

Abstract
We present a threefold contribution to the computational task of motif discovery, a key component in the effort of delineating the regulatory map of a genome: (1) We constructed a comprehensive large-scale, publicly-available compendium of transcription factor and microRNA target gene sets derived from diverse high-throughput experiments in several metazoans. We used the compendium as a benchmark for motif discovery tools. (2) We developed Amadeus, a highly efficient, user-friendly software platform for genome-scale detection of novel motifs, applicable to a wide range of motif discovery tasks. Amadeus improves upon extant tools in terms of accuracy, running time, output information, and ease of use and is the only program that attained a high success rate on the metazoan compendium. (3) We demonstrate that by searching for motifs based on their genome-wide localization or chromosomal distributions (without using a predefined target set), Amadeus uncovers diverse known phenomena, as well as novel regulatory motifs.
One of the main cellular regulatory mechanisms is the transcriptional program, which describes when and to what extent each gene is transcribed to mRNA. Transcription is controlled primarily via transcription factors (TFs)—specialized proteins that bind sequence elements, called binding sites (BSs), which are located mainly in each gene’s promoter sequence upstream its transcription start site (TSS). Another key regulatory effect is controlled by microRNAs (miRNAs), short noncoding RNA molecules. Annealing of a miRNA to its target mRNA, typically in its 3′ untranslated region (UTR), triggers the degradation of the mRNA transcript or inhibits protein translation. Delineating the regulatory program of a species requires the combination of experimental and computational techniques. To this end, huge volumes of experimental data have been generated in the past decade by means of high-throughput technologies, such as gene expression microarrays (Lockhart and Winzeler 2000) and ChIP-chip location analyses (Wu et al. 2006). In parallel, numerous software tools were developed in order to analyze these data and suggest novel biological hypotheses.
A major computational challenge is identifying recurring sequence patterns, or motifs, in cis-regulatory sequences; such motifs represent BSs of TFs/miRNAs. In a typical scenario, given a target set of coregulated genes, one would like to identify TFs whose BSs are statistically overrepresented in the promoters of these genes, compared with some background model or with a supplied reference set of genes. In recent years, a plethora of computational tools have been developed for discovering enriched motifs of known TFs (Elkon et al. 2003; Sharan et al. 2004), as well as for finding novel motifs that represent BSs of yet uncharacterized TFs. The latter task, known as de novo motif discovery, has been tackled using a myriad of algorithmic techniques, such as expectation-maximization (EM) (Bailey and Elkan 1994), Gibbs sampling (Hughes et al. 2000), and efficient enumeration (Pavesi et al. 2001; Sinha and Tompa 2002; Ettwiller et al. 2007). The most common computational models employed by motif finders to describe TF BSs are degenerate (IUPAC) strings (Sinha and Tompa 2002) and position weight matrices (PWMs) (Bailey and Elkan 1994). Commonly used scores for evaluating candidate motifs include likelihood ratio (Bailey and Elkan 1994) and the Z-score (Sinha and Tompa 2002; Ettwiller et al. 2007) and hypergeometric (HG) overrepresentation scores (Eskin and Pevzner 2002).
Most studies that describe novel motif discovery algorithms report their success either on synthetically generated data or on a small ad hoc collection of samples constructed by the investigators for their particular analysis. Obviously, such results do not guarantee equally-good performance in many real-life scenarios. Perhaps the most popular large-scale motif finding benchmark is the yeast ChIP-chip data set of Harbison et al. (2004). To the best of our knowledge, the only large-scale metazoan benchmark constructed to date is that of Tompa et al. (2005). In that study, validated TF BSs from the TRANSFAC (Wingender et al. 1996) database were implanted inside real and synthetic promoter sequences. The benchmark contains 52 data sets (eight from yeast, the rest are from metazoans), with an average of seven sequences per data set. Its main drawback is that it does not reflect many real-life scenarios. For example, one would often like to discover motifs in a cluster of coexpressed genes or in a set of sequences bound by a TF in ChIP-chip. In these scenarios, the analyzed set typically consists of dozens or hundreds of genes, of which only an unknown (often modest) fraction contain BSs; moreover, many of the BSs might reside very far from the TSS or in other types of genomic sequences (introns, UTRs, etc.), and the gene set might be regulated by more than one TF. In this work, we constructed the first publicly-accessible, large-scale compendium of metazoan data sets that were obtained by various experimental techniques and cover a wide-range of real-life motif discovery scenarios.
Despite extensive research, it remains exceedingly difficult to accurately predict BSs and discover novel motifs, especially in metazoan data sets, due to the short and degenerate nature of BSs, the size and complexity of genetic sequences, and the high levels of noise in results obtained by high-throughput technologies (Tompa et al. 2005). Moreover, most motif discovery tools present only a small amount of information for the discovered motifs, usually in textual format, and cannot analyze large sets of genes due to running time or memory limitations. Perhaps most importantly, a user without advanced computer skills would find it quite difficult to execute some of these software tools and interpret their results.
We developed a new software suite for efficient genome-scale detection of known and novel motifs, called Amadeus (a motif algorithm for detecting enrichment in multiple species). Amadeus evaluates the discovered motifs using one or more of several built-in statistical scores, and is suitable to a broad range of motif finding tasks. It has an intuitive, user-friendly, and highly informative graphical interface. We ran Amadeus on the yeast ChIP-chip benchmark and on our metazoan compendium, and compared the results to those found by five popular motif finding tools. In addition, we used it to perform genome-wide discovery of motifs whose occurrences are localized within human and mouse promoters. This analysis uncovered two novel motifs, both of which are supported by multiple independent studies and are thus likely to represent real BSs of yet uncharacterized TFs. Another type of genome-wide search we performed found motifs whose chromosomal distribution is nonrandom. We believe Amadeus sets a new standard for motif discovery software in terms of accuracy, running time, range of application and ease of use.
Results
Overview of Amadeus
We developed a highly accurate, efficient, and user-friendly motif discovery software, called Amadeus, for finding short sequence patterns that are overrepresented in the promoters or 3′ UTRs of a given set of genes with respect to a large background (BG) set, typically the entire genome. The general architecture of Amadeus is a pipeline of filters, or refinement phases, where each phase receives as input a list of motif candidates and applies an algorithm for refining the list and producing a set of improved candidates, which serve as a starting point for the next phase (Fig. 1). The first phases typically work on a very large number of candidates, such as all possible k-mers, and execute simple procedures for choosing the most promising motifs. Successive phases run more complex (and computationally intensive) algorithms in order to converge to better motifs. The default score for evaluating each candidate motif is the HG enrichment score; other scores measure BS localization, strand-bias, and chromosomal preference. Amadeus also searches for pairs of enriched motifs that tend to co-occur in the same sequences and thus represent a putative cooperative cis-regulatory module. Finally, a built-in bootstrapping procedure may be applied to correct for multiple testing. See Methods and Supplemental Notes for a detailed description of the algorithm, scores, and additional features.
Figure 1.

The main components of the Amadeus computational pipeline. The input consists of one or more target gene sets and various parameters such as the score(s) for evaluating the motifs. Starting from all k-mers, the algorithm runs a series of refinement phases that eventually converge to a nonredundant list of high-scoring PWMs. These motifs, together with additional information and analyses, are displayed in the graphical output. For more details, see Methods.
The output of Amadeus is a nonredundant list of top-scoring motifs. For each motif, a wealth of information is displayed, including the motif’s logo, the scores it received, its occurrences localization graph, a list of similar known TF/miRNA motifs from TRANSFAC/miRBase, and the set of genes presumably regulated by the motif (Fig. 2; Supplemental Fig. 2). A graphical TF BS viewer displays the putative BSs of the motifs within the genomic sequences. All these data assist the user in dissecting the regulatory network underlying the studied gene set and in focusing on the most promising motifs for further research.
Figure 2.

Screenshot of Amadeus. The left panel controls the input parameters (organism, target set, promoter region, scores, etc.). Here, Amadeus was executed on the set of genes expressed in G2 and G2/M phases of the human cell cycle (Whitfield et al. 2002). The top-scoring motif shown in the output panel on the right is CHR (cell-cycle genes homology region), a cis-regulatory element that was experimentally found in promoters of several G2/M genes (Zhu et al. 2004), and is not represented in TRANSFAC; the second motif is CCAAT-box (NF-Y). For each motif discovered, the output also lists similar patterns from TRANSFAC, information on the localization of its occurrences, and additional statistics. In agreement with recent studies (Linhart et al. 2005; Tabach et al. 2005), the motif-pairs analysis in Amadeus reports the de novo found CHR and NF-Y motifs as a cis-regulatory module that is highly specific to the G2 and G2/M cell-cycle phases (Supplemental Fig. 1). A screenshot with additional graphical features is shown in Supplemental Figure 2.
Performance on the yeast transcriptional regulatory map
In their seminal paper, Harbison et al. (2004) constructed a nearly complete map of the transcriptional regulatory code of Saccharomyces cerevisiae using ChIP-chip assays. We assessed the performance of Amadeus using the ChIP-chip data of 83 factors that bound more than four promoters (58 on average), and whose binding motifs have been reported in the literature. We executed Amadeus twice on each data set with motif lengths 8 and 10, and compared the two top-scoring motifs obtained from each execution to the corresponding literature motif—as in Harbison et al. (2004), a match was defined if the average Euclidean distance between the columns of the two PWMs, referred henceforth as “PWM divergence,” was below 0.18. Amadeus was said to successfully recover a TF BS pattern if at least one of the four motifs it reported (two for each motif length) matched the literature PWM. Under these strict criteria, Amadeus discovered 54 of the known motifs (65% of 83).
We compared the performance of Amadeus to five popular motif finders that represent an assortment of algorithms and motif evaluation scores—MEME (EM) (Bailey and Elkan 1994), AlignACE (Gibbs sampling) (Hughes et al. 2000), YMF (Sinha and Tompa 2002), Weeder (Pavesi et al. 2001), and Trawler (Ettwiller et al. 2007) (exhaustive search). Of note, Weeder outperformed 13 motif discovery tools by most measures in Tompa’s assessment (Tompa et al. 2005), and Trawler was very recently reported to outperform four tools on mammalian data sets (Ettwiller et al. 2007). As in Tompa’s study, we did not include in our analysis programs that utilize auxiliary information, such as ChIP binding affinities, known TF BS models, or cross-species sequence conservation. Although Amadeus can incorporate some of this information, we wanted to focus on the core functionality of motif detection that is common to the widest possible range of setups. Each program was run with its default parameters using motif lengths 8 and 10, and the two top-scoring motifs were compared to the correct PWM as described earlier. As shown in Supplemental Figure 3, Amadeus recovered the largest number of motifs (65%); in agreement with Tompa et al. (2005), Weeder outperformed MEME, AlignACE, and YMF, successfully recovering 58% of the PWMs. Interestingly, the performance ranking among all five methods remained unchanged for other PWM divergence cutoffs.
Compendium of target sets of metazoan TFs and miRNAs
Ettwiller et al. (2007) tested the performance of their Trawler program using 10 mammalian ChIP-chip data sets. While this benchmark is larger than most data sets used in the literature, it is still relatively small and represents a single experimental technique. As explained earlier, Tompa’s data set (Tompa et al. 2005), the only large-scale metazoan benchmark constructed to date, does not reflect target sets obtained by high-throughput experiments. We therefore set out to construct a comprehensive motif discovery benchmark that is based on a large compendium of experimental studies. We collected diverse types of data sets from several metazoans, as published by independent groups in leading journals. Our compendium, listed in Figure 3, consists of 42 gene sets from human, mouse, fly (Drosophila melanogaster), and worm (Caenorhabditis elegans) and represents a total of 26 TFs and eight miRNAs. The sets were collected from 29 publications and were obtained by various types of technologies, primarily gene expression microarrays and ChIP-chip location analyses. The number of genes in each target set ranges from 25–2338 with mean 400—57-fold larger than Tompa’s sets. For each set, we used the corresponding PWM(s) from TRANSFAC, or the eight-long miRNA seed from miRBase, as the correct motif. A comparison of our compendium to several other motif discovery benchmarks is given in Table 1.
Figure 3.

The metazoan target-set compendium and benchmark results on it. (A) The compendium of metazoan TF/miRNA target sets collected from the literature. The “Source” column indicates the experimental procedure or database from which the target set was derived: gene expression microarrays (Ex), ChIP-chip (CC), ChIP-DSL (C-DSL), DamID (van Steensel et al. 2001), or Gene Ontology (GO) database (Ashburner et al. 2000). For additional information and references, see http://acgt.cs.tau.ac.il/amadeus. (B) Performance of motif finding tools on each target set—each successful motif recovery is marked by a gray-shaded box, according to the PWM divergence (darker shades of gray indicate higher similarity of the recovered motif to the one in the literature); the ∞ symbol marks long executions (>48 h) that were aborted. Here, Amadeus was run with the HG enrichment score. The success-rate patterns of the six motif finders are almost identical when comparing different target sets of the same TF. For example, in all three E2F data sets, Amadeus, Weeder, and Trawler are the only tools that recovered the correct motif; in the two Myod sets, Amadeus and Weeder succeeded with PWM divergence cutoff 0.18, AlignACE succeeded with cutoff 0.24, and MEME and YMF failed with all cutoffs. This consistency, observed for all six TFs that are represented by more than one set in our compendium, is not a result of large overlaps between the target sets, as such overlaps were avoided in the construction of the compendium. Instead, it is likely to stem from properties inherent to the TFs, such as the extent and type of their BSs degeneracy.
Table 1.
Comparison of several medium- and large-scale motif discovery benchmarks

The yeast ChIP-chip data sets (Harbison et al. 2004) are a popular benchmark, but they represent a single, relatively simple species and only one technology. Tompa’s benchmark (Tompa et al. 2005) is based on validated BSs from the TRANSFAC database—the BSs were chosen by the investigators according to various criteria and implanted inside real and synthetic promoters. Very recently, Ettwiller et al. (2007) developed Trawler, a new motif discovery tool for ChIP experiments, and reported its performance on 10 mammalian ChIP-chip data sets. Our compendium is the first large-scale collection of metazoan gene sets derived from high-throughput experiments; it represents diverse technologies and organisms and consists of both TF and miRNA target sets. Of note, the average set size in our compendium is substantially larger than in all other benchmarks.
Results on metazoan benchmark
We executed Amadeus and the five other motif finding tools on the metazoan target-set compendium. Here too, each tool was run with motif lengths 8 and 10, and the two top-scoring motifs were compared to the correct PWM(s). The results of each tool are shown in Figure 3; success rates and running times are summarized in Figure 4. Amadeus significantly outperformed all other programs in terms of motif recovery rate—62% success (with PWM divergence cutoff of 0.18); consistent with recent studies (Tompa et al. 2005; Ettwiller et al. 2007), Weeder and Trawler (43% success) performed better than the rest of the tools (10%–27%). We repeated the benchmark comparison using only the top-scoring motif from each execution and with two other PWM similarity measures and obtained very similar results (Supplemental Fig. 4).
Figure 4.

Performance of six motif finding tools on our compendium of metazoan target sets. (A) Success rates for three PWM divergence cutoffs, indicated by different shades of gray. The light-gray boxes on top of the Amadeus bars show the improved success rates when using the binned enrichment score (instead of the HG score; see Methods). Success rates for other PWM similarity measures and cutoffs are shown in Supplemental Figure 4. (B) Running times in logarithmic scale for the TF target-sets (AlignACE and MEME did not finish within 48 h on several sets). Trawler is a web-based tool so we could not measure its running time. For full results, see Supplemental Table 1 and http://acgt.cs.tau.ac.il/amadeus. A detailed comparison of all tested tools is given in Supplemental Table 2.
We observed a considerable degradation of up to 32% in the success rate of most motif finders on our benchmark relative to their performance on the yeast data sets. Remarkably, the success rate of Amadeus on the metazoan benchmark is comparable to that on the yeast data—62% vs. 65%, respectively. Amadeus is also the fastest tool (10 min per data set, on average); AlignACE and MEME are prohibitively slow on large target sets.
Handling length and GC-content biases
The HG enrichment score might fail to discover the correct motif, or alternatively detect many spurious motifs, when the distribution of the length and/or GC-content of the target set sequences significantly differ from their distribution in the BG set. Biologically meaningful groups of genes with such biases are not uncommon. For instance, genes with GC-rich promoters, such as housekeeping genes, tend to have higher expression rates (Kass et al. 1997; Aerts et al. 2004). Another example is the length bias of 3′ UTRs of tissue-specific genes. For example, genes that are expressed in neuronal tissues have relatively long 3′ UTRs (1300 nucleotides vs. 950 nucleotides in the entire genome) (Sood et al. 2006). To search for enriched motifs in such data sets, we developed a novel score, termed binned enrichment score, which partitions the genes into bins according to the length and GC-content of their cis-regulatory sequences and evaluates the overrepresentation of the motif based on its abundance in each bin (see Methods).
Running Amadeus on our metazoan target-sets compendium using the binned enrichment score further improves over the results of the HG score (Fig. 4). One example is the target set of Mef2 (Blais et al. 2005), for which none of the programs we tested recovered the correct motif. The promoters of these genes are longer than average (972 bp vs. 840 bp after masking out repetitive and coding sequences) and have a higher GC-content (53% vs. 49%). Using the binned enrichment score, Amadeus discovers the Mef2 binding pattern as the top-scoring motif. Additional examples that demonstrate the importance of accounting for length and GC biases are given in the Supplemental material. The improved sensitivity of the binned score remained consistent for other PWM similarity measures and cutoffs (5% improvement, on average) (data not shown).
Genome-wide analyses
Another useful application of motif finding is a genome-wide analysis, targeted to uncover regulatory motifs based on the genome alone, without having at hand a set of coregulated genes. We developed three scores for this type of analysis: localization, strand bias, and chromosomal preference (see Methods).
Localization
Many TFs are known to bind more frequently close to their target genes’ TSSs than in distant promoter regions (Tabach et al. 2007). Some elements that directly interact with or are part of the basal transcriptional machinery, such as TATA-box and Initiator, are found mainly in core promoters, spanning several dozens of bases around the TSS (Smale and Kadonaga 2003). We implemented a localization score that measures the tendency of a motif to occur at specific locations along the promoters. Applying this score on all human and mouse promoter sequences revealed binding patterns of many known TFs, including core promoter elements (e.g., SP1, NF-Y, TATA) and prominent TFs (e.g., MYC, ATF/CREB), as well as two novel motifs. Some of the discovered motifs exhibit a significant strand bias (i.e., they do not appear at similar rates on both strands) or chromosomal preference (i.e., a nonuniform distribution across chromosomes). The main results are listed in Table 2 (see also Discussion). Using a specially tailored method, FitzGerald et al. (2004) reported on nine motifs that localize in human promoters. Eight of these motifs were found by Amadeus. The ninth motif is the ATG start codon, which was not discovered by Amadeus, since we masked out coding sequences.
Table 2.
Main results of human and mouse genome-wide localization analysis

Amadeus was run on all human and mouse promoters and searched de novo for motifs that are significantly localized (i.e., overrepresented at a particular distance from the TSS, measured in bins of size 30) in both species. Approximately 23,000 and 24,000 human and mouse promoters, respectively, were analyzed. Promoters spanned from 500 bp upstream to 100 bp downstream of the TSS. Both known and novel motifs were found. All P-values listed in the table are for human. “Peak” refers to the center(s) of the location bin(s) with the largest motif occurrence rate. Amadeus also tests whether the motif occurrences are distributed nonuniformly between the strands (“Strand bias” column, showing the significance in human) or across the chromosomes (“Chrom pref.” column, showing also the overrepresented chromosome in human in parentheses).
A genome-wide analysis of fly promoters uncovered more than 30 motifs with significant localization (for full results, see the Amadeus website). Ohler et al. (2002) searched for motifs that are enriched in the core promoters of the fly genome. They reported on 10 motifs, all of which are among the top 21 motifs we discovered.
Chromosomal preference
Motivated by the observation that coregulated genes may colocalize (Cohen et al. 2000; Boutanaev et al. 2002), we developed a chromosomal-preference score to discover motifs whose occurrences are not distributed evenly across chromosomes (see Methods). Interestingly, when we applied this score to D. melanogaster promoters, Amadeus found that the Dref binding motif is overrepresented on the X chromosome (Supplemental Fig. 6). Indeed, Dref was recently associated with the dosage compensation complex (DCC) that equalizes the expression levels of X-linked genes in drosophila males and females.
Recently, Ruby et al. (2006) discovered a new class of small 21-nucleotide RNAs in worms, called 21U-RNAs, that reside mainly in introns and intergenic regions on chromosome IV. They also discovered a conserved motif located ∼40 bases upstream to these regions. Running the chromosomal-preference analysis on all C. elegans promoters, Amadeus reported this pattern as the top-ranking motif (Fig. 5). Thus, without any prior knowledge on 21U-RNAs in worm and DCC in fly, Amadeus found motifs known to be associated with them, demonstrating another type of biological signal it can uncover.
Figure 5.

Genome-wide chromosomal preference analysis of C. elegans promoters. (A) Screenshot of Amadeus output, showing the top-scoring motif found in the analysis. The motif is highly overrepresented on chromosome IV (P = 8 × 10−63). (B) The motif reported by Ruby et al. (2006), found upstream of many 21U-RNAs, is nearly identical to the one identified de novo by Amadeus.
Discussion
In this article, we present a compendium of target sets of metazoan TFs and miRNAs that we used as a benchmark for motif finding tools. To the best of our knowledge, our compendium is the first publicly-available large-scale collection of experimentally derived TF and miRNA target sets and thus constitutes a valuable database for studying gene regulation. We believe it is an improvement over previously published benchmarks (Harbison et al. 2004; Tompa et al. 2005), as it more accurately represents a broad range of gene-regulation motif discovery tasks. The yeast ChIP-chip data sets (Harbison et al. 2004) represent only one, relatively simple, species and only a single type of assay. As explained earlier, Tompa’s benchmark (Tompa et al. 2005) is to some extent artificial, or “unrealistically clean”—in the nonsynthetic data sets, each gene has a validated BS in its promoter. As expected in high-throughput techniques, our gene sets are much larger and contain a high rate of false positives, i.e., genes that are not targets of the corresponding TF or that contain a BS further upstream/downstream from the TSS. Moreover, our compendium contains sets of 3′ UTRs targeted by miRNAs; these sets have different statistical properties than promoters bound by TFs (e.g., GC-content and length variance), and thus pose additional computational challenges.
For simplicity of implementation and in order to allow a fair comparison between motif finders and among various types of data sets, our benchmark does not exploit all the available information generated by some of the experimental techniques, such as the binding peaks and affinities derived from ChIP assays. In addition, we did not exploit comparative sequence analysis, a potentially powerful tool that poses additional challenges, on top of the basic motif finding task studied here. For example, recent studies reported a limited cross-species conservation of functional BSs (Borneman et al. 2007; Lin et al. 2007; Odom et al. 2007); thus, in some situations, searching for motifs only within aligned sequences might be unfavorable (for further discussion, see Supplemental material).
We developed Amadeus, a new software platform for de novo motif discovery, and compared its performance to five popular motif finding tools. Amadeus, whose running time depends on the number and length of BG sequences, but not on the size of the target set, was significantly faster than the other programs on most data sets. Unlike the other tools, which performed rather poorly on the metazoan data, Amadeus achieved a high success rate on both the metazoan and yeast benchmarks. We believe this is largely due to the fact that most tools use BG models based on precomputed k-mer counts (k = 1, 4, 4, 8 in AlignACE, YMF, MEME, and Weeder, respectively), whereas Amadeus utilizes the entire set of promoters (or 3′ UTRs) in the genome as a reference set for testing over-representation. This is especially important in higher eukaryotes that have complex signals in their cis-regulatory sequences, which are not likely to be captured by simple BG models. Indeed, on our benchmark, the success rates of extant motif finders correlate with the complexity of their BG models. Trawler is the only extant tool we tested that utilizes a supplied set of BG sequences to assess motif enrichment. However, its BG set is relatively small (it failed to run with more than 2000 BG sequences), which in addition to the algorithm and statistical score it employs may explain its moderate performance on metazoan data sets (for more details, see Supplemental material). In conclusion, the success rate and running time of Amadeus scale up better than extant programs in terms of both the size of the data set and the species complexity. Supplemental Table 2 summarizes the main differences between the tools in terms of algorithms, scores, features, and performance.
The high accuracy of Amadeus remained consistent under various benchmark settings, e.g., evaluating the performance using other common PWM similarity measures or using the top-scoring motif only (Supplemental Fig. 4). Taken together with the fact that our benchmark contains a large number of diverse data sets, our results indicate that the improved performance of Amadeus is inherent, rather than a product of overfitting or biased choice of parameters.
We developed a novel statistical score for evaluating motif overrepresentation in target sets that are biased with respect to the rest of the genome in terms of sequence length or base composition. Although they are quite common, such biases are often ignored, which might lead to false results. This score improved the performance of Amadeus by 5%.
In order to gain insight into the practical limitations of Amadeus, we examined the target sets in which it failed to discover the correct motif. Evidently, in most cases a large fraction of the reported target genes does not contain a BS within the 1200-bp promoter region we analyzed. For example, Boyer et al. (2005) used promoter arrays against the −8-kb to +2-kb region relative to the TSS. Only 30% of the genes they reported as targets of NANOG contain a binding event within 1 kb upstream of the TSS. Another example is HSF (heat-shock factor), which is represented in our compendium by two target sets—human and fly. In both cases, it seems that the overrepresentation of the BS motif is borderline, which explains why none of the tools we tested accurately recovered the motif. Using a combined analysis of both sets together, a unique feature in Amadeus, we were able to successfully discover the correct binding pattern (Supplemental Fig. 7).
In this study, we also demonstrated application of Amadeus to genome-wide motif analysis, which can be applied to any genome with a sufficient number of cis-regulatory sequences without need for target sets from prior experiments. Using various statistical scores, Amadeus discovered an assortment of biological phenomena. Searching for motifs with nonrandom chromosomal distribution in fly and worm revealed the Dref and 21U-RNA–related patterns, respectively, which were found recently using a combination of experimental and computational techniques.
Localization analysis of human and mouse promoters recovered known mammalian TF motifs, the splice donor site, and two novel motifs. The first novel motif (ACTACAWYTC) was also discovered independently by high-throughput location analyses for ESR1 (ER-α) (Kwon et al. 2007), RUNX1, and ETS1 (Hollenhorst et al. 2007). Running Amadeus on these sets reproduced the motif, which apparently has diverse biological functions. Interestingly, the motif has a significant strand bias (the only other localized human TF we found with a strand bias was TATA-box), and like NF-Y, it is over-represented on chromosome 19. Very recently, Sinha et al. (2008) used a decoy corresponding to a variant of this motif, reported in Xie et al. (2005), in order to experimentally validate that it has a regulatory role in cell-cycle progression. The second motif (CTCGCGAGAT), reported also by FitzGerald et al. (2004), was shown to regulate ARF3 in vivo (Haun et al. 1993).
The localization analysis results obtained by Amadeus on both human/mouse and fly promoters compare favorably with other, specially tailored methods (Ohler et al. 2002; FitzGerald et al. 2004): In a single run, Amadeus discovered all the motifs reported by those methods and supplied additional information on their strand and chromosomal distributions. We therefore believe that Amadeus may be used as a general tool for genome-wide motif discovery tasks, aimed at uncovering sequence patterns with various global features.
In addition to sensitivity, efficiency, and supporting multiple target sets and scores, we focused on developing a friendly and informative graphical user interface in order to make Amadeus easily accessible and beneficial to a wide range of users. Built-in algorithmic features, such as pairs analysis and bootstrapping, as well as various graphical and textual displays of the motifs, the scores they attained, and their putative BSs, make it easier for the user to understand the nature of the discovered motifs and highlight the most biologically interesting ones. The Amadeus software (standalone Java application) and our compendium of TF and miRNA target sets are accessible at http://acgt.cs.tau.ac.il/amadeus. We are continuing to implement novel features in Amadeus and to add newly published data sets to the compendium.
Methods
Genomic sequences and binding patterns
Promoter and 3′ UTR sequences (repeat- and coding-sequence-masked) of human, mouse, fly, and worm were extracted from Ensembl (Birney et al. 2004). Yeast promoters were downloaded from SGD (http://www.yeastgenome.org). Binding patterns of TFs and miRNAs were taken from TRANSFAC (Wingender et al. 1996) and miRBase (http://microrna.sanger.ac.uk/sequences), respectively. For more details, see Supplemental material.
Target sets of metazoan TFs and miRNAs
We collected 42 TF/miRNA target sets from the literature, focusing on sets obtained using high-throughput techniques, such as gene expression microarrays and ChIP-chip assays. We included only TFs and miRNAs whose binding patterns are described in TRANSFAC and miRBase, respectively. Genes were mapped to Ensembl gene IDs using Biomart (http://www.biomart.org). In order to avoid strong dependencies between the target sets, we verified that no two sets of the same TF/miRNA have an overlap greater than 30%.
For the TF data sets, we used promoter sequences spanning from 1000 bp upstream to 200 bp downstream of the TSS, a range that covers most of the promoter array sequences and is often used in computational promoter analysis; for the miRNA data sets, we used full-length 3′ UTRs (coding strand only); repetitive and coding sequences were masked out. The total sequence length of the target sets is 383 kbp on average, much larger than the yeast ChIP-chip data sets (35 kbp) and Tompa’s benchmark (8 kbp).
Amadeus software and algorithms
Amadeus executes a series of refinement phases where each phase gets as input a list of motif candidates, applies an algorithm for refining the list, and produces a set of improved candidates, which serves as a starting point for the next phase. Each phase uses a different motif model, which best suits its algorithm and performance requirements. Generally, the first phases use simple motif models and enumerate a very large number of candidates, whereas the final phases evaluate a smaller number of more complex motifs, namely PWMs. Motifs in each phase are evaluated using one or more score functions: enrichment, localization, strand bias, and chromosomal preference, which are combined into a single P-value (see below). The phases of Amadeus, in their running order, are preprocess, mismatch, merge, greedy, postprocess, and pairs analysis (see Fig. 1).
In the preprocess phase, all k-mers are evaluated, where k, the motif length, is a user-defined parameter. In the mismatch phase, the motif model is changed from a k-mer to a list of k-mers by introducing degenerate positions into the k-mers. Afterward, the merge phase combines pairs of similar motifs. This is done recursively until no new high-scoring similar pairs are encountered. The greedy phase constructs a PWM from each motif and optimizes it using a greedy EM-like iterative process: In each iteration, it searches for the PWM cutoff that yields the best score, and then the occurrences in the target set that pass this cutoff are used to build a new, refined PWM; this process is repeated as long as the score improves. Finally, in the postprocess phase, redundancy is eliminated by removing every motif, for which there exists a higher scoring motif, such that more than 5% of their occurrences overlap. The final list of discovered motifs is then compared with a database of known PWMs (TRANSFAC for TFs, miRBase for miRNAs), and all similarities with PWM divergence below 0.24 are reported. Additional statistics and information are provided for each motif to assist the user in evaluating the results (Supplemental Fig. 2). In the optional pairs analysis phase, Amadeus reports pairs of motifs that tend to co-occur within the same cis-regulatory sequences.
Other important features we implemented in Amadeus include automatic removal of redundant sequences (to avoid biases in the analysis due to families of paralogous genes with nearly-identical cis-regulatory sequences) and bootstrapping (to correct the reported P-values for multiple testing, by repeating the entire analysis on randomly selected gene sets). By utilizing highly efficient data structures, designed to minimize running time and memory consumption, Amadeus is able to check a huge number of candidate motifs and analyze quickly whole-genome cis-regulatory sequences. For more details, see Supplemental material.
Scores for evaluating motifs
Amadeus evaluates each candidate motif using one or several model-independent score functions, chosen by the user. The P-values computed for multiple score functions and/or target sets are combined into a single P-value using the Z-transform (Whitlock 2005).
HG enrichment score
Let B and T (T⊆B) denote the BG and target sets, respectively, and let b and t denote the subset of genes from the BG and target set, respectively, that contain at least one occurrence of the motif (hit, in short) in their cis-regulatory sequence. The HG enrichment score computes the probability of observing at least |t| target sequences with a motif occurrence, under the null hypothesis that the genes in the target set were drawn randomly, independently, and without replacement from the BG set (Elkon et al. 2003):
 |
Binned enrichment score
The genes are divided into n bins according to the GC-content and length of their cis-regulatory sequences. Let Bi and Ti be the BG and target set genes, respectively, in the ith bin, and denote by bi the subset of genes from Bi whose sequence contains a hit. The goal of this score is to account for cases where the fraction of targets is uneven across bins. Suppose that targets within each bin are selected uniformly. Then, in bin i the probability that a selected gene will contain a hit (i.e., belong to bi) is |bi|/|Bi|. Since the fraction of targets in bin i is |Ti|/|T|, it follows that the probability that a selected gene will contain a hit is
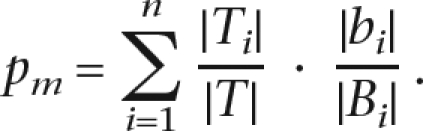 |
Assume now that |T| target genes are sampled with replacement from B. Then, the probability for having at least |t| target genes with a motif occurrence is given by the tail of the following binomial distribution:
Note that this score does not use the number of targets that contain a hit in each bin separately, but rather the total t.
Strand-bias score
The score uses a binomial test to measure the tendency of the motif to occur in one of the strands more often than in the other. A strong strand bias could, for example, indicate that the motif has a post-transcriptional role, as it may be related to the gene’s RNA.
Localization score
The score estimates whether the occurrences of the motif tend to cluster at specific distances from the TSS. The hits are partitioned into bins according to their location; for each bin, a binomial test computes the overrepresentation of hits in that bin under the null hypothesis that the hits are distributed randomly among the bins (i.e., according to the total number of k-mers in each bin); finally, the bin with the lowest P-value is chosen and its score is Bonferroni corrected for multiple testing.
We implemented three variants of the localization score: The “BG” and “Target” variants compute the localization of the hits in the BG and target set, respectively (a motif may exhibit localization across the entire genome, or only for target-set genes); in order to account for global location-dependent biases in the nucleotide composition, the “Target vs. BG” variant checks whether the occurrences of the motif in the target-set tend to localize given the distribution of their locations in the rest of the genome. For a detailed explanation, see Supplemental material.
Chromosomal-preference score
In order to test whether the motif is not distributed evenly among the chromosomes, the enrichment of the motif in each chromosome is evaluated using the HG distribution; the smallest P-value is chosen and Bonferroni corrected for multiple testing.
Pairs of co-occurring motifs
In order to find pairs of cooperative TFs (or miRNAs), Amadeus checks the co-occurrence rate of each pair of motifs by computing the following HG tail probability:
where T is the target set; t1 and t2 are the subsets of target genes that contain at least one occurrence of the first and second motif, respectively; and t12 is the subset of target genes that contain hits for both motifs. Applying an EM-like procedure similar to the one used for single motifs, the PWMs comprising the pair of motifs are tuned in order to optimize the co-occurrence score.
Acknowledgments
We thank Gidi Weber and Adi Maron-Katz for their contributions to the source code. Promoter sequences were downloaded by Ran Elkon. We also thank the anonymous referees for their constructive comments. R.S. was supported in part by the Raymond and Beverly Sackler Chair in Bioinformatics and by a grant from the Wolfson Foundation.
Footnotes
[Supplemental material is available online at www.genome.org. The Amadeus software is available at http://acgt.cs.tau.ac.il/amadeus.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.076117.108.
References
- Aerts S., Thijs G., Dabrowski M., Moreau Y., De Moor B. Comprehensive analysis of the base composition around the transcription start site in Metazoa. BMC Genomics. 2004;5:34. doi: 10.1186/1471-2164-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene Ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L., Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Birney E., Andrews T.D., Bevan P., Caccamo M., Chen Y., Clarke L., Coates G., Cuff J., Curwen V., Cutts T., et al. An overview of Ensembl. Genome Res. 2004;14:925–928. doi: 10.1101/gr.1860604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais A., Tsikitis M., Acosta-Alvear D., Sharan R., Kluger Y., Dynlacht B.D. An initial blueprint for myogenic differentiation. Genes & Dev. 2005;19:553–569. doi: 10.1101/gad.1281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman A.R., Gianoulis T.A., Zhang Z.D., Yu H., Rozowsky J., Seringhaus M.R., Wang L.Y., Gerstein M., Snyder M. Divergence of transcription factor binding sites across related yeast species. Science. 2007;317:815–819. doi: 10.1126/science.1140748. [DOI] [PubMed] [Google Scholar]
- Boutanaev A.M., Kalmykova A.I., Shevelyov Y.Y., Nurminsky D.I. Large clusters of co-expressed genes in the Drosophila genome. Nature. 2002;420:666–669. doi: 10.1038/nature01216. [DOI] [PubMed] [Google Scholar]
- Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G., et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen B.A., Mitra R.D., Hughes J.D., Church G.M. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat. Genet. 2000;26:183–186. doi: 10.1038/79896. [DOI] [PubMed] [Google Scholar]
- Elkon R., Linhart C., Sharan R., Shamir R., Shiloh Y. Genome-wide in silico identification of transcriptional regulators controlling the cell cycle in human cells. Genome Res. 2003;13:773–780. doi: 10.1101/gr.947203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskin E., Pevzner P.A. Finding composite regulatory patterns in DNA sequences. Bioinformatics. 2002;18:S354–S363. doi: 10.1093/bioinformatics/18.suppl_1.s354. [DOI] [PubMed] [Google Scholar]
- Ettwiller L., Paten B., Ramialison M., Birney E., Wittbrodt J. Trawler: De novo regulatory motif discovery pipeline for chromatin immunoprecipitation. Nat. Methods. 2007;4:563–565. doi: 10.1038/nmeth1061. [DOI] [PubMed] [Google Scholar]
- FitzGerald P.C., Shlyakhtenko A., Mir A.A., Vinson C. Clustering of DNA sequences in human promoters. Genome Res. 2004;14:1562–1574. doi: 10.1101/gr.1953904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison C.T., Gordon D.B., Lee T.I., Rinaldi N.J., Macisaac K.D., Danford T.W., Hannett N.M., Tagne J.B., Reynolds D.B., Yoo J., et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haun R.S., Moss J., Vaughan M. Characterization of the human ADP-ribosylation factor 3 promoter. Transcriptional regulation of a TATA-less promoter. J. Biol. Chem. 1993;268:8793–8800. [PubMed] [Google Scholar]
- Hollenhorst P.C., Shah A.A., Hopkins C., Graves B.J. Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes & Dev. 2007;21:1882–1894. doi: 10.1101/gad.1561707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J.D., Estep P.W., Tavazoie S., Church G.M. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- Kass S.U., Pruss D., Wolffe A.P. How does DNA methylation repress transcription? Trends Genet. 1997;13:444–449. doi: 10.1016/s0168-9525(97)01268-7. [DOI] [PubMed] [Google Scholar]
- Kwon Y.S., Garcia-Bassets I., Hutt K.R., Cheng C.S., Jin M., Liu D., Benner C., Wang D., Ye Z., Bibikova M., et al. Sensitive ChIP-DSL technology reveals an extensive estrogen receptor alpha-binding program on human gene promoters. Proc. Natl. Acad. Sci. 2007;104:4852–4857. doi: 10.1073/pnas.0700715104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.Y., Vega V.B., Thomsen J.S., Zhang T., Kong S.L., Xie M., Chiu K.P., Lipovich L., Barnett D.H., Stossi F., et al. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007;3:e87. doi: 10.1371/journal.pgen.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhart C., Elkon R., Shiloh Y., Shamir R. Deciphering transcriptional regulatory elements that encode specific cell cycle phasing by comparative genomics analysis. Cell Cycle. 2005;4:1788–1797. doi: 10.4161/cc.4.12.2173. [DOI] [PubMed] [Google Scholar]
- Lockhart D.J., Winzeler E.A. Genomics, gene expression and DNA arrays. Nature. 2000;405:827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- Odom D.T., Dowell R.D., Jacobsen E.S., Gordon W., Danford T.W., MacIsaac K.D., Rolfe P.A., Conboy C.M., Gifford D.K., Fraenkel E. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat. Genet. 2007;39:730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohler U., Liao G.C., Niemann H., Rubin G.M. Computational analysis of core promoters in the Drosophila genome. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-12-research0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavesi G., Mauri G., Pesole G. An algorithm for finding signals of unknown length in DNA sequences. Bioinformatics. 2001;17:S207–S214. doi: 10.1093/bioinformatics/17.suppl_1.s207. [DOI] [PubMed] [Google Scholar]
- Ruby J.G., Jan C., Player C., Axtell M.J., Lee W., Nusbaum C., Ge H., Bartel D.P. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Sharan R., Ben-Hur A., Loots G.G., Ovcharenko I. CREME: Cis-Regulatory Module Explorer for the human genome. Nucleic Acids Res. 2004;32:W253–W256. doi: 10.1093/nar/gkh385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S., Tompa M. Discovery of novel transcription factor binding sites by statistical overrepresentation. Nucleic Acids Res. 2002;30:5549–5560. doi: 10.1093/nar/gkf669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S., Adler A.S., Field Y., Chang H.Y., Segal E. Systematic functional characterization of cis-regulatory motifs in human core promoters. Genome Res. 2008;18:477–488. doi: 10.1101/gr.6828808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale S.T., Kadonaga J.T. The RNA polymerase II core promoter. Annu. Rev. Biochem. 2003;72:449–479. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- Sood P., Krek A., Zavolan M., Macino G., Rajewsky N. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc. Natl. Acad. Sci. 2006;103:2746–2751. doi: 10.1073/pnas.0511045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabach Y., Milyavsky M., Shats I., Brosh R., Zuk O., Yitzhaky A., Mantovani R., Domany E., Rotter V., Pilpel Y. The promoters of human cell cycle genes integrate signals from two tumor suppressive pathways during cellular transformation. Mol. Syst. Biol. 2005;1:2005.0022. doi: 10.1038/msb4100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabach Y., Brosh R., Buganim Y., Reiner A., Zuk O., Yitzhaky A., Koudritsky M., Rotter V., Domany E. Wide-scale analysis of human functional transcription factor binding reveals a strong bias towards the transcription start site. PLoS One. 2007;2:e807. doi: 10.1371/journal.pone.0000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa M., Li N., Bailey T.L., Church G.M., De Moor B., Eskin E., Favorov A.V., Frith M.C., Fu Y., Kent W.J., et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- van Steensel B., Delrow J., Henikoff S. Chromatin profiling using targeted DNA adenine methyltransferase. Nat. Genet. 2001;27:304–308. doi: 10.1038/85871. [DOI] [PubMed] [Google Scholar]
- Whitfield M.L., Sherlock G., Saldanha A.J., Murray J.I., Ball C.A., Alexander K.E., Matese J.C., Perou C.M., Hurt M.M., Brown P.O., et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock M.C. Combining probability from independent tests: The weighted Z-method is superior to Fisher’s approach. J. Evol. Biol. 2005;18:1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- Wingender E., Dietze P., Karas H., Knuppel R. TRANSFAC: A database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996;24:238–241. doi: 10.1093/nar/24.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Smith L.T., Plass C., Huang T.H. ChIP-chip comes of age for genome-wide functional analysis. Cancer Res. 2006;66:6899–6902. doi: 10.1158/0008-5472.CAN-06-0276. [DOI] [PubMed] [Google Scholar]
- Xie X., Lu J., Kulbokas E.J., Golub T.R., Mootha V., Lindblad-Toh K., Lander E.S., Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W., Giangrande P.H., Nevins J.R. E2Fs link the control of G1/S and G2/M transcription. EMBO J. 2004;23:4615–4626. doi: 10.1038/sj.emboj.7600459. [DOI] [PMC free article] [PubMed] [Google Scholar]