

Abstract
Microtubule-mediated anterograde transport is essential for the transport of herpes simplex virus type 1 (HSV-1) along axons, yet little is known regarding the mechanism and the machinery required for this process. Previously, we were able to reconstitute anterograde transport of HSV-1 on microtubules in an in vitro microchamber assay. Here we report that the large tegument protein UL36p is essential for this trafficking. Using a fluorescently labeled UL36 null HSV-1 strain, KΔUL36GFP, we found that it is possible to isolate a membrane-associated population of this virus. Although these viral particles contained normal amounts of tegument proteins VP16, vhs, and VP22, they displayed a 3-log decrease in infectivity and showed a different morphology compared to UL36p-containing virions. Membrane-associated KΔUL36GFP also displayed a slightly decreased binding to microtubules in our microchamber assay and a two-thirds decrease in the frequency of motility. This decrease in binding and motility was restored when UL36p was supplied in trans by a complementing cell line. These findings suggest that UL36p is necessary for HSV-1 anterograde transport.
All members of the herpesvirus family share four structural components. Each consists of a double-stranded DNA genome enclosed within an icosahedral capsid. The capsid is surrounded by a dense layer of protein termed the tegument, which is bounded by a host-derived membrane that contains virally encoded glycoproteins.
Upon infection, the virion fuses its membrane with that of the host cell (39), and the capsid and tegument are deposited in the cytoplasm. The capsid then traffics to the nucleus along microtubules, with tegument proteins US3, UL36, and UL37 remaining capsid associated as seen in the closely related pseudorabies virus (PrV) (18, 28). After packaging and assembly take place in the nucleus, it is thought that nonenveloped capsids bud into the inner nuclear membrane, acquiring a primary envelope. This primary enveloped particle then fuses its membrane with the outer nuclear membrane, depositing capsids in the cytosol (3, 31, 32, 45). Cytosolic capsids subsequently traffic to the trans-Golgi network (TGN) or endosomes, where they acquire mature tegument and a secondary envelope before exiting the cell (20, 27, 29, 33, 52).
There is currently little known regarding the mechanism that drives anterograde transport of the virus to the cell periphery. While the nature of the particle trafficking to the plasma membrane in nonneuronal cells is believed to be a secondary enveloped mature particle (29), capsids still must travel from the nucleus to the TGN in order to obtain their envelope. Similarly, the structure of the viral particle trafficking in axons is currently the topic of much debate. One school of thought holds that it is the nonenveloped herpes simplex virus type 1 (HSV-1) capsid that is transported along axonal microtubules separately from its envelope, as seen in electron microscopy, immunofluorescence, and explant studies (21, 33, 38, 42, 48, 49). These capsids are thought to acquire their envelope along with outer tegument and glycoproteins upon budding into membranes at the axon terminus. The alternative model is that capsids first acquire their envelope in the cell body before trafficking down the axon as an enveloped or partially enveloped particle (1, 4, 5). Whatever the nature of the particle undergoing transport, the viral structural proteins responsible for motility also remain obscure. Few interactions have been shown between viral proteins and cellular motors (10-12, 25, 29, 30, 37, 54), and only one protein has been identified to be essential for anterograde transport in an alphaherpesvirus, the UL36 protein in PrV (28).
UL36 encodes the largest HSV-1 tegument protein, VP1/2 (UL36p), of 273 kDa. This protein has been implicated in release of the viral genome at the nuclear pore during infection and also in egress (2, 18, 28, 50). It also contains a deubiquitinating domain that is conserved across all members of the herpesvirus family (44). UL36p has been shown to associate with PrV capsids released from the nucleus prior to acquisition of the secondary envelope (23, 28), and for both HSV and PrV, it is thought to comprise the first layer of tegument in the virus, along with the products of the UL37 and US3 genes. It has been shown to immunoprecipitate with the capsid protein VP5 (53), bind the capsid protein UL25p directly in PrV and HSV (7), and interacts with tegument proteins VP16 and UL37p (23, 53). The absence of UL36p from HSV-1 and PrV results in the accumulation of nonenveloped capsids in the cytoplasm and nucleus that appear unable to traffic to the site of secondary envelopment and to acquire tegument (9, 16, 29).
We have previously described an in vitro assay that allows us to replicate anterograde transport of green fluorescent protein (GFP)-labeled membrane-associated HSV-1 along rhodamine-labeled microtubules (27). Because of the in vivo phenotypes of the UL36 null HSV-1 virus, we tested the behavior of such a null strain in our assay system. We made use of the HSV-1 strain KΔUL36GFP (9), which carries a large deletion in the UL36 gene and has GFP fused to the capsid protein VP26.
Surprisingly, we found membrane-associated capsids in the cytoplasm of cells infected with KΔUL36GFP which contained at least some of the components of outer tegument. These particles also differed morphologically from wild-type HSV-1. Strikingly, they were capable of binding to microtubules in vitro but demonstrated a severe defect in motility.
MATERIALS AND METHODS
Cells and viruses.
Vero cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% penicillin-streptomycin (PS) and 10% newborn calf serum (Gibco Laboratories). HS30 cells (9) were grown in DMEM supplemented with 1% PS, 10% fetal calf serum (Gibco Laboratories), and Geneticin (Invitrogen). K26GFP (8) and KΔUL36GFP (9) stocks were grown, and the titers were determined by plaque assay on Vero and HS30 cell monolayers as previously described (6).
Single-step growth curves.
Vero and HS30 cells were infected with K26GFP and KΔUL36GFP at a multiplicity of infection (MOI) of 10. After 1 h at 37°C, cells were acid washed, in which cells were washed twice with ice-cold phosphate-buffered saline (PBS), treated for 30 seconds with ice-cold glycine-buffered saline (136 mM NaCl, 5 mM KCl, 100 mM glycine, pH 2.8), and then washed twice with PBS. Cells were then overlaid with prewarmed DMEM-newborn calf serum-PS. Cells were either harvested immediately or at 12 or 18 h postinfection, and the titers were determined by plaque assay on Vero and HS30 cell monolayers as previously described (6).
Isolation of membrane-associated HSV.
Vero or HS30 cells were infected with K26GFP or KΔUL36GFP at an MOI of 10 or 50 for 1 h at 37°C. After 1 h, the cells were acid washed, as described above. Infected cells were then overlaid with fresh prewarmed medium and incubated at 37°C for 11 h. Cells were then washed once with ice-cold MEPS buffer [5 mM MgSO4, 5 mM EGTA, 0.25 M sucrose, 35 mM piperazine-N,N′-bis(2-ethanesulfonic acid), pH 7.1] (36), scraped and collected, and resuspended in MEPS buffer containing 2 mM phenylmethylsulfonyl fluoride, 2% (vol/vol) protease inhibitor cocktail (Sigma), and 4 mM dithiothreitol (27). Postnuclear supernatant (PNS) and a membrane-associated fraction were then isolated as previously described (27).
Western blotting and antibodies.
PNS was isolated as previously described (27). Membrane-associated fractions of virus were resuspended in MEPS and centrifuged in a TLS55 rotor (Beckman) for 60 min at 101,000 × g. Pellets were resuspended in PBS, stored overnight at 4°C, and sonicated. All samples were then boiled in Laemmli buffer and electrophoresed in a denaturing 8% resolving sodium dodecyl sulfate-polyacrylamide gel. Samples were transferred to a polyvinylidene difluoride membrane (Bio-Rad) and blocked in a 5% milk Tris-buffered saline solution supplemented with Tween (26). Membranes were incubated overnight with the appropriate primary antibodies and for 1 hour with alkaline phosphatase-conjugated goat anti-rabbit antibody (Chemicon, Pittsburgh, PA) or alkaline phosphatase-conjugated goat anti-mouse antibody (Antibodies Incorporated, Davis, CA) as appropriate. Secondary antibodies were detected with alkaline phosphatase, as previously described (34). Primary antibodies were obtained as follows: VP5 was from Genetech, VP16 was from Santa Cruz Biotechnology, vhs antiserum was raised as previously described (26), and anti-VP22 rabbit antiserum was raised against a synthetic peptide corresponding to the carboxy-terminal 21 amino acids of HSV-1 VP22 (S. Smith and D. Wilson, unpublished data).
Preparation of fluorescent microtubules.
Fluorescent microtubules were prepared as previously described (27) with unlabeled and rhodamine-labeled tubulins (Cytoskeleton) mixed at concentrations of either 10:1 or 31:1 (total concentration, 6.5 or 6.22 μg/μl, respectively).
Construction of optical microchamber.
Optical microchambers were assembled as previously described (27, 35, 36). Two pieces of double-sided tape (Scotch 3M) were laid parallel on a large coverslip (Corning) coated with 20 μg/ml of DEAE-dextran (Pharmacia) and covered with a piece of cut glass, creating a chamber holding 3 to 5 μl.
Binding and motility assays.
Motility assays were performed as previously described (27, 36). Binding studies were performed similarly to motility assays, but omitting ATP. Membrane-associated virions were thawed and allowed to flow into the optical microchamber containing prebound microtubules. Images were taken, and the chambers were washed with assay buffer [35 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), 5 mM MgSO4, 1 mM EGTA, 0.5 mM EDTA, 20 μM taxol, 2 mg/ml bovine serum albumin, 4 mM dithiothreitol, 2 mg/ml ascorbic acid, pH 7.4]. Images were then taken again. Binding percentages were calculated, comparing the total number of vesicles present before the wash to those that were microtubule bound after the wash.
Light microscopy and image analysis.
All imaging was done at the Analytical Imaging Facility of the Albert Einstein College of Medicine, and images were saved as .tif files and processed using ImageJ 1.36 software as previously described (27).
Electron microscopy.
Vero and HS30 cells were infected with KΔUL36GFP or K26 at an MOI of 10. Cells were acid washed after 1 h. At 12 h postinfection, samples were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, postfixed with 1% osmium tetroxide followed by 2% uranyl acetate, dehydrated through a graded series of ethanol, and embedded in LX112 resin (LADD Research Industries, Burlington, VT). Ultrathin sections were cut on a Reichert Ultracut UCT, stained with uranyl acetate followed by lead citrate, and viewed on a JEOL 1200EX transmission electron microscope at 80 kV.
RESULTS
Characterization of KΔUL36GFP.
Desai (9) reported the construction of HSV-1 strain KΔUL36GFP, which carries a large deletion in the UL36 gene and GFP fused to VP26, and kindly made this virus available to us. However, to our knowledge the growth properties of this virus have not been published. To establish these properties for this strain, HS30 cells, which produce the UL36 protein in trans (9), were infected with either KΔUL36GFP or the wild-type K26GFP strain at an MOI of 0.01. At full cytopathic effect, the cells were harvested and titers were determined on both Vero and HS30 cells. KΔUL36GFP exhibited wild-type titers when grown on HS30 cells but showed a 3-log decrease in infectivity on Vero cells (Table 1).
TABLE 1.
Viral yields following low-MOI infections on HS30 cells
| Virusa | Cell line for titer determination | PFU/ml |
|---|---|---|
| K26GFP | Vero | 7 × 108 |
| K26GFP | HS30 | 3.2 × 108 |
| KΔUL36GFP | Vero | 3.2 × 105 |
| KΔUL36GFP | HS30 | 1.72 × 108 |
K26GFP and KΔUL36GFP were isolated from HS30 cells.
Similarly, K26GFP and KΔUL36GFP (prepared from HS30 cells) were subjected to a single round of replication on either HS30 or Vero cells, and then titers were determined on each cell line (Fig. 1). KΔUL36GFPH (KΔUL36GFP isolated from HS30 cells) titers on HS30 cells showed wild-type growth kinetics and yields (Fig. 1B); however, KΔUL36GFPH showed a 3-log decrease in titer on Vero cells (Fig. 1A). KΔUL36GFPV (KΔUL36GFP isolated from Vero cells) titers on HS30 cells showed 1,000- and 10,000-fold-lower PFU yields than KΔUL36GFPH when titers were determined on HS30 (Fig. 1B) and Vero cells (Fig. 1A), respectively.
FIG. 1.
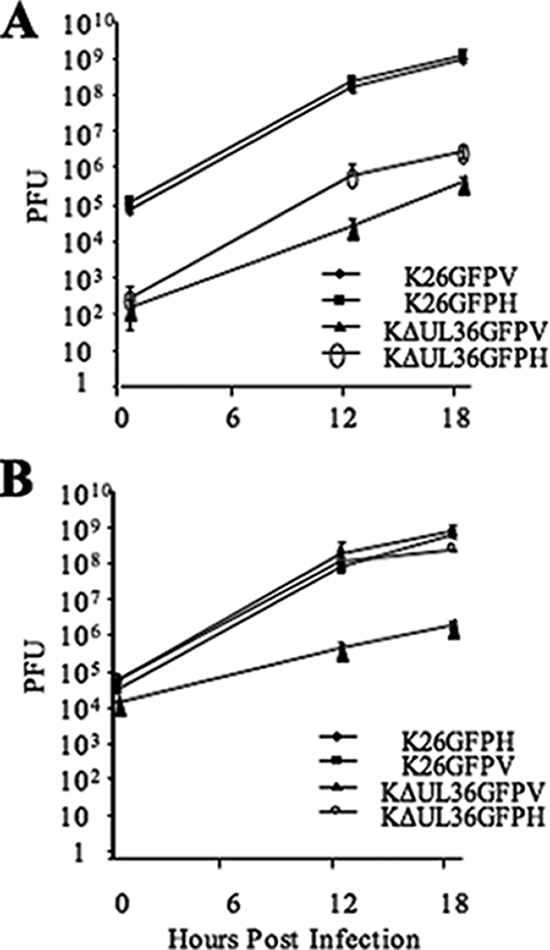
Single-step growth curves of K26GFP and KΔUL36GFP. Vero (V) and HS30 (H) cells were infected with K26GFP and KΔUL36GFP at an MOI of 10 and acid washed 1 h postinfection. Cells were harvested after 1, 12, or 18 h at 37°C and titers were determined on Vero (A) or HS30 (B) cells. Error bars represent standard deviations from the means.
KΔUL36GFP is capable of assembling an enveloped, partially tegumented particle which is not infectious.
In order to study how the absence of UL36p affects motility, PNS was isolated from both K26GFP- and KΔUL36GFP-infected cells and then tested in our standard microchamber assay along with rhodamine-labeled microtubules (27, 35, 36). However, both wild-type and UL36 null virus fractions showed low levels of binding and little to no motility on microtubules. Similar results have been reported for endosomal trafficking in this system and have been shown to be due to the presence of abundant cellular ATPases in the PNS (A. Wolkoff, personal communication).
We have previously performed motility studies using a membrane-associated K26GFP float-up fraction from infected cells (27). Previous studies reported only nonenveloped capsids present in the cytoplasm and nucleus of KΔUL36GFP-infected cells, suggesting that no KΔUL36GFP should be present in such a gradient float-up fraction (9, 16). Nevertheless, we collected such gradient fractions from KΔUL36GFP-infected cells and loaded them into microchambers along with microtubules to test for the presence of fluorescent vesicles (27, 36). GFP-fluorescing vesicles were readily apparent in the microchambers and were bound to microtubules (Fig. 2), indicating that, unexpectedly, there is a population of KΔUL36GFP capsids which acquire a secondary envelope (9, 16, 23).
FIG. 2.

KΔUL36GFP was present in a membrane-associated float-up fraction. Vero cells were infected with KΔUL36GFP at an MOI of 50. At 12 h postinfection cells were harvested and then PNS was prepared and loaded at the bottom of a sucrose gradient. A membrane-associated buoyant fraction was isolated and flowed into a microchamber containing rhodamine-labeled microtubules. HSV membrane-associated particles are shown in green and microtubules are in red. Bar, 10 μm.
The infectivities of these membrane-associated KΔUL36GFP and K26GFP particles were determined by assessing titers on both Vero and HS30 cells (Fig. 3). Membrane-associated KΔUL36GFPH (isolated from HS30 cells) showed wild-type yields when titers were determined on HS30 cells, with titers reaching ca. 3.1 × 106 PFU, but displayed a 2- to 3-log decrease on Vero cells. KΔUL36GFPV (isolated from Vero cells) displayed a 2- to 3-log decrease in infectivity compared to K26GFP when titers were measured on either cell line. Thus, while it appears that KΔUL36GFP acquires a secondary envelope, there appears to be some defect that results in a failure to generate infectious particles.
FIG. 3.
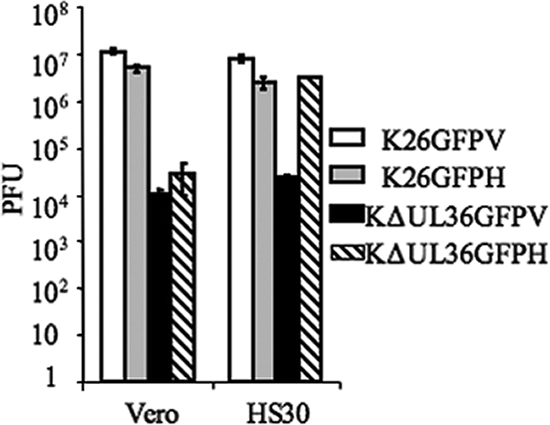
Membrane-associated fractions of KΔUL36GFP show a greatly diminished infectivity compared to the wild type. Membrane-associated fractions of both K26GFP and KΔUL36GFP were isolated from Vero (V) and HS30 (H) cells as indicated. Titers for these fractions were then determined on either Vero or HS30 cells, as shown. Error bars represent standard deviations from the means.
The membrane-associated fraction containing KΔUL36GFP has a similar tegument composition to wild-type HSV-1.
In previous studies of UL36 null HSV-1, it was reported that tegument proteins such as VP16 are absent from cytoplasmic capsids (9, 23, 24). Thus, we performed Western blot assays on membrane-associated K26GFP and KΔUL36GFP isolated from Vero and HS30 cells using antibodies against the tegument proteins VP16, vhs, VP22, and the capsid protein VP5 (Fig. 4). Levels of VP5 were used to control for the numbers of viral capsids. There appeared to be similar amounts of VP5 present in the membrane-associated particles of KΔUL36GFPV and K26GFP, and there was no significant difference in the levels of tegument proteins VP16, VP22, and vhs in either fraction. Thus, the capsid protein VP5 and some tegument proteins appear to associate with membranes even in the absence of full-length UL36p.
FIG. 4.

Buoyant membranes from KΔUL36GFP-infected cells are associated with HSV capsids and some outer tegument proteins. PNS and membrane-associated fractions from K26GFP- and KΔUL36GFP-infected Vero or HS30 cells were prepared. They were then subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotted for the capsid protein VP5 or tegument proteins VP16, vhs, and VP22 as indicated. Lanes 1 and 3 represent K26GFP isolated from Vero or HS30 cells, and lanes 2 and 4 represent KΔUL36GFP isolated from Vero or HS30 cells, respectively.
Loss of UL36 leads to a defect in microtubular motility.
UL36 has been implicated in egress in both HSV-1 and PrV, and it is essential for anterograde transport of capsids in PrV (9, 28, 29). In order to examine the motility of KΔUL36GFP enveloped particles, optical microchambers containing rhodamine-labeled microtubules were constructed as previously described (27, 36). Membrane-associated K26GFP or KΔUL36GFP was allowed to flow into the chambers, and approximately 77% of the K26GFP particles isolated from Vero cells were able to bind microtubules (Fig. 5A). Similar results were seen for K26GFP isolated from HS30 cells (data not shown). KΔUL36GFP isolated from complementing HS30 cells had a similar binding efficiency to wild type, 79%. However, KΔUL36GFP particles isolated from Vero cells showed somewhat reduced binding.
FIG. 5.
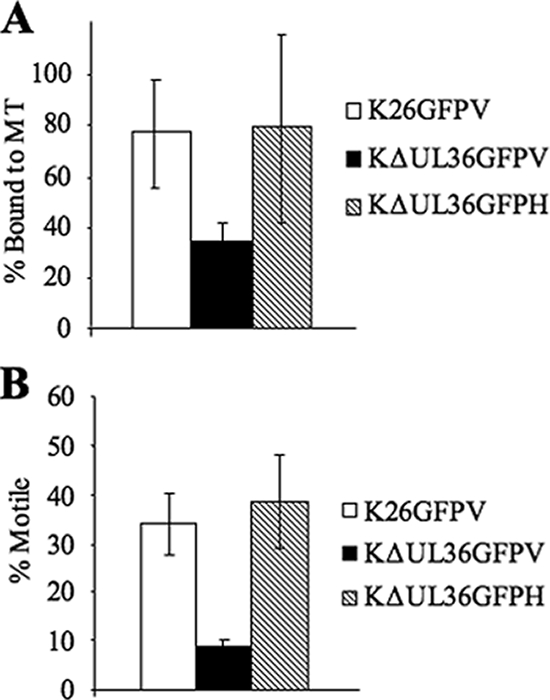
KΔUL36GFP shows decreased binding and motility compared to wild-type virus. (A) Microtubule binding efficiency was calculated for the membrane-associated populations of K26GFP and KΔUL36GFP prepared from Vero (V) or HS30 (H) cells. Membrane-associated fractions were flowed into a microchamber along with rhodamine-labeled microtubules (MT) as described for Fig. 2. Binding efficiency was calculated based on the total number of vesicles flowed into the chamber compared to the number of vesicles remaining bound to microtubules after washing. (B) Motility efficiency was calculated based on the total number of particles that were bound to microtubules compared to the number that became motile upon addition of ATP. Error bars represent standard deviations from the means.
Upon addition of ATP, K26GFP particles isolated from either Vero or HS30 cells began to move along the microtubules, and in each case, approximately 34% of microtubule-bound virions exhibited motility (Fig. 5B and data not shown). Similarly, 38% of KΔUL36GFP particles isolated from HS30 cells exhibited motility. In contrast, KΔUL36GFP isolated from Vero cells and, hence, lacking UL36p, exhibited a profound defect in motility, with only approximately 9% of bound viral particles trafficking on microtubules. In all cases, the average velocity of these particles (0.71 μm/s) was consistent with velocities previously described for transport of HSV (27, 38, 43) but slower than reported for PrV (46, 47).
Enveloped KΔUL36GFP particles are morphologically distinct from wild-type HSV-1.
Electron microscope images taken 12 h postinfection showed differences in morphology between KΔUL36GFPV and the wild-type control K26GFPV. We observed accumulations of capsids both in the nucleus and cytoplasm of KΔUL36GFP-infected cells (Fig. 6D), as previously described for this virus and for UL36 null PrV (9, 16, 29). However, membrane-associated KΔUL36GFP capsids were also found to be present in the cytoplasm, consistent with our biochemical findings (Fig. 6C to E). Occasionally, enveloped virus was also seen on the surface of the plasma membrane. Enveloped K26GFP viral particles found in the cytoplasm of infected cells appeared to have a somewhat irregular and nonsymmetrical envelope and tegument characteristic of mature wild-type HSV-1 virions (Fig. 6A and B). In contrast, cytoplasmic enveloped KΔUL36GFP particles (Fig. 6C to E) displayed a more symmetrical envelope, and the tegument appeared to form a dense, smooth, rather homogenous ring lining the inner surface at the envelope, rather similar to primary enveloped virions in the perinuclear space (17, 40, 41).
FIG. 6.

Enveloped K26GFP and KΔUL36GFP particles are present in the cytoplasm of infected Vero cells. Enveloped HSV-1 particles were present in the cytoplasm of both K26GFP-infected (A and B) and KΔUL36GFP-infected (C to E) Vero cells. (A) Cytoplasmic enveloped K26GFP particles can be observed in the cytoplasm (black arrow), with a number of enveloped capsids contained within one organelle. Extracellular capsids are also present (black arrowhead), attached to the plasma membrane. (B) An enveloped K26GFP particle is present in the cytoplasm. (C to E) Enveloped KΔUL36GFP particles are present in the cytoplasm (black arrows), and nonenveloped capsids (C and D) were also observed (white arrows). Bars, 500 nm (A to C); 200 nm (D and E).
DISCUSSION
The purpose of this study was to determine whether UL36p plays a role in microtubule-mediated transport of HSV-1. Using an in vitro assay that reconstitutes viral trafficking along microtubules (27), we found that in the absence of UL36p, a membrane-associated HSV-1 particle forms which has greatly reduced infectivity and a decreased ability to both bind and traffic along microtubules. These defects are restored when UL36p is provided in trans by a complementing cell line. Although the outer tegument composition of UL36p-deficient particles does not appear to differ from the wild type with respect to VP22, vhs, or VP16, these particles do show a difference in their morphology, displaying a symmetrically circular-shaped membrane with a dense layer of smooth homogenous tegument lining the inner surface of the envelope.
KΔUL36GFP prepared from a complementing cell line displays a 3- to 4-log decrease in titer when plated on noncomplementing Vero cells (Fig. 1A). Interestingly, it appears that KΔUL36GFP is able to enter Vero cells with greater efficiency than is K26GFP. All cells were acid washed 1 hour postinfection, inactivating any virus remaining on the cell surface. The K26GFP titers seen at the 1-hour time point after the acid wash correspond to the approximately 0.1% of input virus not inactivated, which is a routine figure. However, titers of KΔUL36GFP on Vero cells at 1 h were 2 to 3 logs lower (Fig. 1A), suggesting an increased rate of entry on this cell line. Alternatively, this may be due to greater sensitivity of the null virus to acid washing, due to incomplete complementation in the HS30 cell line.
Note that following the replication of KΔUL36GFP in Vero cells, the resulting cell extracts contained infectious particles capable of subsequent replication on Vero cells (Table 1; Fig. 1A). These particles result from the well-documented high rate of reversion of KΔUL36GFP when stocks are prepared on the complementing cell line (9).
Previous ultrastructural studies reported that an HSV-1 strain carrying the same deletion present in the KΔUL36GFP virus accumulated nonenveloped and apparently nontegumented naked capsids in the cytoplasm of infected cells (9). These observations were consistent with the finding that, following density gradient centrifugation, capsids prepared from such infected cells failed to cofractionate with envelope proteins and the tegument protein VP16. In contrast we found that the tegument proteins VP16, VP22, and vhs were present at normal levels in the buoyant capsid-containing fractions of our gradients (Fig. 4). This apparent contradiction is easily explained by differences in the cell lysis conditions. Desai (9) broke infected cells by several freeze-thaw cycles followed by sonication, whereas in our studies cells were broken under less harsh conditions by shear forces in a syringe needle.
We propose that in the absence of UL36p, the capsid is weakly attached to outer tegument components, perhaps via the network of interactions between UL25p and UL17p (51), UL17p, VP11/12, and VP13/14 (J. D. Baines, personal communication), VP11/12 and VP13/14 with VP16 (53), and finally between VP16 and gH (19, 22). Additional interactions between VP16 and VP22, and between VP22 and gE and gM, have also been reported for HSV and PrV (13-15). Under our conditions of cell breakage, these interactions maintain a stable enveloped particle which we can isolate by density centrifugation, although such particles are noninfectious (Fig. 3), nonmotile on microtubules (Fig. 5), and morphologically distinct from the wild type (Fig. 6). This also supports our electron microscopy data (Fig. 6), in which the tegument appears to be located in a dense, homogenous layer at the inner surface of the envelope instead of forming an amorphous layer between the capsid and membrane. The outer tegument can still assemble at the TGN and endosomes, while the UL36p-dependent inner tegument is lost. The discrepancy in ultrastructural studies (9, 16, 29) can be explained by the fact that naked capsids present in the cytoplasm far outnumber enveloped capsids, and the latter are more difficult to observe.
We have shown in this study that UL36p is involved in the binding and motility of enveloped HSV-1 on microtubules. Membrane-associated KΔUL36GFP particles showed a slight decrease in their ability to bind microtubules, and their motility was reduced by more than two-thirds (Fig. 5). When UL36p was added back using a complementing cell line, both binding and motility were restored.
How might UL36p, which is normally thought to lie between the capsid and envelope, influence the interactions between microtubules and the surface of the organelle within which the enveloped virions reside? We propose two possible explanations, as summarized in Fig. 7. First, the capsids, though attached to their surrounding membrane by tegument, may be only partially enveloped. Thus, the capsid surface and UL36p would be exposed to cytoplasmic components, and thus to microtubules, motors, and accessory proteins necessary for microtubule transport. Such partially enveloped structures have recently been proposed to exist in vivo (G. A. Smith, personal communication), and we have observed them in our in vitro system (27).
FIG. 7.
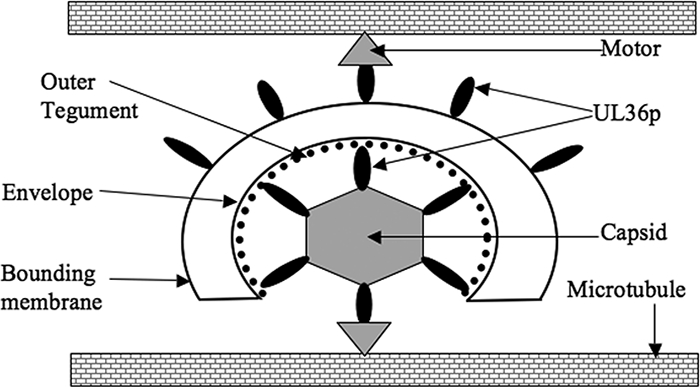
Model for the role of UL36p in microtubule-mediated HSV motility. Based on our data and those of others, we propose that UL36p (black oval) is exposed to the cytoplasm when present on incompletely enveloped capsids (lower part of figure) and/or on the surface of a bounding organellar membrane (upper part of figure). These populations of UL36p directly or indirectly recruit motor proteins (gray triangles) which mediate movement along the microtubules. There must also be additional viral or cellular factors (not shown) that tether membrane-associated capsids to the microtubules in the absence of UL36p but which are not sufficient to confer motility.
An alternative explanation is that UL36p also resides on the surface of the bounding organelle (Fig. 7). This is consistent with the findings of Desai and colleagues, who reported that UL37p binds to the surface of cytoplasmic organelles in a UL36p-dependent fashion (P. Desai, personal communication). In both of these models, UL36p would then directly or indirectly recruit kinesin motors to the particle to enable motility. Note that while UL36p may stabilize attachment to microtubules, it does not appear to be essential. However, it is possible that the remaining 400 codons encoding the amino terminus of UL36p generate a gene product with some remaining biological activity, such as microtubule attachment.
Finally, since we prepared buoyant, membrane-associated particles in this study, it is clear that enveloped or partially enveloped HSV-1 capsids are capable of microtubule-mediated motility, consistent with earlier reports (27, 28, 33, 38). However, nonenveloped capsids would be lost under our preparation conditions, and so we can reach no conclusion concerning the motility of naked HSV-1 capsids.
Acknowledgments
This work was supported by Molecular Pathogenesis Training Grant 5 T32 AI007506, as well as by NIH program project grant DK41918 and by RO1 AI38265 (to D.W.W.).
HSV strains K26GFP and KΔUL36GFP and cell line HS30 were kind gifts from Prashant Desai. We thank Allan Wolkoff and John Murray for their helpful discussions and Lily Huang for excellent technical assistance.
Footnotes
Published ahead of print on 21 May 2008.
REFERENCES
- 1.Antinone, S. E., and G. A. Smith. 2006. Two modes of herpesvirus trafficking in neurons: membrane acquisition directs motion. J. Virol. 8011235-11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batterson, W., D. Furlong, and B. Roizman. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol. 45397-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Browne, H., S. Bell, T. Minson, and D. W. Wilson. 1996. An endoplasmic reticulum-retained herpes simplex virus glycoprotein H is absent from secreted virions: evidence for reenvelopment during egress. J. Virol. 704311-4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ch'ng, T. H., and L. W. Enquist. 2005. Efficient axonal localization of alphaherpesvirus structural proteins in cultured sympathetic neurons requires viral glycoprotein E. J. Virol. 798835-8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ch'ng, T. H., and L. W. Enquist. 2005. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J. Virol. 7910875-10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Church, G. A., and D. W. Wilson. 1997. Study of herpes simplex virus maturation during a synchronous wave of assembly. J. Virol. 713603-3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coller, K. E., J. I. Lee, A. Ueda, and G. A. Smith. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 8111790-11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desai, P., and S. Person. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 727563-7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai, P. J. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 7411608-11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diefenbach, R. J., E. Diefenbach, M. W. Douglas, and A. L. Cunningham. 2004. The ribosome receptor, p180, interacts with kinesin heavy chain, KIF5B. Biochem. Biophys. Res. Commun. 319987-992. [DOI] [PubMed] [Google Scholar]
- 11.Diefenbach, R. J., M. Miranda-Saksena, E. Diefenbach, D. J. Holland, R. A. Boadle, P. J. Armati, and A. L. Cunningham. 2002. Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain. J. Virol. 763282-3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Douglas, M. W., R. J. Diefenbach, F. L. Homa, M. Miranda-Saksena, F. J. Rixon, V. Vittone, K. Byth, and A. L. Cunningham. 2004. Herpes simplex virus type 1 capsid protein VP26 interacts with dynein light chains RP3 and Tctex1 and plays a role in retrograde cellular transport. J. Biol. Chem. 27928522-28530. [DOI] [PubMed] [Google Scholar]
- 13.Elliott, G., G. Mouzakitis, and P. O'Hare. 1995. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol. 697932-7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farnsworth, A., K. Goldsmith, and D. C. Johnson. 2003. Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J. Virol. 778481-8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuchs, W., B. G. Klupp, H. Granzow, C. Hengartner, A. Brack, A. Mundt, L. W. Enquist, and T. C. Mettenleiter. 2002. Physical interaction between envelope glycoproteins E and M of pseudorabies virus and the major tegument protein UL49. J. Virol. 768208-8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuchs, W., B. G. Klupp, H. Granzow, and T. C. Mettenleiter. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 7811879-11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granzow, H., B. G. Klupp, W. Fuchs, J. Veits, N. Osterrieder, and T. C. Mettenleiter. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 753675-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Granzow, H., B. G. Klupp, and T. C. Mettenleiter. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 793200-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gross, S. T., C. A. Harley, and D. W. Wilson. 2003. The cytoplasmic tail of herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 3171-12. [DOI] [PubMed] [Google Scholar]
- 20.Harley, C. A., A. Dasgupta, and D. W. Wilson. 2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J. Virol. 751236-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holland, D. J., M. Miranda-Saksena, R. A. Boadle, P. Armati, and A. L. Cunningham. 1999. Anterograde transport of herpes simplex virus proteins in axons of peripheral human fetal neurons: an immunoelectron microscopy study. J. Virol. 738503-8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamen, D. E., S. T. Gross, M. E. Girvin, and D. W. Wilson. 2005. Structural basis for the physiological temperature dependence of the association of VP16 with the cytoplasmic tail of herpes simplex virus glycoprotein H. J. Virol. 796134-6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klupp, B. G., W. Fuchs, H. Granzow, R. Nixdorf, and T. C. Mettenleiter. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 763065-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klupp, B. G., H. Granzow, E. Mundt, and T. C. Mettenleiter. 2001. Pseudorabies virus UL37 gene product is involved in secondary envelopment. J. Virol. 758927-8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koshizuka, T., Y. Kawaguchi, and Y. Nishiyama. 2005. Herpes simplex virus type 2 membrane protein UL56 associates with the kinesin motor protein KIF1A. J. Gen. Virol. 86527-533. [DOI] [PubMed] [Google Scholar]
- 26.Lee, G. E., G. A. Church, and D. W. Wilson. 2003. A subpopulation of tegument protein vhs localizes to detergent-insoluble lipid rafts in herpes simplex virus-infected cells. J. Virol. 772038-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee, G. E., J. W. Murray, A. W. Wolkoff, and D. W. Wilson. 2006. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J. Virol. 804264-4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luxton, G. W., S. Haverlock, K. E. Coller, S. E. Antinone, A. Pincetic, and G. A. Smith. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 1025832-5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luxton, G. W., J. I. Lee, S. Haverlock-Moyns, J. M. Schober, and G. A. Smith. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80201-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez-Moreno, M., I. Navarro-Lerida, F. Roncal, J. P. Albar, C. Alonso, F. Gavilanes, and I. Rodriguez-Crespo. 2003. Recognition of novel viral sequences that associate with the dynein light chain LC8 identified through a pepscan technique. FEBS Lett. 544262-267. [DOI] [PubMed] [Google Scholar]
- 31.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 761537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mettenleiter, T. C., T. Minson, and P. Wild. 2006. Egress of alphaherpesviruses. J. Virol. 801610-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miranda-Saksena, M., P. Armati, R. A. Boadle, D. J. Holland, and A. L. Cunningham. 2000. Anterograde transport of herpes simplex virus type 1 in cultured, dissociated human and rat dorsal root ganglion neurons. J. Virol. 741827-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukhopadhyay, A., G. E. Lee, and D. W. Wilson. 2006. The amino terminus of the herpes simplex virus 1 protein vhs mediates membrane association and tegument incorporation. J. Virol. 8010117-10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray, J. W., E. Bananis, and A. W. Wolkoff. 2002. Immunofluorescence microchamber technique for characterizing isolated organelles. Anal. Biochem. 30555-67. [DOI] [PubMed] [Google Scholar]
- 36.Murray, J. W., E. Bananis, and A. W. Wolkoff. 2000. Reconstitution of ATP-dependent movement of endocytic vesicles along microtubules in vitro: an oscillatory bidirectional process. Mol. Biol. Cell 11419-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogawa-Goto, K., S. Irie, A. Omori, Y. Miura, H. Katano, H. Hasegawa, T. Kurata, T. Sata, and Y. Arao. 2002. An endoplasmic reticulum protein, p180, is highly expressed in human cytomegalovirus-permissive cells and interacts with the tegument protein encoded by UL48. J. Virol. 762350-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Penfold, M. E., P. Armati, and A. L. Cunningham. 1994. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. USA 916529-6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reske, A., G. Pollara, C. Krummenacher, B. M. Chain, and D. R. Katz. 2007. Understanding HSV-1 entry glycoproteins. Rev. Med. Virol. 17205-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roller, R. J., Y. Zhou, R. Schnetzer, J. Ferguson, and D. DeSalvo. 2000. Herpes simplex virus type 1 UL34 gene product is required for viral envelopment. J. Virol. 74117-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryckman, B. J., and R. J. Roller. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 78399-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saksena, M. M., H. Wakisaka, B. Tijono, R. A. Boadle, F. Rixon, H. Takahashi, and A. L. Cunningham. 2006. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J. Virol. 803592-3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Satpute-Krishnan, P., J. A. DeGiorgis, and E. L. Bearer. 2003. Fast anterograde transport of herpes simplex virus: role for the amyloid precursor protein of Alzheimer's disease. Aging Cell 2305-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlieker, C., G. A. Korbel, L. M. Kattenhorn, and H. L. Ploegh. 2005. A deubiquitinating activity is conserved in the large tegument protein of the Herpesviridae. J. Virol. 7915582-15585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skepper, J. N., A. Whiteley, H. Browne, and A. Minson. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment → deenvelopment → reenvelopment pathway. J. Virol. 755697-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith, G. A., S. P. Gross, and L. W. Enquist. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. USA 983466-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith, G. A., L. Pomeranz, S. P. Gross, and L. W. Enquist. 2004. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. USA 10116034-16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Snyder, A., B. Bruun, H. M. Browne, and D. C. Johnson. 2007. A herpes simplex virus gD-YFP fusion glycoprotein is transported separately from viral capsids in neuronal axons. J. Virol. 818337-8340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Snyder, A., T. W. Wisner, and D. C. Johnson. 2006. Herpes simplex virus capsids are transported in neuronal axons without an envelope containing the viral glycoproteins. J. Virol. 8011165-11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sodeik, B., M. W. Ebersold, and A. Helenius. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1361007-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trus, B. L., W. W. Newcomb, N. Cheng, G. Cardone, L. Marekov, F. L. Homa, J. C. Brown, and A. C. Steven. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-Filled HSV-1 capsids. Mol. Cell 26479-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turcotte, S., J. Letellier, and R. Lippe. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 798847-8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vittone, V., E. Diefenbach, D. Triffett, M. W. Douglas, A. L. Cunningham, and R. J. Diefenbach. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 799566-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ye, G. J., K. T. Vaughan, R. B. Vallee, and B. Roizman. 2000. The herpes simplex virus 1 UL34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J. Virol. 741355-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]