

Abstract
Background
The purpose of this study was to determine whether pairs of compounds, including general anesthetics, could simultaneously modulate receptor function in a synergistic manner, thus demonstrating the existence of multiple intra-protein anesthetic binding sites.
Methods
Using standard electrophysiologic methods, we measured the effects of at least one combination of benzene, isoflurane, halothane, chloroform, flunitrazepam, zinc and pentobarbital on at least one of the following ligand gated ion channels: N-methyl-D-aspartate receptors (NMDARs), glycine receptors (GlyRs) and γ-aminobutyric acid type A receptors (GABAARs).
Results
All drug-drug-receptor combinations were found to exhibit additive, not synergistic modulation. Isoflurane with benzene additively depressed NMDAR function. Isoflurane with halothane additively enhanced GlyR function, as did isoflurane with zinc. Isoflurane with halothane additively enhanced GABAAR function as did all of the following: halothane with chloroform, pentobarbital with isoflurane, and flunitrazepam with isoflurane.
Conclusions
The simultaneous allosteric modulation of ligand gated ion channels by general anesthetics is entirely additive. Where pairs of general anesthetic drugs interact synergistically to produce general anesthesia, they must do so on systems more complex than a single receptor.
Keywords: GABA, glycine, NMDA, receptor, isoflurane, halothane, chloroform, pentobarbital, zinc, additivity, anesthetic mechanism
Introduction
The precise molecular mechanisms of general anesthetic action are not yet fully understood. Many neuronal ion channels have been identified that are sensitive to several general anesthetic drugs (1) but the locations of drug binding sites on these receptors and the molecular events that follow anesthetic binding are still under investigation. An important question central to this endeavor is: Do receptors contain multiple anesthetic binding sites or do all anesthetics modulate an individual receptor via action at a single site?
We have chosen to address this question in the present study by investigating the phenomenon known as “synergy”. If two drugs, when applied at equi-effective concentrations have significantly smaller effects on a target than a combination of the two drugs, applied at one half of an equi-effective concentration, then the two drugs are said to be synergistic. This effect cannot occur if the two drugs both act at the same site. Synergism requires that the two drugs act at different sites. Therefore, the detection of a synergistic effect between two drugs on a given target would reveal the existence of multiple drugs binding sites on a single receptor. However, when two drugs combine to give non-synergistic effects, less can be said about the number of drug binding sites on the receptor. Additive and antagonistic (sub-additive or infra-additive) effects can both occur when drugs compete for the same site, or when they modulate a receptor via separate sites.
In this study we sought to determine whether we could detect synergism between drugs with known or suspected separate binding sites. We also sought to determine if we could detect synergism between drugs with unknown binding sites, thereby defining multiple drug binding sites on the N-methyl-D-aspartate receptor (NMDAR), the glycine receptor (GlyR) and the γ-aminobutyric acid type A receptor (GABAAR)
Methods
In this collaborative study, we carried out 4 groups of electrophysiologic experiments in 3 different laboratories in order to detect synergistic modulation of ligand gated ion channels. All experiments were performed at 22–24°C.
Receptor and Drug selection
NMDARs are an important class of fast excitatory ligand gated channels found throughout the central nervous system. They have a complex pharmacology and are known to harbor multiple binding sites for agonists, co-agonists, metal ions, drugs of abuse and general anesthetics (2). Two NMDAR modulators, benzene and isoflurane, are thought to act via different binding sites (3) and were therefore selected in this study since simultaneous modulation of the receptor at both sites could potentially produce a synergistic inhibition of NMDAR function.
GlyRs are fast inhibitory ligand gated channels found throughout the central nervous system that are positively modulated (potentiated) by zinc, many anesthetics and alcohols (4). Zinc is thought to allosterically enhance receptor function via an N-terminal binding site (5) and isoflurane is thought to stabilize the open state of the receptor by acting at an intrasubunit pocket defined by the 4 transmembrane segments of each subunit (6). The precise site of action of halothane is unknown. Therefore, two pairs of compounds were selected for study: i. isoflurane and zinc could potentially produce a synergistic potentiation of GlyR function via their different sites and ii. isoflurane and halothane would interact synergistically if halothane has a separate and novel binding site from that of isoflurane.
GABAARs are the most common fast inhibitory ligand gated ion channels found in the central nervous system and are potentiated by a diverse group of sedative and hypnotic compounds including benzodiazepines and most general anesthetics. Benzodiazepines enhance receptor function via a well characterized N-terminal binding site (7) while intravenous anesthetics, including pentobarbital, are thought to interact with the transmembrane domain of the β subunit (8). Halogenated inhaled anesthetics are thought to act and perhaps compete with one another within a cavity defined by the transmembrane segments of the α subunit (6,9,10). The cavity has been hypothesized to be ~210 Å3 in size, thus accommodating either one isoflurane molecule, one halothane molecule or two chloroform molecules. A cavity of this size would also be able to simultaneously accommodate a molecule of halothane and chloroform at the same time which might bind more tightly than one halothane or two chloroform molecules. Therefore, the following 4 pairs of compounds were selected for study: isoflurane with flunitrazepam, isoflurane with pentobarbital, isoflurane with halothane and halothane with chloroform. Due to their proposed different binding sites, isoflurane plus flunitrazepam could potentially combine to synergistically potentiate GABAAR function, as could isoflurane with pentobarbital. Furthermore, isoflurane and halothane are thought to compete for the same site and could produce an additive or infra-additive effect. Finally, chloroform and halothane, which are also thought to bind to the same site (9), may combine to interact with different components of the binding cavity and produce a “superligand” that would produce greater potentiation and, therefore, synergy.
Oocytes - NMDAR experiments
We have previously described the molecular biologic and electrophysiologic methods used to characterize the molecular pharmacology of NMDARs in great detail (11). Briefly, NR1 and NR2B subunit mRNAs were injected into Xenopus laevis oocytes at least 72 hours before conducting 2-electrode voltage clamp experiments. Electrophysiologic solutions, electrodes, hardware and software were the same as previously published (12).
Isoflurane solutions were prepared by diluting a saturated solution using gas-tight syringes. Benzene solutions were prepared from a concentrated stock solution using gas-tight syringes (12,13).
For each experiment, the oocyte was perfused for 30 seconds with buffer solution containing the agonist mixture (100 μM NMDA and 10 μM glycine) to generate a control current. After at least 5 minutes of recovery, the oocyte was first perfused with buffer solution containing the test anesthetic for 30 seconds, and then perfused with buffer solution containing both the agonist mixture and the test anesthetic for 30 seconds. After another 5-minute recovery period, the agonist mixture was again applied to the cell for 30 seconds to ensure reversibility of any anesthetic-induced change in current response. Peak current responses were recorded, and the magnitude of anesthetic-induced current inhibition was determined using the average of the two control experiments (before and after application of anesthetic).
Oocytes – Glycine receptor and GABAA receptor experiments
We have previously described the methods used to characterize the function of glycine and GABAA receptors in great detail (14). Briefly, GlyR α1 or GABAAR α1:β2:γ2s subunit cDNAs were injected into Xenopus laevis oocyte nuclei at least 72 hours before conducting 2-electrode voltage clamp experiments. Electrophysiologic solutions, electrodes, hardware, software and sources of chemicals were identical to our previously published work (15).
1 mM GABA or glycine was applied for 20 s to test the maximal response, and lower concentrations were applied for 30 s to reach a peak response for that concentration. After determining the maximal current, the EC5–10 of GABA or glycine was determined for each expressing oocyte, where EC5 is the effective concentration that elicits 5% of the maximal response and EC10 is the effective concentration that elicits 10% of the maximal response. After ten minutes, the anesthetic solutions were applied as a one-minute preincubation in extracellular saline alone followed by a 30-second co-application of the anesthetic in an EC5–10 solution of GABA or glycine. After a ten-minute washout, a second GABA or glycine EC5–10 test pulse was applied. Potentiation by drugs was calculated by dividing the drug-induced current by the average EC5–10 GABA- or glycine-induced currents applied 10 minutes before and after each drug application.
Concentrations of anesthetics were chosen that elicited approximately 100% potentiation (i.e., a doubling) of the EC5–10 GABA- and glycine-induced response. For GABA experiments these concentrations were 120 μM isoflurane, 114 μM halothane, 15 μM sodium pentobarbital, and 0.5 μM flunitrazepam. For glycine experiments the concentrations were 75 μM isoflurane, 62.5 μM halothane, 0.1 μM zinc. Drugs were dissolved in solution immediately before application to the oocytes. For each experiment, isoflurane was paired with one of the other three anesthetics, and recordings were made in a single cell as follows: 1) Potentiation by isoflurane [ISO] was determined at the full concentration (for 100% potentiation), 2) Potentiation by the full concentration of the paired anesthetic e.g.: [HAL] was determined 3) The potentiation by half the concentration of isoflurane [ISO]/2 was determined. 4) The potentiation by half the concentration of the paired anesthetic [HAL]/2 was determined). 5) Finally, the potentiation by mixed solutions of concentrations of the drugs in steps 3 and 4 were tested ([ISO]/2+[HAL]/2). The Student’s paired t-test was used to compare the potentiation measured in step 5 with step 1 and step 2. The same procedure was used to examine additivity between isoflurane and sodium pentobarbital on GABAARs, between isoflurane and flunitrazepam on GABAARs, between isoflurane and halothane on GlyRs and between isoflurane and zinc on GlyRs.
GABAAR patch clamp experiments
We have previously published the detailed methods used to measure the effects of general anesthetic drugs on the concentration-effect relationship of GABAARs (16). Briefly, HEK293 cells transiently expressing human α1, β2 and γ2s subunits were whole cell patch clamped at −60mV and superfused with 18 different solutions containing 0.3 – 1,000 μM GABA and a combination of 0 – 10 MAC halothane and 0 – 10 MAC chloroform. For each GABA exposure, the peak current amplitudes were measured. Stock solutions of GABA were diluted in extracellular solutions shortly before use. Halothane and chloroform solutions were prepared by injection of liquid anesthetic with a gas-tight syringe as previously described (16).
In all experiments, the concentrations of the selected drugs were chosen to give the desired modulations as described, but were also chosen to be in the clinically relevant range. Aqueous human MACs for isoflurane, halothane and chloroform were taken to be 310 μM, 220 μM and 900 μM respectively and the human EC50 for pentobarbital anesthesia was taken to be 50 μM (17,18). All concentrations less than four times these values can be considered to be clinically relevant and all concentrations above this can be assumed to be toxic and are used here simply to complete concentration response relationships.
Analysis
Concentration – response relationships were fit to the Hill equation:
where I is the peak current of each response, Imax is the maximum response elicited, C50 is the concentration eliciting half maximal effect (IC50 for the blockade of NMDARs – the concentration that elicits 50% inhibition, and EC50 for the activation of GABAARs – the effective concentration of GABA that elicits 50% of maximal activation), [D] is the concentration of the ligand and n is the Hill coefficient.
For GlyR and GABAAR potentiation experiments, if the effect of each drug when applied alone (the first two modulations) were both significantly less than the combination (the final modulation), then the two drugs were said to be synergistic. If the effect of the drug combination was less than that of each drug when applied alone, then the two drugs were said to be infra-additive. Drug pairs that fell into neither of these categories were said to be additive. Significance between different experimental conditions was assessed using a Students t-test.
By adapting the method of Minto & Shafer (19), we have previously used a response surface method to interpret the modulation of receptor function by a pair of general anesthetic drugs. Briefly, the reduction of GABA EC50 that underpins the potentiation by a pair of general anesthetics can be fitted to a response surface. The midpoint of this surface [described here as C50, previously described as U50(θ)] and slope [described here as n and previously described as γ(θ)] can be approximated by a pair of parabolas, the curvatures of which are an indication of synergy or infra-additivity (16,19).
Results
NMDA receptors
Isoflurane and benzene both inhibited NMDA receptor-mediated currents. Figure 1a shows a typical set of current traces demonstrating the inhibition of NMDA receptor-mediated current by a mixture of 1,200 μM isoflurane and 170 μM benzene. In this oocyte, the anesthetic mixture inhibited NMDA receptor-mediated current by 51.3%. When applied individually, both isoflurane and benzene inhibited NMDA receptor function in a concentration-dependent fashion, as shown in Figure 1b. For isoflurane the IC50 was 2,400 ± 300 μM and the Hill coefficient −1.0 ± 0.1. For benzene the IC50 was 340 ± 40 μM and the Hill coefficient was −0.96 ± 0.09.
Figure 1.
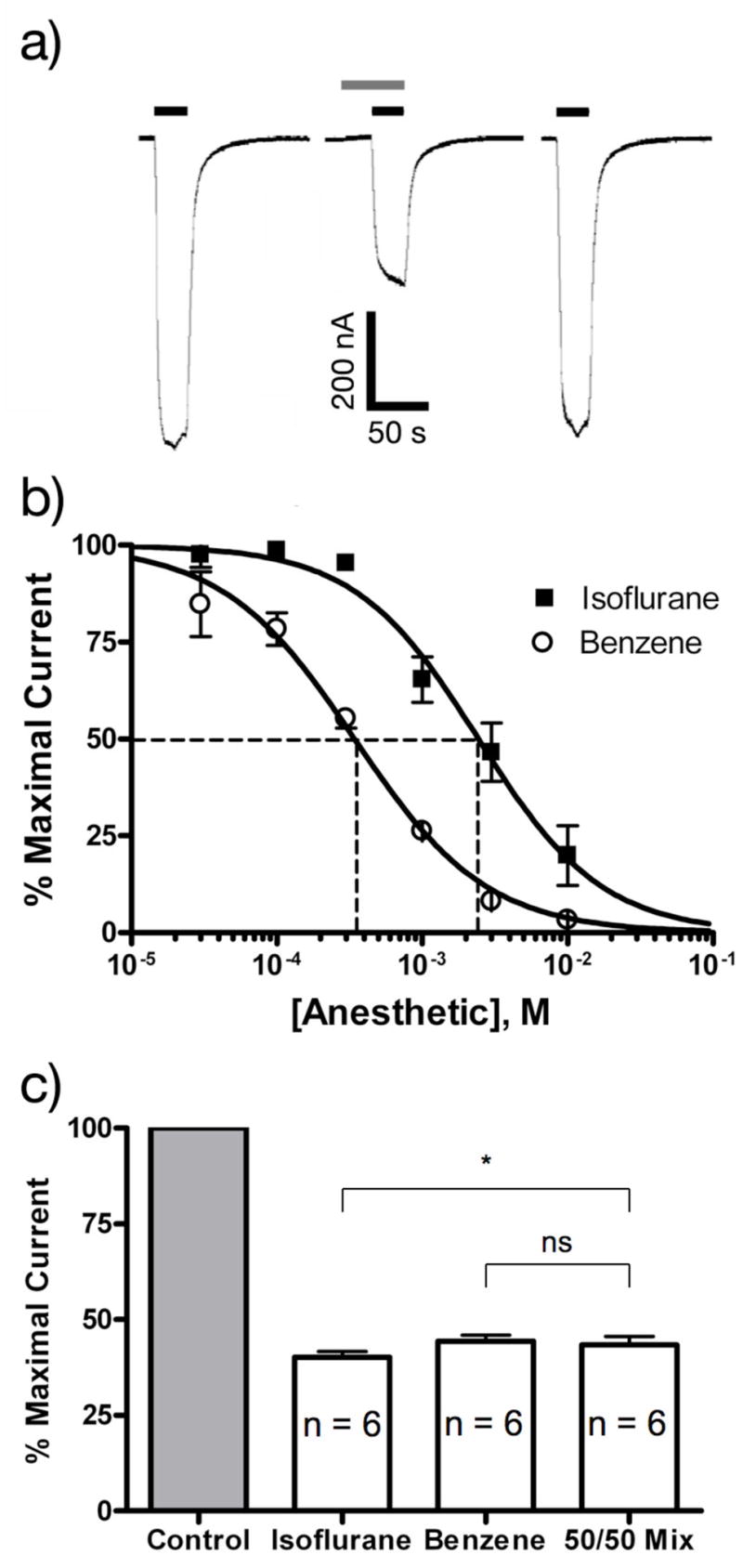
Inhibition of NMDA receptor function by isoflurane and benzene. a) 2-electrode voltage clamp recordings of NMDA/glycine responses (100 μM NMDA and 10 μM glycine) in Xenopus laevis oocytes before during and after the application of 1 mM isoflurane + 150 μM benzene. Bars above the current traces indicate the duration of agonists application (black) and the duration of the modulator application (gray). Calibration bars indicate the amplitude and duration of the responses. b) Normalized concentration response relationship for the inhibition of NMDA receptor function by isoflurane and benzene. Symbols represent the mean±SEM inhibition of responses to 100 μM NMDA and 10 μM glycine determined from 6 cells. The IC50s and Hill coefficients for isoflurane and benzene were 2.4 ± 0.3 mM; −1.0 ± 0.1 and 0.34 ± 0.04; −0.96 ± 0.09. respectively. c) Inhibition of NMDA receptor function by a combination of isoflurane and benzene did not differ for 300 μM benzene and a 50/50 mixture (1 mM isoflurane + 150 μM benzene). Isoflurane (2mM) alone was slightly more effective as an inhibitor than the 50/50 mixture.
* P < 0.05
We then tested whether isoflurane and benzene have additive inhibitory effects on NMDA receptor-mediated currents by determining NMDA receptor inhibition by an anesthetic mixture containing 1,200 μM isoflurane and 170 μM benzene (i.e. a mixture containing one-half the IC50 concentration of each drug). The experiment shown in Figure 1a was repeated in 6 different oocytes yielding an average inhibition of 56 ± 5% (see Figure 1c), suggesting that isoflurane and benzene have additive inhibitory effects on human NR1/NR2B NMDA receptors.
Glycine receptors
Isoflurane and halothane both potentiated glycine receptor function. This enhancement is shown in Figure 2a where applications of 37.5 μM, 75 μM isoflurane ([ISO]/2, [ISO]), 31.2 μM, 62.5 μM halothane ([HAL]/2, [HAL]) and a combination of 37.5 μM isoflurane and 31.2 μM halothane ([ISO]/2+[HAL]/2) all enhanced the amplitude of EC5–10 responses to glycine. Figure 2b shows that when the two drugs were applied at half this concentration (37.5 μM isoflurane and 31.2 μM halothane) the amplitude of the modulation was indistinguishable from the modulation produced by 75 μM isoflurane or 62.5 μM halothane suggesting purely additive actions of isoflurane and halothane.
Figure 2.
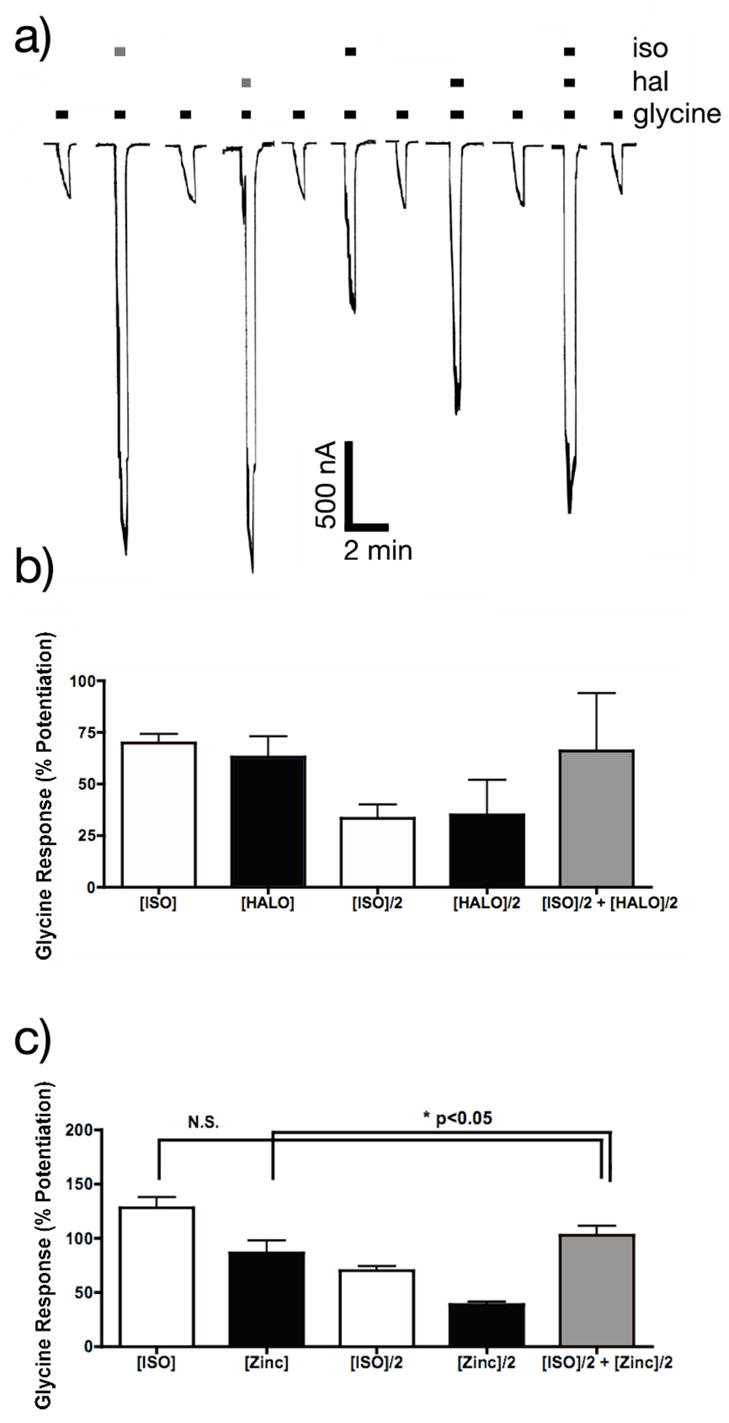
Potentiation of glycine receptor function by isoflurane, halothane and zinc. a) 2-electrode voltage clamp recordings of glycine responses EC5–10 responses in Xenopus laevis oocytes before, during and after the application of isoflurane and/or halothane. Bars above the current traces indicate the duration of agonist application. Grey represents the duration of the anesthetic (75 μM isoflurane or 62.5 μM halothane) application and black represents the duration of the anesthetic (37.5 μM isoflurane and/or 31.2 μM halothane). Calibration bars indicate the amplitude and duration of the responses. b) Potentiation of glycine receptor function by isoflurane ([ISO] = 75 μM) and halothane, ([HAL] = 62.5 μM) did not differ from [ISO]/2+[HAL]/2. c) Potentiation of glycine receptor function by a combination of isoflurane ([ISO] = 75 μM) and zinc ([Zinc] = 0.1 μM): There was no significant difference between the potentiation by [ISO] and [ISO]/2+[Zinc]/2, but [ISO]/2+[Zinc]/2 was slightly more effective as a potentiator than [Zinc] alone.
* P < 0.05.
Because zinc is also known to enhance glycine receptor function, we examined whether combinations of 50 nM zinc and 37.5 μM isoflurane synergistically modulated glycine receptor function. We found no significant difference in the modulation by [ISO] and [ISO]/2+[Zinc]/2 (see Figure 2c). However the potentiation by [ISO]/2+[Zinc]/2 versus [Zinc] did differ significantly.
GABAA receptors
The methodology used in the glycine receptor experiments was repeated with GABAA receptors, using isoflurane in combination with either halothane or flunitrazepam. Figure 3a shows the modulation of responses to EC5–10 concentrations of GABA by isoflurane and halothane. In all cases, the anesthetic applications potentiated receptor function. The enhancements of receptor function by [ISO]/2+[HAL]/2 or [ISO]/2+[FNZ]/2 did not differ from that produced by [ISO] (Figures 3b and 3c), indicating additive effects of isoflurane with halothane and flunitrazepam. For combinations of isoflurane and pentobarbital the effects of [ISO] and [ISO]/2+[PB]/2 were indistinguishable, but there was a small but significant difference for [PB] and [ISO]/2+[PB]/2 (Figure 3d).
Figure 3.
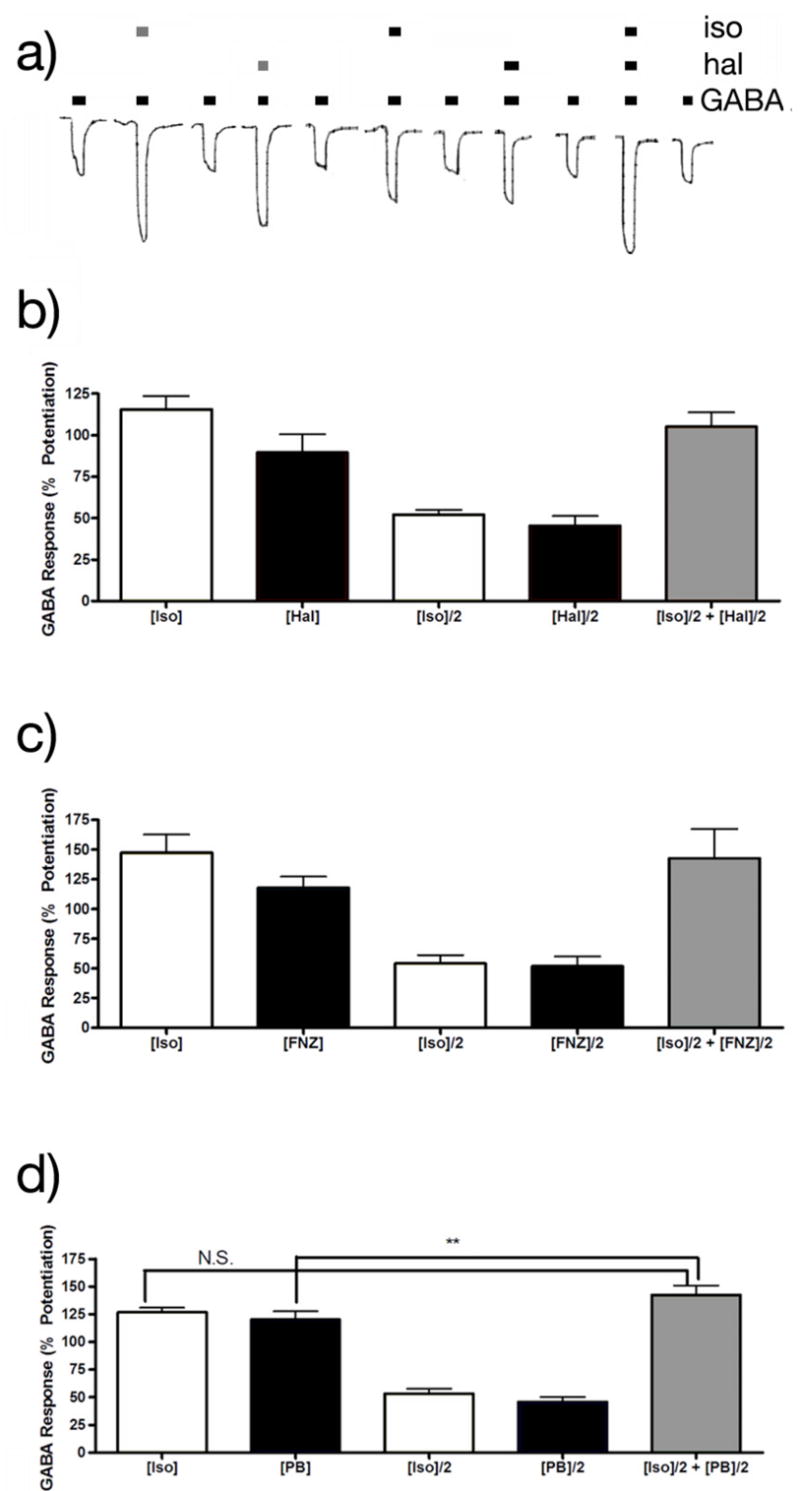
Potentiation of GABAA receptor function by isoflurane, halothane, flunitrazepam and pentobarbital. a) 2-electrode voltage clamp recordings of GABA responses EC5–10 responses in Xenopus laevis oocytes before during and after the application of isoflurane and/or halothane. Bars above the current races indicate the duration of agonist application. The grey bars above these represent the duration of 120 μM isoflurane or 114 μM halothane application; and the black bars indicate the duration of 60 μM isoflurane and/or 57 μM halothane. b) Potentiation of GABAA receptor function by a combination of isoflurane and halothane: [ISO] = 120 μM, [HAL] = 114 μM. The potentiation by [ISO] or [HAL] did not differ significantly from that by [ISO]/2+[HAL]/2. c) Potentiation of GABA receptor function by a combination of isoflurane and flunitrazepam: [ISO] = 120 μM, [FNZ]=500nM. There was no significant difference between the potentiation by [ISO] and [FNZ] and [ISO]/2+[FNZ]/2. d) Potentiation of GABA receptor function by a combination of isoflurane and pentobarbital: [ISO] = 120 μM, [PB]=15μM. [PB] did not differ from potentiation by [ISO]=120μM and [ISO]/2+[PB]/2, but [ISO]/2+[PB]/2 was slightly more effective as potentiator than [PB] alone.
*** P = 0.007
Finally, in order to understand the effect of halothane and chloroform on GABAA receptor function, a full concentration response surface was constructed for the actions of these two anesthetics on the GABA concentration response relationship. Application of 1 MAC halothane mixed with 1 MAC chloroform enhanced currents elicited by low (<10μM) concentrations of GABA but decreased the amplitudes of responses to higher (>10μM) concentrations (Figure 4a). Figures 4b–e highlight these effects for each drug when applied alone. Both halothane and chloroform decreased the GABA EC50 in a dose dependent and saturable manner (Figures 4b & c) and both halothane and chloroform decreased the maximal current elicited by GABA, with chloroform being more efficacious than halothane (Figures 4d & e). Using the methods described, the effects of 17 halothane-chloroform combinations on the fractional change in GABA EC50 were determined and a response surface fitted to the data. The functions C50 and n [previously defined as U50(θ) and γ(θ)] (16,19) did not differ significantly from unity across the surface, indicating that halothane and chloroform were additive in their ability to enhance GABAA receptor function (Figures 4f & g).
Figure 4.
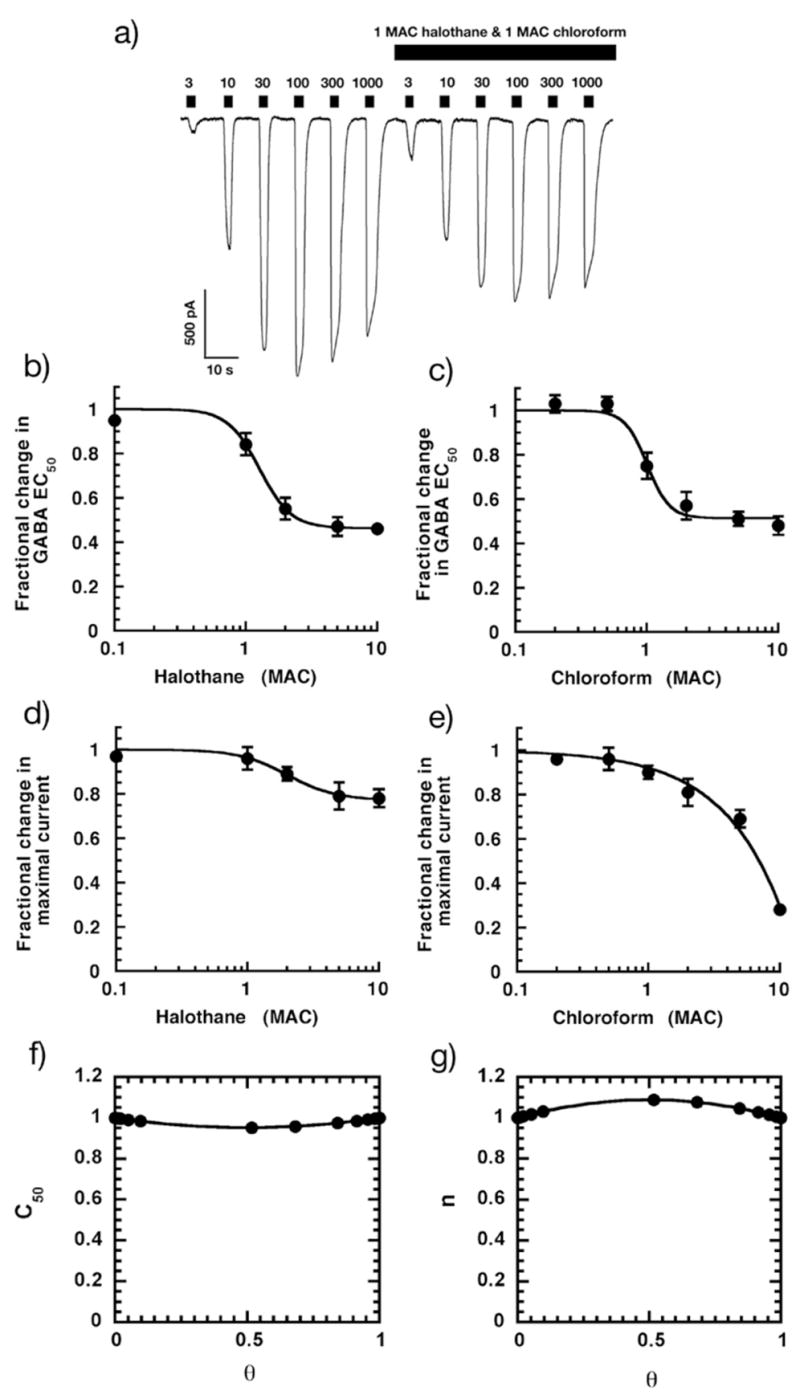
Responses in HEK-293 cells voltage clamped at −60mV expressing α1β2γ2s GABAA receptor subunits to 0.3 – 1000 μM GABA are modulated by 220 μM halothane and 900 μM chloroform (1 MAC of each). a) Halothane and chloroform enhance the amplitude of currents when applied together at lower (<10 μM) but depressed at greater concentrations of GABA. Calibration bars indicate the amplitude and duration of the responses. b) Halothane enhances GABAA receptor function by reducing the GABA EC50. c) Cloroform enhances GABAA receptor function by reducing GABA EC50. d) Halothane reduces maximal GABAA receptor function. e) Chloroform reduces maximal GABAA receptor function. f) & g) Halothane and chloroform are additive in their ability to decrease GABA EC50. C50 and n were determined using the methods described and plotted in the range 0 < θ ≤ 1. Both C50 and n did not significantly deviate from unity indicating that there is no significant synergism or antagonism between the two anesthetics in the reduction of GABAAR EC50.
Discussion
In this study, we tested the ability of general anesthetic combinations to modulate three neuronal receptors, anticipating that the detection of synergy would demonstrate that the two agents under examination acted via dissimilar sites. The three receptors investigated are prototypic allosteric proteins; the binding of a ligand at one site alters the binding of a ligand at another site on the same protein. Therefore, we were interested to see if there was allosteric linkage between these proposed separate modulatory sites. A priori, we expected a certain degree of synergism since for some combinations there is published data that suggest that our drug pairs bind to different amino acids on the receptor (20)(21)(7). However, in all three preparations tested, synergistic receptor modulation by drug was not detected.
One can argue that absence of synergism implies a unified common site of action as the only mechanism of action (22). However, as we have demonstrated here, two compounds that likely bind at different locations on a receptor do not necessarily produce synergism. Thus our additive data would infer that rather than there being a unitary site of action, there seems instead to be a missing allosteric link between the two intramolecular targets. Moreover, this supports the absence of an imaginary mechanism whereby infinitesimally small molecular effects can be amplified to generate a significant response (23). However an interesting consideration is the allosteric and highly synergistic interaction between the general anesthestic and the neurotransmitter molecule. Isoflurane, halothane, pentobarbital and chloroform all enhance glycine and GABA activated responses at concentrations at which they are without intrinsic efficacy. However, they are agonists in their own right at much higher concentrations. In light of this, one prediction we can make is that maybe for these drugs, maximal synergism has been reached with potentiation alone, with nothing left to be done by the second drug.
General anesthetic synergism is common for many drug pairs acting on different target receptors (22,24,25). The results presented here suggest that when synergism for one of the drug pairs tested in this study occurs in an animal, it must do so as a result of action at different receptors in different parts of the neuronal circuitry that underpin the response being measured. A companion study to this report describes how the amplitudes of simple activating and inactivating stimuli can be integrated within neuronal circuits to give very different combinatorial effects (22). It is noteworthy that similar methods have been used before (26) and could be used extensively in the future to predict the temporal effects of simultaneously enhancing hyperpolarizing and inhibiting depolarizing inputs within neuronal circuits. The results of such models would be of great interest to the present debate. However, by comparing the molecular studies shown here with the results of animal studies (24), where quantal responses were being measured with the full spectrum of potential target sites present, and the findings of a meta-analysis (25), it is now apparent that general anesthetic drugs often have different effects at different sites yet these effects simply add up linearly in the circuitry within which they are embedded. This appears to especially true for the inhaled anesthetics.
In summary, synergism was not observed in the modulation of NMDA receptors, glycine receptors or GABAA receptors by pairs of general anesthetics. Our results are in agreement with recent mutagenesis experiments that indicate that isoflurane, flunitrazepam and pentobarbital all have separate sites of action. Yet these drugs share a converging mechanism of action, namely the prolongation of the open time of ligand gated ion channels.
Acknowledgments
The authors would like to thank Meagan A. Jenkins, Carrie Williams, Robert S. Harris, Peter Sebel, Jay Johansen, M. Bruce MacIver, Jan Hendrickx, Pamela Flood & Steve Shafer for helpful discussions.
Funding: Supported by NIH GM073959(AJ), NIH GM04718(EIE & RAH) and institutional and/or departmental resources.
Contributor Information
Andrew Jenkins, The Department of Anesthesiology, Emory University, Atlanta, GA 30322.
Ingrid A. Lobo, Waggoner Center for Alcohol and Addiction Research, Section of Neurobiology and Institute for Cellular and Molecular Biology, The University of Texas at Austin, Austin, TX 78712.
Diane Gong, Waggoner Center for Alcohol and Addiction Research, Section of Neurobiology and Institute for Cellular and Molecular Biology, The University of Texas at Austin, Austin, TX 78712.
James R. Trudell, Department of Anesthesiology, Stanford University, Stanford CA.
Ken Solt, Department of Anesthesia and Critical Care, Massachusetts General Hospital and Department of Anesthesia, Harvard Medical School, Boston MA.
R. Adron Harris, Waggoner Center for Alcohol and Addiction Research, Section of Neurobiology and Institute for Cellular and Molecular Biology, The University of Texas at Austin, Austin, TX 78712.
Edmond I Eger, Department of Anesthesia and Perioperative Care, University of California, San Francisco, CA 94143-0464.
References
- 1.Hemmings HC, Jr, Akabas MH, Goldstein PA, et al. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–10. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Harris RA, Mihic SJ, Dildy-Mayfield JE, Machu TK. Actions of anesthetics on ligand-gated ion channels: role of receptor subunit composition. Faseb J. 1995;9:1454–62. doi: 10.1096/fasebj.9.14.7589987. [DOI] [PubMed] [Google Scholar]
- 3.Ogata J, Shiraishi M, Namba T, et al. Effects of anesthetics on mutant N-methyl-D-aspartate receptors expressed in Xenopus oocytes. J Pharmacol Exp Ther. 2006;318:434–43. doi: 10.1124/jpet.106.101691. [DOI] [PubMed] [Google Scholar]
- 4.Laube B, Kuhse J, Rundstrom N, et al. Modulation by zinc ions of native rat and recombinant human inhibitory glycine receptors. J Physiol. 1995;483(Pt 3):613–9. doi: 10.1113/jphysiol.1995.sp020610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller PS, Da Silva HM, Smart TG. Molecular basis for zinc potentiation at strychnine-sensitive glycine receptors. J Biol Chem. 2005;280:37877–84. doi: 10.1074/jbc.M508303200. [DOI] [PubMed] [Google Scholar]
- 6.Mihic SJ, Ye Q, Wick MJ, et al. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature. 1997;389:385–9. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- 7.Rudolph U, Crestani F, Benke D, et al. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- 8.Amin J. A single hydrophobic residue confers barbiturate sensitivity to gamma-aminobutyric acid type C receptor. Mol Pharmacol. 1999;55:411–23. [PubMed] [Google Scholar]
- 9.Jenkins A, Greenblatt EP, Faulkner HJ, et al. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci. 2001;21:RC136. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenkins A, Andreasen A, Trudell JR, Harrison NL. Tryptophan scanning mutagenesis in TM4 of the GABA(A) receptor alpha 1 subunit: implications for modulation by inhaled anesthetics and ion channel structure. Neuropharmacology. 2002;43:669–78. doi: 10.1016/s0028-3908(02)00175-2. [DOI] [PubMed] [Google Scholar]
- 11.Raines DE, Claycomb RJ, Scheller M, Forman SA. Nonhalogenated alkane anesthetics fail to potentiate agonist actions on two ligand-gated ion channels. Anesthesiology. 2001;95:470–7. doi: 10.1097/00000542-200108000-00032. [DOI] [PubMed] [Google Scholar]
- 12.Solt K, Eger EI, 2nd, Raines DE. Differential modulation of human N-methyl-D-aspartate receptors by structurally diverse general anesthetics. Anesth Analg. 2006;102:1407–11. doi: 10.1213/01.ane.0000204252.07406.9f. [DOI] [PubMed] [Google Scholar]
- 13.Kelly EW, Solt K, Raines DE. Volatile aromatic anesthetics variably impact human gamma-aminobutyric acid type A receptor function. Anesth Analg. 2007;105:1287–92. doi: 10.1213/01.ane.0000282829.21797.97. table of contents. [DOI] [PubMed] [Google Scholar]
- 14.Mascia MP, Gong DH, Eger EI, 2nd, Harris RA. The anesthetic potency of propanol and butanol versus propanethiol and butanethiol in alpha1 wild type and alpha1(S267Q) glycine receptors. Anesth Analg. 2000;91:1289–93. doi: 10.1097/00000539-200011000-00044. [DOI] [PubMed] [Google Scholar]
- 15.Lobo IA, Trudell JR, Harris RA. Accessibility to residues in transmembrane segment four of the glycine receptor. Neuropharmacology. 2006;50:174–81. doi: 10.1016/j.neuropharm.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 16.Sebel LE, Richardson JE, Singh SP, et al. Additive effects of sevoflurane and propofol on gamma-aminobutyric acid receptor function. Anesthesiology. 2006;104:1176–83. doi: 10.1097/00000542-200606000-00012. [DOI] [PubMed] [Google Scholar]
- 17.Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–14. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 18.Krasowski MD, Harrison NL. General anaesthetic actions on ligand-gated ion channels. Cellular and Molecular Life Sciences (CMLS) 1999;55:1278–303. doi: 10.1007/s000180050371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minto CF, Schnider TW, Short TG, et al. Response surface model for anesthetic drug interactions. Anesthesiology. 2000;92:1603–16. doi: 10.1097/00000542-200006000-00017. [DOI] [PubMed] [Google Scholar]
- 20.Dalziel JE, Cox GB, Gage PW, Birnir B. Mutant human alpha(1)beta(1)(T262Q) GABA(A) receptors are directly activated but not modulated by pentobarbital. Eur J Pharmacol. 1999;385:283–6. doi: 10.1016/s0014-2999(99)00710-4. [DOI] [PubMed] [Google Scholar]
- 21.Harvey RJ, Thomas P, James CH, et al. Identification of an inhibitory Zn2+ binding site on the human glycine receptor alpha1 subunit. J Physiol. 1999;520(Pt 1):53–64. doi: 10.1111/j.1469-7793.1999.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shafer SL, Hendrickx JFA, Flood P, et al. Additivity, Synergy and Anesthetic Mechanisms. Submitted to Anesth Analg. doi: 10.1213/ane.0b013e31817b7140. [DOI] [PubMed] [Google Scholar]
- 23.Eger EI, 2nd, Fisher DM, Dilger JP, et al. Relevant concentrations of inhaled anesthetics for in vitro studies of anesthetic mechanisms. Anesthesiology. 2001;94:915–21. doi: 10.1097/00000542-200105000-00032. [DOI] [PubMed] [Google Scholar]
- 24.Eger EI, Tang M, Liao M, et al. Inhaled Anesthetics Do Not Combine to Produce Synergistic Effects Regarding MAC. Submitted to Anesth Analg. doi: 10.1213/01.ane.0000295805.70887.65. [DOI] [PubMed] [Google Scholar]
- 25.Hendrickx JFA, Eger EI, Sonner J, Shafer SL. Is Synergy the Rule? A review of Anesthetic Interactions Producing Hypnosis and Immobility. Submitted to Anesth Analg. doi: 10.1213/ane.0b013e31817b859e. [DOI] [PubMed] [Google Scholar]
- 26.Dutton RC, Zhang Y, Stabernack CR, et al. Temporal summation governs part of the minimum alveolar concentration of isoflurane anesthesia. Anesthesiology. 2003;98:1372–7. doi: 10.1097/00000542-200306000-00011. [DOI] [PubMed] [Google Scholar]