produce detectable quantities of infectious virus (<10 FFU/ml). (B) Core antigen as measured by quantitative ELISA in each fraction. (C) Semiquantitative RT-PCR assay of HCV RNA in fractions from gradients loaded with cell culture supernatant fluids (top panels) or cell lysates (bottom panels). The open and closed arrows indicate two distinct slowly sedimenting (open) and rapidly sedimenting (closed) species of HCV RNA-containing particles present in supernatants.
To determine if the absence of infectivity in the HJ3 gradient fractions reflected an absence of high-density particles similar to those associated with infectivity in the HJ3/QL lysates (Fig. 5A, left versus right panels), we tested these fractions for the presence of core protein using a sensitive ELISA (Fig. 5B) and for HCV RNA using a semiquantitative RT-PCR assay (Fig. 5C). Core protein was not detectable in any fraction of gradients loaded with supernatant fluids from HJ3-transfected cells (Fig. 5B, left panel) but was present in the same fractions of gradients loaded with supernatants from HJ3/QL-transfected cells that contained infectious virus (particularly in fraction 6, ∼1.12 g/cm3) (Fig. 5B, right panel). The more sensitive RT-PCR analysis of these fractions was consistent with these results, revealing a large peak of viral RNA in fraction 6 of gradients loaded with supernatant fluids from HJ3/QL-transfected cells (Fig. 5C, top panels). Since infectivity peaked in fractions 5 and 6 (Fig. 5A, right panel), while core and viral RNA were greatest in fractions 6 and 7 of gradients loaded with the HJ3/QL supernatant fluids (Fig. 5B, right panel, and C), these data confirm previous reports suggesting that the most dense HCV particles produced in cell culture have relatively low specific infectivity (26, 45). Importantly, the small amount of RNA detectable in supernatant fluids from HJ3-transfected cells banded at a low density, peaking in fraction 2 (Fig. 5C, top panel). Thus, there was no evidence of noninfectious virus particles being released from the HJ3-transfected cells.
Consistent with the high abundance of intracellular infectious particles found in lysates of HJ3/QL-transfected cells (Fig. 1 and 5A, right panel), most of the core protein that was present in lysates of these cells banded at higher densities (∼1.09 to 1.12 g/cm3) (Fig. 5B, right panel). In sharp contrast, the core protein present in HJ3-transfected cells was mostly present within low-density fractions, with a major peak in fraction 2 and possibly a minor secondary peak in fraction 5 (Fig. 5B, left panel). The RNA present in lysates of these cells also banded at lower densities and was predominantly in fractions 2 to 5 (Fig. 5C, lower panels). Much of this RNA is likely to be present in replication complexes, rather than assembled particles. There was relatively little RNA detectable by this semiquantitative assay in high-density fractions of HJ3 lysates, in contrast to gradients loaded with lysates of HJ3/QL-transfected cells in which there was substantially more RNA in fraction 6 (Fig. 5C, lower panels). Taken together, the data shown in Fig. 5 indicate that replication of the HJ3 RNA does not lead to the assembly of high-density intracellular particles containing viral RNA and core protein in the absence of the compensatory Q221L mutation.
The distribution of core protein that we observed in isopycnic gradients loaded with lysates from HJ3-transfected cells is consistent with it being localized to low-density lipid droplets, as suggested by the immunofluorescence microscopy results in Fig. 3C. To further characterize its biophysical properties, we compared its sedimentation rate with that of core present in lysates of HJ3/QL-transfected cells in rate zonal sucrose gradients (Fig. 6). Under these conditions, infectious particles present within lysates of HJ3/QL- and JFH1/QL-transfected cells formed a peak centered in fraction 14 (Fig. 6, top panel). Immunoblotting revealed a distinct peak of core protein in these same fractions, as well as a second, more abundant peak of core protein with a broad peak in fractions 6 to 7 (Fig. 6, bottom panel). The sedimentation properties of core protein present in lysates of HJ3-transfected cells was significantly different. Core was most abundant in fractions 6 and 7, and while substantial amounts of core were found to extend into fraction 12, there was no evidence of the rapidly sedimenting core species that peaked in fractions 14 and 15 of gradients loaded with lysates of HJ3/QL- or JFH1/QL-transfected cells. These results thus indicate that core protein fails to be assembled into rapidly sedimenting particles in the absence of the Q221L mutation.
FIG. 6.

Rate zonal centrifugation of cell lysates derived from FT3-7 cells transfected with the HJ3, HJ3/QL, or JFH1/QL RNAs or Huh7/1-191 cells which conditionally express only the HCV core protein (23). Cell lysates were prepared by multiple rounds of freeze-thawing and centrifuged in preformed sucrose density gradients. The sedimentation profiles of infectious virus present in lysates of HJ3/QL- and JFH1/QL-transfected cells are shown at the top. No infectious virus was present in lysates from the HJ3-transfected cells. The immunoblots of core protein present in the top 20 fractions of the gradients are shown at the bottom. No detectable core protein was present in fractions beyond fraction 20 (data not shown). Fractions prepared from the four gradients depicted had density profiles indistinguishable from that shown.
To better understand the nature of the core protein present in these gradient fractions, we also studied core protein expressed in Huh7/191-20 cells. These cells express the full-length core protein (191 residues) in a tetracycline-dependent manner in the absence of other HCV proteins (23). The protein is processed upstream of the E1 signal sequence by signal peptide peptidase and is associated with cytoplasmic lipid droplets in these cells. Immunoblot assays demonstrated that the greatest abundance of core protein was present in fractions 2 to 4 of rate zonal gradients loaded with lysates of Huh7/191-20 cells (Fig. 6, lower panel). Thus, the rate zonal gradients shown in Fig. 6 demonstrate at least three distinct species of core protein with different sedimentation velocities: soluble core protein (fractions 2 to 4), medium size aggregates (fractions 6 and 7), and larger, more rapidly sedimenting infectious particles (fractions 14 and 15). It is interesting that there was little assembly of core into medium-size aggregates in the absence of expression of the other viral proteins (Huh7/191-20 cells), suggesting that the medium-sized aggregates might represent complexes of core with other viral proteins. The most significant feature of the data shown in Fig. 6, however, is the absence of larger, rapidly sedimenting particles in cells transfected with HJ3 RNA. Taken together with the data from the isopycnic gradients, these results show that the HJ3 chimera is defective in an early-intermediate step in virus assembly that follows the recruitment of NS5A to lipid droplets and precedes the formation of dense intracellular viral particles. This defect is corrected by the Q221L mutation within the helicase domain of NS3.
The Q221L mutation has no apparent effect on NS3-associated enzyme activities.
Gln-221 is located within the segment of the NS3 protein that contains NTPase and helicase activities. More specifically it is located between the highly conserved motif I and II of the helicase, the so-called Walker motifs A and B, that are involved in NTP binding and hydrolysis (25, 46). Since Gln is highly conserved at residue 221 across all six HCV genotypes, it was of interest to determine the effect of the Leu substitution on NS3 NTPase and helicase activities. To assess this, we expressed JFH1 NS3 as a GST-fused protein, with and without the Q221L substitution, in bacteria (Fig. 7A). Helicase activities, determined using a 32P-labeled double-stranded DNA oligonucleotide substrate, were similar for both proteins, while GST alone did not possess any unwinding activity (Fig. 7B). Similarly, the presence of the Q221L substitution had no affect on NTPase activity as assessed with a colorimetric assay measuring released phosphate (Fig. 7C). Thus, within the limit of these assays with GST-fused NS3 proteins, the ability of the Q221L mutation to rescue infectious virus production by the HJ3 chimera could not be related to an increase (or decrease) in NS3 helicase activity.
FIG. 7.

Lack of effect of the Q221L mutation on NS3-associated enzyme activities. (A) SDS-PAGE separation of purified GST-fused JFH1 NS3 proteins with or without the Q221L mutation. *, a truncated NS3 protein that was reactive with anti-NS3 antibody. (B) Helicase (unwindase) activities were measured with a radiolabeled double-stranded oligonucleotide substrate using a range of concentrations of the indicated GST-NS3 proteins or GST alone. (C) NTPase activities of the GST-NS3 proteins were measured by determining release of free phosphate. (D) The protease activity of GST-NS3 proteins (or GST alone) was assessed in the presence or absence of added NS4A cofactor peptide in a FRET assay. The velocity of the reaction (Vmax) was calculated for a range of GST-NS3 protein concentrations from measurements of the product of the reaction taken at 5-min intervals over a 30-min period. (E) In vitro cis-processing of 35S-labeled JFH1 polyprotein segments representing NS2-NS3-NS4A and containing either the wild-type Gln-221 residue or the mutant Leu-221 residue. Samples were collected from a cell-free translation reaction carried out in rabbit reticulocyte lysates at 15, 30, 60, and 180 min and subjected to SDS-PAGE analysis followed by autoradiography.
Since it is possible that substitutions within the helicase domain may affect the catalytic activity of the upstream NS3 protease (33), we also determined the effect of the Q221L substitution on NS3/4A protease activity using a FRET-based assay incorporating a peptide substrate. The GST-fused JFH1 NS3 protein showed activity in this assay only in the presence of the NS4A cofactor peptide (Fig. 7D, compare wt and wt+4A), consistent with proper folding of the protease domain. Importantly, there was no significant difference in the proteolytic activities of the NS3 proteins with the wild-type Gln or mutant Leu residues at position 221. Finally, we assessed the impact of the Q221L mutation on cis-cleavage of the NS2-NS3 junction directed by the NS2/NS3 autoprotease in cell-free translation reactions programmed with RNAs encoding the NS2-NS4A segment of JFH1, with and without the NS3 Q221L mutation. Again, we observed no significant differences in the self-cleavage activities of the wild-type and Q221L mutant polyproteins (Fig. 7E). Thus, the substitution of Gln-221 with Leu has no apparent impact on any NS3-associated enzymatic activity and is thus likely to exert its effect on virus assembly by another mechanism.
The Gln-221 mutation acts at the level of protein, not nucleic acid sequence.
We next considered the possibility that the Q221L mutation might act at the level of the cognate nucleotide substitution and that an important RNA packaging signal might reside within this segment of the RNA encoding NS3. The Q221L mutation we identified in the chimeric virus is caused by a single nucleotide substitution (CAG to CUG). To determine whether it promotes HJ3 virus production by altering the RNA sequence or, alternatively, through a specific change in the amino acid sequence of NS3, we mutated all three nucleotides encoding Gln-221 in the HJ3 plasmid, changing the wild-type CAG to UUA, thereby recreating the Q221L substitution with a completely different codon sequence. Transfection of this mutant chimeric RNA resulted in the production of infectious virus in a manner indistinguishable from the original NS3 Q221L mutant (Fig. 8A), suggesting that the NS3 mutation acts at the level of protein and not the underlying RNA sequence.
FIG. 8.
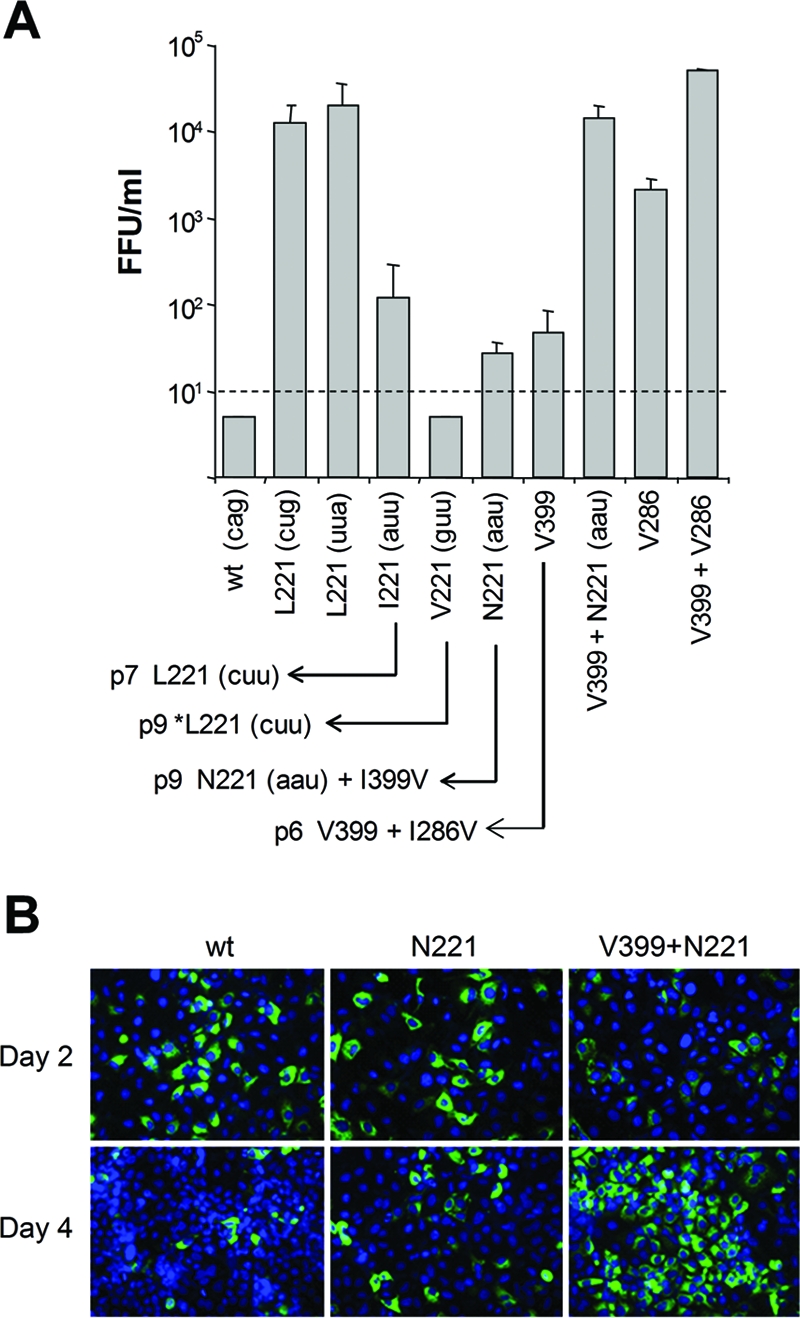
(A) Impact of substitutions within the NS3 221 codon on release of infectious virus by cells transfected with HJ3 RNA or various mutants derived from it. The data shown represent the means plus standard deviations of virus released 2 days after transfection of the indicated RNAs in replicate experiments (uppercase letters refer to the amino acid present at the NS3-221 position, while lowercase letters in parentheses indicate the nucleotide sequence of the NS3 codon 221 in each). The defect in virus production that is evident in the HJ3 chimera containing NS3-Q221 was rescued by introduction of a Q221L mutation (irrespective of the nucleotide sequence of codon 221) or to a lesser extent by the Q221I and Q221N mutations. Although there was no detectable virus produced by an RNA encoding Val-221, continued passage of transfected cells gave rise to a mutated virus encoding Leu-221, as shown by an arrow in the lower part of the figure. Virus yield from other mutants, and second-site mutations arising from continued passage of transfected cells, are as shown. *Leu-221 indicates that in addition to the mutation of Val-221 to Leu-221, the Val-221 mutant also acquired a Leu-to-Ser substitution at residue 66 of the NS2 sequence (L875S in the H77 polyprotein). The V286 and V399+V286 mutants encode the wild-type Gln-221 residue. Infectious virus was measured in supernatant fluids 2 days after RNA electroporation. The detection limit was 10 FFU/ml. (B) Core protein expression as determined by immunofluorescence 2 and 4 days after transfection of cells with the indicated RNAs.
To assess the dependence on Leu in this position, we made additional mutations within this codon in the HJ3 background, resulting in RNAs encoding Ile-221, Val-221, and Asn-221 (HJ3/QI, HJ3/QV, and HJ3/QN, respectively). Interestingly, both the Q221I and Q221N substitutions were capable of rescuing infectious virus production by HJ3, although at a substantially reduced level compared with Q221L (Fig. 8A). In contrast, there was no detectable release of infectious virus by cells transfected with the Q221V mutant (<10 FFU/ml). This suggests that the length of the side chain (which is similar for Leu, Ile, and Asn) (10) may be more important at this position for viral assembly than hydrophobicity or charge. Val has a shorter side chain than Leu, Ile, or Asn, while the Gln side chain is significantly longer. Since Gln-221 is located on the solvent-exposed surface of NS3 (Fig. 9), it is tempting to speculate that these differences in the nature of the residue 221 side chain may be an important determinant in the interaction of NS3 with a second viral (or possibly cellular) molecule involved in virus assembly (see Discussion).
FIG. 9.

Location of mutations promoting assembly of HJ3 virus within the crystallographic structure of the NS3 helicase from the genotype 1b BK strain of HCV (PDB 1CU1) (48). (A) Two views of the major NS3 helicase subdomains, showing the side chains in space-filling view for residues Gln-221 (red), Ile-286 (orange), and Val-399 (yellow). Gln-221 and Ile-286 are conserved between the JFH1 and BK virus NS3 molecules, while residue 399 is Ile in JFH1 and Val in the BK virus. The protein backbone of subdomain 1 (NTPase) is displayed in blue, with the Walker A and Walker B motifs highlighted in green, while the backbone of subdomain 2 (RNA binding activity) is displayed in brown. (B) Expanded view of subdomain 1, showing the relationship of the side chains of Ile-286 (orange, space-filling view), Gln-221 (red, stick-and-ball view), and the flanking tyrosines Tyr-223 and Tyr-118 (green and yellow, respectively; stick-and-ball view).
A second-site mutation within the NS3 helicase enhances virus production by HJ3/Q221N.
Since the additional RNAs shown in Fig. 8A were replication competent but produced only low titers of infectious virus following transfection, we attempted the isolation of second-site mutations that would further promote the production of infectious virus in the anticipation that this might provide additional insight into the assembly defect in HJ3. To accomplish this, we serially passaged the transfected cells, monitoring the release of infectious virus. By the seventh and ninth cell passage, respectively, the Ile-221 and Val-221 mutants had each acquired a single nucleotide substitution so that each now encoded Leu at residue 221 and produced abundant infectious virus (Fig. 8A, bottom). This outcome suggests that Leu-221 is optimal for promotion of HJ3 assembly. The Val-221 mutant also acquired a Leu-to-Ser substitution at residue 66 of the NS2 sequence (L875S in the H77 polyprotein). Although we have shown previously that mutations in NS2 may promote the production of infectious virus by the HJ2 chimera (in which H77 and JFH1 sequences are fused in the middle of NS2) (51), the contribution of the NS2-L66S mutation is uncertain, since the NS3-Q221L mutation is sufficient for efficient HJ3 virus release (Fig. 1B and 8A).
While single nucleotide changes in HJ3/QI and HJ3/QV are capable of altering NS3-Gln-221 to Leu, the construction of HJ3/QN required two base changes and it could not revert to Leu-221 with a single base change, as was the case for Ile-221 and Val-221 (Fig. 8A). Nonetheless, by the ninth passage, cells transfected with this RNA also began to release substantially increased titers of infectious virus. Interestingly, sequencing of the released virus revealed no changes in the codon encoding Asn-221 but identified a second site mutation, NS3-I399V (Fig. 8A, bottom). To assess its contribution to infectious virus release, we introduced the I399V mutation into the wild-type HJ3 (HJ3/IV) as well as the HJ3/QN construct (HJ3/QN/IV). Cells transfected with HJ3/QN/IV RNA released high titers of infectious virus by 2 days posttransfection in a manner that was fully comparable to the release of virus by HJ3/QL-transfected cells (Fig. 8A, N221+V399). Like HJ3/QL-transfected cells (Fig. 2A), cells transfected with HJ3/QN/IV also demonstrated a reduced intensity of labeling with core antibody and rapid spread of virus within the culture compared to HJ3 (Fig. 8B). Cells transfected with HJ3/IV (containing only the I399V mutation, with the wild-type Gln residue at position 221) released only small amounts of infectious virus, similar to those transfected with HJ3/QN RNA (Fig. 8A, V399). Thus, the combination of the Q221N and I399V mutations is necessary to rescue infectious virus production as fully as the Q221L mutation.
The NS3 residue 399 is located within subdomain 2 of the NS3 helicase, which is associated with RNA binding activity. Although it is Ile in the JFH1 strain of HCV, this is atypical, as a Val residue is conserved at this position in most HCV sequences. The I399V mutation acquired by HJ3/QN thus changes this residue to the Val present in the H77 NS3 protein. Importantly, this residue is not positioned close to NS3-221, which is within subdomain 1 of the helicase (Fig. 9A). Interestingly, further passage of cells transfected with the V399 mutant led to the acquisition of yet another mutation, NS3-I286V (Fig. 8B). Residue 286 of NS3 is located close to residue Gln-221 in subdomain 1 of the helicase (Fig. 9A). The introduction of I286V by itself resulted in the release of substantial amounts of infectious virus 2 days after transfection of the RNA; however, the combination of I286V and I399V resulted in 10-fold higher yields of infectious virus (Fig. 8A, compare V286 and V286+V399). Thus, efficient intracellular virus assembly and release can be rescued by any of three distinct sets of mutations within the helicase domain of NS3: Q221L alone, Q221N with I399V, or I286V with I399V.
DISCUSSION
NS3 is a multifunctional protein. It is an essential component of the membrane-associated viral replicase complex and contains protease as well as helicase and NTPase activities that are necessary for viral RNA replication (8, 19, 21). The data presented here demonstrate a novel and previously unsuspected role for the NS3 helicase domain in the intracellular assembly of HCV particles. The mutations that we identified are located within the helicase domain of NS3 (Fig. 9) and compensate for a profound defect in infectious virus production by an intergenotypic chimeric viral RNA, HJ3, which contains the core-NS2 segment of the genotype 1a H77 virus placed within the background of the genotype 2a JFH1 genome (51). A single mutation, NS3-Q221L, was selected in multiple experiments involving transfection of the HJ3 RNA and led to efficient release of infectious virus particles (Fig. 1B and Table 1). This mutation substitutes a Leu residue for a highly conserved Gln within subdomain 1 of the helicase, which contains several sequence motifs involved in NTP binding and NTPase activity (41, 46, 48). However, we also found that equivalently efficient production of infectious virus could be conferred by two other combinations of mutations in the helicase involving either NS3-Q221N or NS3-I286V (both in subdomain 1 of the helicase, which is associated with NTPase activity) and NS3-I399V (which is located within subdomain 2, associated with RNA binding activity) (Fig. 8A). These mutations promote the assembly of infectious intracellular virus particles (Fig. 5 and 6) but, at least in the case of Q221L, have no discernible impact on enzymatic activities associated with NS3, including protease, helicase, and NTPase activities (Fig. 7). In addition, the Q221L mutation did not promote replication of a subgenomic JFH1 replicon that is incapable of producing infectious virus (Fig. 2D). Thus, the data we present here provide novel and compelling genetic evidence for the involvement of both of the major subdomains of the NS3 helicase in infectious virus assembly, independent of the role of this protein in HCV RNA replication.
Although a role for NS3 in virus assembly has not been suspected previously for HCV, several studies have suggested that the related NS3 proteins of classical flaviviruses, including both KUN and YFV (13, 20, 27), as well as pestiviruses such as bovine viral diarrhea virus (1), are involved in the formation of infectious virus particles. It is not clear how NS3 functions in assembly of these viruses, but the helicase domain in particular seems to be involved. Deletions and double frameshift mutations in the KUN genome that knock out expression of a functional NS3 helicase can be complemented in trans to restore RNA replication, but they are also associated with a defect in infectious virus production that cannot be trans-complemented (27, 35). Further studies have shown that the noncomplemented defect in KUN assembly is not related to a requirement for specific RNA sequence in this region of the genome (35). This is consistent with our findings with HCV, in which the Q221L mutation appears to function at the level of the NS3 protein, not the RNA encoding it (Fig. 8). Other evidence suggests a role for the YFV NS3 helicase in assembly of YFV. Second-site mutations at Asp-435 within the helicase domain of the YFV NS3 protein compensate for a defect in NS2A that does not impair processing of the structural proteins or RNA replication but prevents the production of infectious virus particles (20). In contrast to the studies with KUN, however, a recent report that appeared while the manuscript was in preparation showed that the defect in infectious YFV particle assembly can be complemented by NS3 expressed in trans, albeit with low efficiency (34). It is not clear whether these differences in the ability to trans-complement NS3-related defects in YFV versus KUN assembly are due to fundamentally different mechanisms of assembly or technical differences in the experimental approaches taken. It is interesting, however, that we have been unable to trans-complement the assembly defect in the HCV HJ3 RNA by overexpression of NS3 protein containing the Q221L mutation using a recombinant VEE replicon vector (M. K. Yi and S. M. Lemon, unpublished data).
Studies with other viruses have shown that viral helicase and/or NTPase activities can contribute to the packaging of some viral RNAs and DNAs into preformed structures. For example, DNA helicase activity is required for packaging of the adenovirus type 2 genome into preformed empty capsids (18). Also, the rotavirus NSP2 protein, which is associated with NTPase and helix-destabilizing activities, has been suggested to function as a molecular motor in packaging template mRNAs into replication intermediate precursors (42). However, it is unlikely that the HCV NS3 helicase plays such a role in the assembly of infectious HCV particles, since the Q221L mutation, as mentioned above, resulted in no change in the NTPase or helicase activities of the JFH1 NS3 protein (Fig. 7B and C). Moreover, the ability of the YFV NS3 protein to trans-complement an NS3-related defect in assembly was not eliminated by mutations that knock out NS3 helicase or NTPase activities (34). Thus, although all of the mutations that we identified in the HCV NS3 protein that enable the formation of infectious HJ3 virus particles (Fig. 8) are located within the helicase domain, it is likely that they do not influence the enzymatic activities of the protein but rather alter some other function of the protein.
It is tempting to speculate that such a function might be the ability of the helicase domain to participate in a protein-protein interaction that is required for assembly of infectious intracellular particles. The nature of this putative protein-protein interaction is uncertain, but it occurs after the recruitment of NS5A to the core protein-lipid droplet complex, an essential early step in virus assembly (32), as the presence or absence of the Q221L mutation did not influence the colocalization of core and NS5A (or NS3) on the surface of lipid droplets in cells transfected with HJ3 RNA (Fig. 4). However, the involvement of NS3 in the assembly process precedes the intracellular formation of rapidly sedimenting, dense particles containing core protein and HCV RNA, as such particles were not found in cells transfected with HJ3 RNA lacking the Q221L mutation (Fig. 5B and C and 6). In contrast to its dramatic effect in rescuing production of infectious particles by the chimeric HJ3 RNA, the NS3-Q221L mutation had no appreciable effect on infectious virus assembly when placed within the HJ2 chimera (Fig. 1B). The HJ2 chimera differs in its nucleotide sequence from the HJ3 chimera only within the RNA segment encoding the C-terminal 77 residues of NS2 (51) (Fig. 1). This suggests that the NS3 helicase may interact with the C-terminal domain of NS2 during a critical step required for the assembly of infectious particles (5, 15). This provides an interesting parallel to YFV, in which the second site mutations at Asp-435 of the NS3 helicase rescue a defect in particle assembly caused by a mutation in NS2A (20).
The fact that the NS3-Q221L mutation, in addition to rescuing infectious virus production caused by an incompatibility within the genotype 1a and 2a segments of the HJ3 polyprotein (51), leads to a large increase in infectious virus production by JFH1 RNA (which shares the C-terminal NS2 sequence in common with HJ3) (Fig. 1B), suggesting that it is also a cell culture-adaptive mutation. In fact, the NS3-I286V mutation, which like NS3-Q221L rescues particle assembly by HJ3 RNA (Fig. 8), was recently identified as an adaptive mutation during passage of JFH1 virus in cell culture (14). Thus, it is possible that these mutations promote the association of NS3 with an unknown host cell protein involved in particle assembly. Interestingly, the NS3 Gln-221 residue is absolutely conserved in the curated NS3 sequences of all six major genotypes of HCV in the online Los Alamos National Laboratory HCV database (http://hcv.lanl.gov/content/hcv-index). Nonetheless, we have recently recovered infectious HJ3/QL virus from a chimpanzee following intravenous virus challenge (N. Bourne, M. K. Yi, R. Veselenak, R. L. Lanford, and S. M. Lemon, unpublished data), suggesting that the Q221L mutation is not incompatible with virus replication in vivo within the liver.
Although the structure of the genotype 2a JFH1 NS3 protein (which is present in HJ3) has not been determined by crystallography, the mutations we identified involve residues that are easily aligned with sequences of genotype 1 viruses for which such structures are available (4, 17, 48). The positions of these residues within the folded helicase structures are consistent with the mutations potentially influencing the interaction of the helicase domain with NS2 or another viral or possibly cellular protein. Gln-221 is located within subdomain 1 of the NS3 helicase. Its side chain is solvent accessible, on the surface of the subdomain opposite the Walker A and Walker B motifs, which face the interdomain cleft and are involved in NTP binding (Fig. 9A). Ile-286 is within the core of subdomain 1, in relatively close proximity to Gln-221 and with its side chain oriented toward the aromatic rings of Tyr-218 and Tyr-223, which are positioned beneath Gln-221 (Fig. 9B). It is thus possible that the substitution of Ile with the smaller Val side chain at residue 286 could influence the position of the overlying, solvent-exposed Gln-221. While speculative, this could explain the similar positive effects of mutations at Ile-286 and Gln-221 on assembly of HJ3 virus (Fig. 8). Residue 399, on the other hand, is located within subdomain 2 of the helicase. It is Ile in JFH1 virus but Val in H77 and most other HCV sequences. Val-399 is partially solvent exposed, with its side chain oriented toward the interior of the molecule (Fig. 9A). Ile-286 and Ile-399 are thus located at a considerable distance from each other within the structure of the helicase. It is difficult to envision how substitutions at these two positions might act cooperatively to promote viral assembly (Fig. 8), but this could occur by promoting a protein-protein interaction involving the relevant solvent-exposed regions of the two helicase subdomains.
In summary, the data presented here provide compelling genetic evidence for the essential involvement of the two major subdomains of the HCV NS3 helicase in an early step in the assembly of infectious intracellular particles. The helicase domain appears to participate in particle assembly in a fashion that is independent of its associated enzymatic activities, as has recently been shown to be the case for the YFV NS3 helicase (34). Its role in particle assembly occurs after the recruitment of NS5A and NS3 to core protein decorating intracellular lipid droplets, but prior to the formation of dense intracellular particles containing core protein and viral RNA. We believe that its role in assembly may involve critical interactions of the solvent-exposed surfaces of the NTPase and RNA binding domains of the helicase with one or more other viral or cellular proteins involved in this process. However, the nature of these interactions and the identity of the NS3 binding partner(s) remain to be determined.
Acknowledgments
We thank Takaji Wakita for JFH1 cDNA, Charles Rice for Huh7.5 cells, Masaru Enomoto and Kui Li for Huh7/FT3-7 cells, Michinori Kohara for the gift of NS3 antibody, and Craig Cameron for antibody to NS5A.
This work was supported in part by National Institute of Allergy and Infectious Diseases grants U19-AI40035, R21-AI063451, and N01-AI25488.
Footnotes
Published ahead of print on 28 May 2008.
REFERENCES
- 1.Agapov, E. V., C. L. Murray, I. Frolov, L. Qu, T. M. Myers, and C. M. Rice. 2004. Uncleaved NS2-3 is required for production of infectious bovine viral diarrhea virus. J. Virol. 782414-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alter, H. J. 2005. HCV natural history: the retrospective and prospective in perspective. J. Hepatol. 43550-552. [DOI] [PubMed] [Google Scholar]
- 3.Bartenschlager, R. 1999. The NS3/4A proteinase of the hepatitis C virus: unravelling structure and function of an unusual enzyme and a prime target for antiviral therapy. J. Viral Hepat. 6165-181. [DOI] [PubMed] [Google Scholar]
- 4.Cho, H. S., N. C. Ha, L. W. Kang, K. M. Chung, S. H. Back, S. K. Jang, and B. H. Oh. 1998. Crystal structure of RNA helicase from genotype 1b hepatitis C virus. A feasible mechanism of unwinding duplex RNA. J. Biol. Chem. 27315045-15052. [DOI] [PubMed] [Google Scholar]
- 5.Dimitrova, M., I. Imbert, M. P. Kieny, and C. Schuster. 2003. Protein-protein interactions between hepatitis C virus nonstructural proteins. J. Virol. 775401-5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gastaminza, P., G. Cheng, S. Wieland, J. Zhong, W. Liao, and F. V. Chisari. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation and secretion. J. Virol. 822241-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gastaminza, P., S. B. Kapadia, and F. V. Chisari. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 8011074-11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gosert, R., D. Egger, V. Lohmann, R. Bartenschlager, H. E. Blum, K. Bienz, and D. Moradpour. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 775487-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffin, S. D., L. P. Beales, D. S. Clarke, O. Worsfold, S. D. Evans, J. Jaeger, M. P. Harris, and D. J. Rowlands. 2003. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, amantadine. FEBS Lett. 53534-38. [DOI] [PubMed] [Google Scholar]
- 10.Hirel, P. H., M. J. Schmitter, P. Dessen, G. Fayat, and S. Blanquet. 1989. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc. Natl. Acad. Sci. USA 868247-8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang, H., F. Sun, D. M. Owen, W. Li, Y. Chen, M. Gale, Jr., and J. Ye. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 1045848-5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones, C. T., C. L. Murray, D. K. Eastman, J. Tassello, and C. M. Rice. 2007. Hepatitis C virus p7 and NS2 proteins are essential for infectious virus production. J. Virol. 818374-8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones, C. T., C. G. Patkar, and R. J. Kuhn. 2005. Construction and applications of yellow fever virus replicons. Virology 331247-259. [DOI] [PubMed] [Google Scholar]
- 14.Kaul, A., I. Woerz, P. Meuleman, G. Leroux-Roels, and R. Bartenschlager. 2007. Cell culture adaptation of hepatitis C virus and in vivo viability of an adapted variant. J. Virol. 8113168-13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiiver, K., A. Merits, M. Ustav, and E. Zusinaite. 2006. Complex formation between hepatitis C virus NS2 and NS3 proteins. Virus Res. 117264-272. [DOI] [PubMed] [Google Scholar]
- 16.Kim, D. W., Y. Gwack, J. H. Han, and J. Choe. 1995. C-terminal domain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Biochem. Biophys. Res. Commun. 215160-166. [DOI] [PubMed] [Google Scholar]
- 17.Kim, J. L., K. A. Morgenstern, J. P. Griffith, M. D. Dwyer, J. A. Thomson, M. A. Murcko, C. Lin, and P. R. Caron. 1998. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure 689-100. [DOI] [PubMed] [Google Scholar]
- 18.King, J. A., R. Dubielzig, D. Grimm, and J. A. Kleinschmidt. 2001. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J. 203282-3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolykhalov, A. A., K. Mihalik, S. M. Feinstone, and C. M. Rice. 2000. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 742046-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kummerer, B. M., and C. M. Rice. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 764773-4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam, A. M., and D. N. Frick. 2006. Hepatitis C virus subgenomic replicon requires an active NS3 RNA helicase. J. Virol. 80404-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, K., E. Foy, J. C. Ferreon, M. Nakamura, A. C. Ferreon, M. Ikeda, S. C. Ray, M. Gale, Jr., and S. M. Lemon. 2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 1022992-2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, K., T. Prow, S. M. Lemon, and M. R. Beard. 2002. Cellular response to conditional expression of hepatitis C virus core protein in Huh7 cultured human hepatoma cells. Hepatology 351237-1246. [DOI] [PubMed] [Google Scholar]
- 24.Li, X. D., L. Sun, R. B. Seth, G. Pineda, and Z. J. Chen. 2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 10217717-17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin, C., and J. L. Kim. 1999. Structure-based mutagenesis study of hepatitis C virus NS3 helicase. J. Virol. 738798-8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309623-626. [DOI] [PubMed] [Google Scholar]
- 27.Liu, W. J., P. L. Sedlak, N. Kondratieva, and A. A. Khromykh. 2002. Complementation analysis of the flavivirus Kunjin NS3 and NS5 proteins defines the minimal regions essential for formation of a replication complex and shows a requirement of NS3 in cis for virus assembly. J. Virol. 7610766-10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285110-113. [DOI] [PubMed] [Google Scholar]
- 29.Lorenz, I. C., J. Marcotrigiano, T. G. Dentzer, and C. M. Rice. 2006. Structure of the catalytic domain of the hepatitis C virus NS2-3 protease. Nature 442831-835. [DOI] [PubMed] [Google Scholar]
- 30.McLauchlan, J., M. K. Lemberg, G. Hope, and B. Martoglio. 2002. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 213980-3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meylan, E., J. Curran, K. Hofmann, D. Moradpour, M. Binder, R. Bartenschlager, and J. Tschopp. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 4371167-1172. [DOI] [PubMed] [Google Scholar]
- 32.Miyanari, Y., K. Atsuzawa, N. Usuda, K. Watashi, T. Hishiki, M. Zayas, R. Bartenschlager, T. Wakita, M. Hijikata, and K. Shimotohno. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 91089-1097. [DOI] [PubMed] [Google Scholar]
- 33.Morgenstern, K. A., J. A. Landro, K. Hsiao, C. Lin, Y. Gu, M. S. Su, and J. A. Thomson. 1997. Polynucleotide modulation of the protease, nucleoside triphosphatase, and helicase activities of a hepatitis C virus NS3-NS4A complex isolated from transfected COS cells. J. Virol. 713767-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patkar, C. G., and R. J. Kuhn. 2008. Yellow Fever virus NS3 plays an essential role in virus assembly independent of its known enzymatic functions. J. Virol. 823342-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pijlman, G. P., N. Kondratieva, and A. A. Khromykh. 2006. Translation of the flavivirus Kunjin NS3 gene in cis but not its RNA sequence or secondary structure is essential for efficient RNA packaging. J. Virol. 8011255-11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reed, K. E., A. Grakoui, and C. M. Rice. 1995. Hepatitis C virus-encoded NS2-3 protease: cleavage-site mutagenesis and requirements for bimolecular cleavage. J. Virol. 694127-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shepard, C. W., L. Finelli, and M. J. Alter. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5558-567. [DOI] [PubMed] [Google Scholar]
- 38.Shi, S. T., S. J. Polyak, H. Tu, D. R. Taylor, D. R. Gretch, and M. M. Lai. 2002. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology 292198-210. [DOI] [PubMed] [Google Scholar]
- 39.Shimotohno, K., Y. Tanji, Y. Hirowatari, Y. Komoda, N. Kato, and M. Hijikata. 1995. Processing of the hepatitis C virus precursor protein. J. Hepatol. 2287-92. [PubMed] [Google Scholar]
- 40.Steinmann, E., F. Penin, S. Kallis, A. H. Patel, R. Bartenschlager, and T. Pietschmann. 2007. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 3e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tai, C. L., W. C. Pan, S. H. Liaw, U. C. Yang, L. H. Hwang, and D. S. Chen. 2001. Structure-based mutational analysis of the hepatitis C virus NS3 helicase. J. Virol. 758289-8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taraporewala, Z. F., and J. T. Patton. 2004. Nonstructural proteins involved in genome packaging and replication of rotaviruses and other members of the Reoviridae. Virus Res. 10157-66. [DOI] [PubMed] [Google Scholar]
- 43.Thibeault, D., R. Maurice, L. Pilote, D. Lamarre, and A. Pause. 2001. In vitro characterization of a purified NS2/3 protease variant of hepatitis C virus. J. Biol. Chem. 27646678-46684. [DOI] [PubMed] [Google Scholar]
- 44.Timpe, J. M., Z. Stamataki, A. Jennings, K. Hu, M. J. Farquhar, H. J. Harris, A. Schwarz, I. Desombere, G. L. Roels, P. Balfe, and J. A. McKeating. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 4717-24. [DOI] [PubMed] [Google Scholar]
- 45.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walker, J. E., M. Saraste, M. J. Runswick, and N. J. Gay. 1982. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1945-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Welbourn, S., R. Green, I. Gamache, S. Dandache, V. Lohmann, R. Bartenschlager, K. Meerovitch, and A. Pause. 2005. Hepatitis C virus NS2/3 processing is required for NS3 stability and viral RNA replication. J. Biol. Chem. 28029604-29611. [DOI] [PubMed] [Google Scholar]
- 48.Yao, N., P. Reichert, S. S. Taremi, W. W. Prosise, and P. C. Weber. 1999. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure 71353-1363. [DOI] [PubMed] [Google Scholar]
- 49.Yi, M., F. Bodola, and S. M. Lemon. 2002. Subgenomic hepatitis C virus replicons inducing expression of a secreted enzymatic reporter protein. Virology 304197-210. [DOI] [PubMed] [Google Scholar]
- 50.Yi, M., and S. M. Lemon. 2004. Adaptive mutations producing efficient replication of genotype 1a hepatitis C virus RNA in normal Huh7 cells. J. Virol. 787904-7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yi, M., Y. Ma, J. Yates, and S. M. Lemon. 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81629-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yi, M., R. A. Villanueva, D. L. Thomas, T. Wakita, and S. M. Lemon. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. USA 1032310-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 1029294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhong, J., P. Gastaminza, J. Chung, Z. Stamataki, M. Isogawa, G. Cheng, J. A. McKeating, and F. V. Chisari. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J. Virol. 8011082-11093. [DOI] [PMC free article] [PubMed] [Google Scholar]