

Abstract
In recent years, there have been major developments of population genetics methods to estimate both rates of recombination and levels of natural selection. However, genomic variants subject to positive selection are likely to have arisen recently and, consequently, had less opportunity to be affected by recombination. Thus, the two processes have an intimately related impact on genetic variation, and inference of either may be vulnerable to confounding by the other. We illustrate here that even modest levels of positive selection can substantially reduce population-based recombination rate estimates. We also show that genome-wide scans to detect loci under recent selection in humans have tended to highlight loci in regions of low recombination, suggesting that confounding by recombination rate may have reduced the power of these studies. Motivated by these findings, we introduce a new genome-wide approach for detecting selection, based on the ratio of pedigree-based to population-based estimates of recombination rate. Simulations suggest that our “Ped/Pop” method, which is designed to capture completed sweeps, has good power to discriminate between neutral and adaptive evolution. Unusually for a multimarker method, our approach performs well in regions of high recombination and also has good power for many generations after the fixation of an advantageous variant. We apply the method to human HapMap and Perlegen data sets, finding confirmation of reported candidates as well as identifying new loci that may have undergone recent intense selection.
The processes of recombination and natural selection have an important influence on genetic variation, observable in patterns of linkage disequilibrium (LD) (Hinds et al. 2005; The International HapMap Consortium 2005). Recombination tends to reduce LD between segregating sites, whereas selection can produce a footprint of high LD. Numerous methods have been developed to exploit the features of each mechanism to infer their intensities from population data (Sabeti et al. 2002; Li and Stephens 2003; McVean et al. 2004; Carlson et al. 2005). However, since the impact of the two processes on patterns of genetic variation is closely related, it is important to account for one when making inferences about the other (Stumpf and McVean 2003; McVean and Spencer 2006).
Here, we focus on recent positive selection since this is biologically interesting and offers a realistic prospect of detection using population data. With the advent of genome-wide population data in humans, this has been the focus of much research (Carlson et al. 2005; Voight et al. 2006; Sabeti et al. 2007; Williamson et al. 2007). When an allele conveys an advantage over alternatives to the survival and reproductive success of its host, its frequency in the population is expected to increase rapidly, resulting in reduced opportunity for recombination relative to a neutral locus. Combined with a similar reduction in mutations (since the most recent common ancestor), this can result in high levels of LD, long high-frequency haplotypes, a large proportion of common derived alleles, a loss of genetic variation at the locus following fixation, and many geographically differentiated allele frequencies. Population genetics methods for detecting recent selection in humans are based on exploiting one or more of these factors (Sabeti et al. 2006). However, a neutral locus with a low recombination rate may also exhibit these characteristics and resemble a locus having undergone a selective sweep. Conversely, a locus that has been subject to selection could generate variation data suggesting a low recombination rate.
In this article, we first illustrate the extent to which positive selection can bias downward population-based estimates of the recombination rate. We then review existing genome-wide studies aimed at detecting loci subject to selection, and highlight their potential for confounding by recombination rate. For example, we show that 25 selection candidates identified by the HapMap and Chimpanzee consortia tend to be located in regions of low recombination. Next, we propose a novel genome-wide method for detecting selection (“Ped/Pop”) that identifies genomic regions with a markedly lower population-based than pedigree-based estimate of recombination rate. We illustrate the power of the Ped/Pop method using simulation, and also conduct genome-wide scans to detect human genetic loci subject to recent selection using the HapMap and Perlegen data sets (Hinds et al. 2005; The International HapMap Consortium 2005). We find that the method can replicate well-established candidates for selection and identify new candidates. As well as addressing the problem of confounding by recombination rate, particular strengths of the method are that it maintains good power many generations after fixation of a selected variant and performs well in regions of high recombination rate. Genetic maps and population variation resources are available, and hence the Ped/Pop method may be useful, for many species of scientific or economic importance, such as cows, pigs, dogs, chickens, tomatoes, maize, and rice (Speed and Zhao 2007).
Results
Positive selection reduces population-based estimates of recombination rates
Population variation data can be exploited to infer sex-averaged recombination rates over the genome at a fine-scale (Stumpf and McVean 2003). Ideally this inference would be achieved within a full-likelihood framework, but since this is computationally infeasible for large data sets (Fearnhead and Donnelly 2001), several approximate-likelihood methods have been developed based on the standard coalescent model (Li and Stephens 2003; Fearnhead et al. 2004; McVean et al. 2004). One such method, LDhat (McVean et al. 2004), has been employed to estimate fine-scale recombination rates across the human genome, validate a hotspot model of recombination, and assess concordance of hotspots between humans and chimpanzees (Myers et al. 2005, 2006; Winckler et al. 2005).
Approximate-likelihood methods for estimating recombination rates have been shown to be robust to various neutral deviations from the standard coalescent model and to SNP ascertainment (Li and Stephens 2003; McVean et al. 2004; Smith and Fearnhead 2005), but here we explore the effect of deviations from the neutrality assumption. These methods infer historical recombinations from LD patterns in population data, so that for loci subject to recent selection we may expect a reduced recombination rate estimate because the MRCA (most recent common ancestor) is relatively recent. Figure 1 shows the very low recombination rate estimated using LDhat at the LCT locus (the Lactase gene), one of the most replicated candidates for recent selection in humans (Bersaglieri et al. 2004; The International HapMap Consortium 2005; Tishkoff et al. 2007). In fact, the LCT locus has the second lowest recombination rate estimate over chromosome 2 using this method, yet the pedigree-based estimate indicates that the recombination rate is not unusual (0.58 cM/Mb) (also see Table 1). This suggests that LDhat and related methods may produce biased estimates at loci subject to positive selection.
Figure 1.

The reciprocal of recombination estimates from the population-based method LDhat taken from the HapMap website (see Methods). The lactase (LCT) gene (136.26–136.31 Mb), lies under the second-highest peak; in the inset, the gene is indicated with a tab.
Table 1.
Chromosome and physical position (Mb) of 25 selection candidate regions identified by the HapMap (H) and Chimpanzee (C) consortia, and empirical quantile of recombination rate

aThe percentile of the pedigree-based recombination rate in the region, relative to the genome-wide distribution for intervals of the same size.
bAutosomal region showing no evidence of recombination in the pedigree data; these represent 11.2% of all 0.5-Mb regions, and are assigned a percentile of 5.6.
cThe LCT locus is at 136.3 Mb.
dX chromosome region showing no evidence for recombination in the pedigree data.
To investigate further the robustness of LDhat to deviations from neutrality, we used SelSim (Spencer and Coop 2004) to simulate population data under various intensities of positive selection, s, and then applied LDhat to attempt to recover the recombination rate used in the simulations. The results (Fig. 2; Supplemental Fig. S1) indicate that the sensitivity of LDhat to selection is detectable even for s = 0.01; actual estimates of s in humans typically range up to 0.1, the approximate intensity of selection reported at LCT (Bersaglieri et al. 2004; Tishkoff et al. 2007), although estimates of s up to 0.3 have been reported at other loci (Schliekelman et al. 2001). These findings have analytical support from Campbell (2007), who estimates that the total branch length of the coalescent tree, which is approximately proportional to the number of detectable recombinations, at a locus subject to selection of strength s = 0.01 in a population of size 104 is 49% of that under neutrality.
Figure 2.

Boxplots summarizing the distribution of LDhat recombination rate estimates from 100 chromosomes sequenced in 100-kb regions, simulated using SelSim to have at the center a positively selected variant with (unscaled) selection coefficient s. The pairwise algorithm of LDhat was used; 250 regions were simulated for each value of s; the effective population size was 104 diploid individuals; and the scaled recombination and mutation rates in each region were Rho = 50 and Theta = 100. The individual estimates underlying the s = 0 and s = 0.1 boxplots are displayed in Supplemental Figure S1.
Recombination confounds genome-wide methods for detecting selection
Prior to the availability of extensive genetic variation data, the effects of selection were usually sought by comparing a statistic evaluated at a candidate locus with its null distribution under the standard neutral model (Nielsen 2001). An assumption of no recombination can be made in this approach, which has a conservative effect on the null distribution of the test statistic. Genome-wide data now permit the identification of outliers from an empirical distribution of the statistic, which conveys the advantage of eliminating the genome-wide confounding effects of demographic history, such as population bottlenecks and expansions (Lewontin and Krakauer 1973). However, recombination is a locus-specific confounder, and an assumption of no recombination is not available under this approach. If low recombination rates are more frequent in the genome than loci under strong selection, then many of the outliers in the empirical distribution may reflect the former more than the latter.
Statistics to detect selection usually aim to capture one or more of the following characteristics associated with recent positive selection: (1) high levels of LD or unexpectedly long haplotypes, (2) a skewing of the site frequency spectrum toward low frequency alleles, (3) a high proportion of derived alleles, and (4) high variability in allele frequencies between populations (for a general review, see Sabeti et al. 2006). We now briefly review examples of these four types of test statistic used in genome-wide studies for detecting selection, and examine the potential for confounding due to variable recombination rate.
Carlson et al. (2005) search for “Contiguous Regions of Tajima’s D Reduction” (CRTR) and interpret these as candidates for selection. CRTR are defined as genomic regions of at least 20 overlapping 100-kb windows (total >290 kb) where more than 75% of the windows are in the bottom 1% of the empirical distribution for Tajima’s D. A similar genome scan by Kelley et al. (2006) focused on locally clustered Tajima’s D outliers. The latest pedigree-derived human genetic map, derived from several populations of European origin (Matise et al. 2007), suggests that there may be many regions up to 300 kb under little recombination. Supplemental Figure S2 shows the distribution of Tajima’s D for 300-kb regions evolving under neutrality with no recombination (solid green curve), compared with that under various intensities of positive selection with the genome-average recombination rate. These simulations tend to exaggerate the power of Tajima’s D to detect selection because there is no ascertainment bias (problematic for Tajima’s D; Kelley et al. 2006) and because the advantageous allele has just reached fixation.
Both Carlson et al. (2005) and Kelley et al. (2006) note that regions that they highlight as being subject to selection have a significantly lower than average rate of recombination. Both suggest that this may be due to signatures of selection being more pronounced in regions of low recombination. However, in some cases the low rate of recombination could have contributed to a spurious signal of positive selection. A statistic sensitive to recombination rate will incorrectly estimate the selection coefficient at every locus due to dependence on the local recombination rate. Therefore, the ranking of loci from least to greatest evidence for selection implied by the statistic is reordered, resulting both in a reduction of overall statistical power and an increased potential for false positives.
Weir et al. (2005) apply several strategies to exploit the standard measure of population differentiation, FST, to detect selection via unusual allele-frequency differences across the HapMap and Perlegen populations. Because these investigators found FST to be highly variable over loci, they used mean FST over 5-Mb windows. However, the confounding effect of recombination rate, which is absent at a single SNP, is introduced by this averaging due to the correlation between neighboring SNPs. When a SNP happens to generate a large value of FST under drift in a region of low recombination, many flanking SNPs will also show high FST, which may be misinterpreted as strong evidence for selection. While 5-Mb windows ensure that variability in recombination rates is reduced, the impact that selection has on such large regions is likewise reduced.
The HapMap and Chimpanzee consortia combined several test statistics to identify a set of selection candidates (The Chimpanzee Sequencing and Analysis Consortium 2005; The International HapMap Consortium 2005). The six genomic regions highlighted by the Chimpanzee Consortium showed unexpectedly low genetic diversity in humans relative to the divergence from the chimpanzee sequence. The proportion of high-frequency derived alleles in each of the six regions was in the top 10% of the genome-wide distribution, and three were in the top 1%. In the HapMap study, 19 selection candidates were identified as significant outliers in the joint distribution of heterozygosity and either population differentiation or a skewing of the allele frequency toward rare alleles. Additional regions were identified using the long range haplotype (LRH) test (see Discussion).
For each of the 25 selection candidates reported by the HapMap and Chimpanzee consortia, Table 1 reports the recombination rate estimated from the best available pedigree-based genetic map (Matise et al. 2007), expressed as a percentile of the genome-wide distribution of estimates in regions of the same size. There is a significant bias toward regions of low recombination (Kolmogorov-Smirnov P = 1.9 × 10−8; Fig. 3).
Figure 3.

Cumulative distribution functions of the uniform distribution (diagonal line) and of the values in the final column of Table 1. The Kolmogorov-Smirnov statistic D is the maximum vertical distance between the two. Here, D = 0.61 which gives P = 1.9 × 10−8.
Both studies highlight a region close to 50 Mb on chromosome 1. Figure 4 shows that the pedigree-estimated recombination rate in this region is particularly low. Also noteworthy is the candidate in the lowest percentile of recombination density (chr X: 61.8–64.5 Mb), since this is one of only three candidates replicated by three different methods in the HapMap study (another at 20 Mb on chromosome X is in the fourth lowest percentile and the other is LCT). The enrichment of selection candidates on chromosome X in the HapMap study (six of 19 candidates) may reflect the reduced sex-averaged recombination rate.
Figure 4.

Pedigree-estimated (relative) recombination rates in 4-Mb sliding windows along human chromosome 1. The vertical dotted line indicates a 4-Mb window encompassing two candidate regions identified by the Chimpanzee and HapMap consortia and coinciding with a recombination desert.
The Ped/Pop statistic to detect selection
Our Ped/Pop approach consists of dividing the recombination rate estimate for a locus obtained from pedigree data by the corresponding estimate from a population genetics method (here LDhat but other methods could be used). Large signals result if the LDhat estimate of recombination rate is low relative to the pedigree estimate, and are interpreted as indicating candidates for recent selection. The intuition is that LDhat and related methods effectively measure the breakdown of LD at a given locus, which reflects the amount of recombination since the MRCA at the locus. When a beneficial variant reaches fixation, the time since the MRCA is typically very low (Campbell 2007; Hoggart et al. 2007). Therefore, when the per-generation recombination rate is controlled via pedigree-based estimates, an aberrantly low estimate from LDhat is likely to reflect intense recent selection. The Ped/Pop statistic is comparable to Tajima’s D for detecting selection, which compares two estimates for the mutation rate expected to be equal under neutrality.
We conducted a simulation study to assess the performance of the Ped/Pop statistic in detecting loci at which a beneficial variant has recently reached fixation. SelSim (Spencer and Coop 2004) was used to generate 500-kb regions with genome-wide average rates of mutation and recombination, under neutral evolution and several intensities of selection. The population-based recombination rate was estimated using LDhat in 100-kb sliding windows over each region, while the pedigree-based estimates were sampled from a Poisson distribution with mean 1.59 (see Methods). The window size and Poisson mean reflect the latest genetic map (Matise et al. 2007). This procedure was repeated 250 times under each scenario. The results, illustrated in Figure 5 and Table 2, suggest that the Ped/Pop ratio has good power to discriminate between neutral and adaptive evolution for many generations after the fixation of a beneficial variant. Supplemental Figure S3 gives results from repeating the analysis with low and high rates of recombination. The statistical power at the high rate of recombination appears similar to that at the average rate. At the low recombination rate, the neutral density has a larger upper tail, corresponding to a greater possibility for false positive results, but the densities with selection also show more extreme values. These simulations provide only a limited guide to the potential power of the Ped/Pop approach applied to real data due to the simplistic model assumptions made. Nevertheless, it is encouraging that the distribution of the Ped/Pop statistic appears to be highly sensitive to the value of s, even in regions of high recombination.
Figure 5.

Density of log10(Ped/Pop) at fixation under neutrality and positive selection, estimated from the SelSim simulation study described in the text.
Table 2.
Key tail probabilities for the Ped/Pop statistic, each estimated from the simulation study of Figure 5 (described in the text)

Neutmax denotes the largest value of the statistic obtained under the neutral simulations. Also shown are the same tail probabilities 10,000 and 20,000 yr after fixation (20-yr generations).
Using the Ped/Pop statistic, we scanned the HapMap and Perlegen data (Hinds et al. 2005; The International HapMap Consortium 2005) for signals of recent selection (see Methods; for full results, see Supplemental Figs. S4, S5). Following the method of Myers et al. (2005), to improve the accuracy of recombination rate estimates, the scans were performed using averages over the worldwide populations for each data set and are therefore less likely to identify selection operating in just one population. There are 52 extreme outliers with Ped/Pop > 104 arising from four distinct genomic loci. One of these loci corresponds to a candidate identified by the HapMap study on chromosome 16 and is the only candidate locus from Table 1 with recombination rate above the median (row 17). Another Ped/Pop extreme outlier locus is within 1.5 Mb of the CCR5 gene on chromosome 3, a proposed candidate for recent selection in humans (Carrington et al. 1997), which did not yield a signal of selection using the LRH test (Sabeti et al. 2005). One of the remaining two extreme outlier loci is within 5 Mb of another strong candidate for selection, CD40LG, and lies in a region rich in OMIM genes (chr X, near 130 Mb), whereas the final outlier is located within the METT5D1 gene (chr 11, near 28 Mb), for which there does not appear to be a prior report of a signal of recent selection. Of the 25 selection candidates from the HapMap and Chimpanzee consortia (Table 1), 11 have Ped/Pop >20 (the upper 0.6% point of the empirical distribution) and four have Ped/Pop >50 (upper 0.2% point). Figure 6 depicts the results of the Ped/Pop scans of three of the chromosomes: highlighting the extreme outliers described above on chromosomes 3 and 16, as well as the HapMap candidate from chromosome 14 (with Ped/Pop > 300). Supplemental Table S1 lists the 100 strongest candidates for selection from these scans.
Figure 6.

Ped/Pop values over three human chromosomes. Chromosomes 16 and 14 show replications of HapMap candidates from Table 1 (highlighted with dashed line) from the Perlegen scan (chr 16 panel includes inset of HapMap scan), while the chromosome 3 plot shows the close proximity of the CCR5 gene (dashed lines) with the extreme outlier signal from the HapMap scan (Perlegen scan, inset).
We also conducted scans for selection over the separate HapMap populations (separate LDhat rates courtesy of Gil McVean, Oxford University, UK). Supplemental Table S2 presents the strongest 20 candidates for each population. Supplemental Figure S6 shows that the density of the Ped/Pop statistic is similar across populations (after standardization of total map length), indicating that the European genetic map provides reliable recombination estimates for Africans and Asians as well. However, a difference between the densities does occur in the upper tail, which we expect harbors loci under recent positive selection. Interestingly, the African tail is markedly lighter, implying that the African LDhat recombination rate estimates are more concordant with the European genetic map than their European equivalent here; at Ped/Pop >20 there are 40% fewer African outliers than European and 11% fewer Asian. This otherwise surprising result is, however, consistent with non-African populations having had more completed selective sweeps in recent history after being exposed to new environments (Sabeti et al. 2007; Williamson et al. 2007; Hancock et al. 2008).
In order to assess more carefully whether extreme Ped/Pop values genuinely reflect loci under selection, we performed a simulation study to produce a more realistic null distribution. Schaffner et al. (2005) calibrate a population genetics model to generate simulated data consistent with empirical data across a wide range of characteristics for three major worldwide human populations. Their model incorporates variation in recombination rates at both broad and fine scales, as well as a complex demographic model reflecting the main features of the evolution of modern human populations. By using their software, we simulated 400-Mb regions corresponding to Perlegen and HapMap data sets. We then used LDhat to obtain population recombination estimates across the simulated regions. Finally, we constructed a 400-Mb pseudo genetic map by first laying down markers with marker spacings randomly drawn from the actual genetic map, and then obtained corresponding pedigree-based recombination estimates by drawing from an appropriate Poisson distribution (see Methods). Supplemental Figure S7 compares the resulting “Schaffner null distribution” of Ped/Pop with the empirical distribution from the HapMap and Perlegen data sets. The latter shows heavier tails, consistent with the effects of recent positive selection (upper tail), and possibly also balancing selection (lower tail). Figure 7 displays the right extreme of the empirical Ped/Pop distribution (Ped/Pop > 20). Comparison with the Schaffner null distribution (Fig. 7, inset) suggests that ∼90% of Ped/Pop values exceeding 20 correspond to true positives.
Figure 7.

The main histogram represents the right-extreme of the empirical log10(Ped/Pop) distribution (Ped/Pop >20) from the combined scans for selection (HapMap and Perlegen data sets). The inset depicts the corresponding histogram for the Schaffner null distribution.
Despite these promising findings, there are well-established candidates for selection that do not have extreme Ped/Pop values. The LCT locus is a particular omission, which happens to have widely spaced flanking pedigree markers that span regions of high recombination in addition to the LCT locus, which has a low population-based recombination estimate (Fig. 1). However, in a preliminary scan performed using 100-kb sliding windows over the genome, involving linear interpolation to impute recombination rate estimates for each interval, LCT gave the most extreme Ped/Pop ratio on chromosome 2. To limit false positives, however, we suggest that the Ped/Pop statistic be calculated between loci that span pedigree markers as in our genome-wide scans here.
A note on selection and recombination in genic regions
One of the findings of McVean et al. (2004) (see also Myers et al. 2005) is that recombination is low within genes, while recombination hotspots tend to exist marginally outside rather than within genes. Eberle et al. (2006) report high levels of LD in genic as opposed to intergenic regions and, after further analysis, suggest that this is primarily due to positive selection. Thus, essentially the same phenomenon has been attributed to low recombination in genes and to a tendency for positive selection to occur in genic regions. Both interpretations have good intuitive support (low recombination rate in genes to avoid disruption of function; selection in genes due to beneficial mutations), and it may be that both are correct: Recombination may be suppressed in some genes, while positive selection explains the high LD in others. In order to test such hypotheses, methods that can isolate the effects of selection and recombination are required.
Discussion
When estimating recombination rates or inferring selection from population genetic data, it is important to consider the influence that both processes have on genetic variation. Ideally when studying one of these processes, the confounding effect of the other would be eliminated. We have shown that methods for estimating recombination rates can be sensitive to deviations from neutrality and that methods for detecting selection may be confounded by variable recombination rates. Furthermore, we proposed the ratio of pedigree-based and population-based estimates of recombination as a statistic to identify positive selection, in particular recently completed sweeps. We demonstrated that this Ped/Pop approach performs well in simulation studies and illustrated its application to human genome-wide data.
Nevertheless, the Ped/Pop method is subject to several potential limitations. First, reliance on pedigree-based recombination rates assumes that recombination rates are constant over time. Several studies using LDhat suggest that recombination hotspots may evolve quickly. However, recombination is thought to be constrained over large regions (Myers et al. 2005, 2006; Winckler et al. 2005), and we found concordance of LDhat estimated rates in the HapMap populations (Supplemental Fig. S5). If recombination rates do change significantly over short evolutionary time-scales, then this would pose a problem for our method, and many others. Second, our approach is limited by the accuracy of the methods for estimating recombination that we use. Population-based estimates of recombination have been found to be robust to factors such as ascertainment bias and demography (McVean et al. 2004; Smith and Fearnhead 2005). The accuracy of pedigree-based estimates is limited by the numbers of meioses and markers. However, improvements will be realized as the genetic map is progressively updated.
We have highlighted several genome-wide studies that may be confounded by recombination, but other studies have taken recombination into account. Nielsen et al. (2005) construct a likelihood framework to deduce selection from the site-frequency spectrum. However, in attempting to estimate the recombination rate and selection coefficient simultaneously from the same population data and in using a composite likelihood whose accuracy declines with linkage, this approach is also susceptible to confounding: The investigators note that incorrect assumptions about the recombination rates lead to biased estimates of the selection coefficients. Furthermore, we investigated their CLR statistic using data simulated under neutrality according to the null model of Schaffner et al. (2005) and found evidence for sensitivity to demography. Williamson et al. (2007), who perform a genome-wide scan using the CLR statistic, found that the CLR had more extreme values under the Schaffner model than the standard equilibrium model, but we also found substantially more extreme values for Asians than Europeans (see Supplemental Fig. S8; Methods). This dependence on demography may contribute to the large proportion of Asian putative selection loci identified by Williamson et al. (2007).
The LRH approach (Sabeti et al. 2002) explicitly attempts to eliminate the confounding effects of recombination by comparing the conservation of different haplotypes at a locus. Candidates for selection listed by the HapMap Consortium extracted using the LRH approach are not overrepresented in regions of low recombination (data not shown), unlike those obtained via other statistics (Fig. 3). However, there are several limitations to the LRH approach. First, it assumes that recombination rates are not sequence-specific, whereas there is evidence that different haplotypes can have different recombination rates (Jeffreys and Neumann 2002; Yauk et al. 2003; Kong et al. 2008). Inversions are a genome feature that result in SNPs having alleles that are associated with different local recombination rates: A mutant allele tagging an inversion event should be on a background with comparatively less “haplotype breakdown” than the alternative allele due to suppression of recombination between inverted and noninverted haplotypes (assuming that the inverted is the minor type). Although sequence-specific recombination presents a challenge to many methods for detecting selection, a statistic based on a direct comparison of haplotypes may be particularly sensitive to this problem. Second, methods based on the LRH approach are only likely to have good power to detect incomplete selective sweeps within the population considered (Voight et al. 2006). The implementation of the approach by Voight et al. (2006) has maximal power when the beneficial (genotyped) allele has a frequency of ∼0.7, and power declines rapidly above a frequency of 0.8. The logistic selective sweep model (Kaplan et al. 1989; Stephan et al. 1992) implies that a sweep of intensity s = 0.1 spends only 26 generations between frequencies of 40% and 90% (see Methods). A recent modification to the LRH approach compares the relevant statistic, EHH, in the population being tested with another population, so that sweeps at or near fixation can be detected (Sabeti et al. 2007; Tang et al. 2007); however, this may not always be an option. Also, the problem of sequence-specific recombination rate may be exacerbated by using another population, and many sweeps of interest may be shared by (or complete in) both populations (Barreiro et al. 2008).
A final limitation of the LRH approach relates to “soft sweeps,” which are caused by selection via standing variation, migration, or recurrent (or physiologically equivalent) mutations (Przeworski et al. 2005; Pennings and Hermisson 2006a, b). Soft sweeps are likely to go undetected by the LRH approach because several haplotypic backgrounds are associated with the adaptive substitution (Sabeti et al. 2007), and by statistics such as Tajima’s D because genetic variation is maintained around the selected locus. However, overall LD can be even stronger under soft sweeps than for classical selection (Pennings and Hermisson 2006b), so we expect that the Ped/Pop statistic has good power to detect soft sweeps.
Use of pedigree data as an independent source for controlling for variability in recombination rate could be exploited in other methods for detecting selection. A specific strength of our approach is its relatively good power in regions of high recombination rate, while a limitation is the present accuracy of pedigree-based recombination estimates. Our scans for selection replicated almost half of the candidates established by the HapMap and Chimpanzee consortia, and identified many new loci that may have been subject to recent selection. In addition to humans, the Ped/Pop method should be applicable to many species of scientific or economic interest.
Scanning the human genome for signals of selection is motivated not only by a desire to understand the recent evolution of our species but also by the intuition that loci subject to selection may also be strong candidates for disease-causing variants (Bamshad and Wooding 2003; Bustamante et al. 2005; Barreiro et al. 2008; Hancock et al. 2008). Given the inherent difficulty of the task, we hope that an additional approach to the problem, and an extra avenue for investigation, may prove useful to this end.
Methods
Simulations
The population SNP data relating to Figures 2 and 5, Supplemental Figure S2, and Table 2 were simulated under neutral and adaptive evolution using the coalescent-based package SelSim (Spencer and Coop 2004). Samples of 100 sequences were simulated to reflect 100 kb (Fig. 2), 300 kb (Supplemental Fig. S2), and 500 kb (Fig. 5; Table 2) regions under uniform mutation and recombination rates of μ = 2.5 × 10−8 and r = 1.25 × 10−8, corresponding to approximate genome-wide average rates (Jobling et al. 2004; Matise et al. 2007). All SNPs were used for the analyses. Under adaptive evolution, the advantageous allele arises in the center of each region, with relative selection coefficients of 1 + s and 1 + 2s for the heterozygote and the derived homozygote, respectively. Data were simulated at fixation of a selected variant and, for the simulation study of Figure 5, at 500 and 1000 generations following fixation also. For each scenario, 250 replicates were simulated.
In the analysis of the sensitivity of LDhat to deviations from neutrality (Fig. 2), the pairwise algorithm of LDhat was used to estimate recombination rates from the samples of 100 sequences described above. In Supplemental Figure S2, Tajima’s D values were calculated from the data using the convert algorithm from the LDhat package.
For the power analysis of the Ped/Pop statistic (Fig. 5; Table 2), the interval algorithm of LDhat was used to estimate mean recombination rates in 100-kb sliding windows, centered on each SNP across each 500-kb region. Pedigree-based recombination rates were simulated by first sampling from the Poisson distribution with mean 1.59, since the genetic map was constructed from data with an average of 1.59 informative recombinations per 100 kb, and then scaling each value appropriately to provide corresponding rates. Because we are interested in extreme values of the Ped/Pop statistic and because the pattern of LD at the selected locus may sometimes be complex (Reed and Tishkoff 2006; McVean 2007), we used the largest value of the ratio from any 100-kb interval within the 500-kb regions.
Recombination rate estimates
Pedigree-based recombination rate estimates were obtained using Rutgers combined linkage-physical map of the human genome (Matise et al. 2007). This map represents the merger of several sources of pedigree data and is the highest resolution pedigree genetic map published to date. The latest release is available for download at http://compgen.rutgers.edu/maps/. Recombination rate estimates were deduced from the map by taking the difference in genetic position between adjacent markers and dividing by the intervening physical distance.
Population-based recombination rates were obtained from two resources. HapMap based rates were downloaded from www.hapmap.org and estimated from phase I data (release 16a) using the composite likelihood method, LDhat, described in McVean et al. (2004). Estimates for the main scans for selection were gained by applying LDhat to each of the HapMap populations (CEU, YRI, CHB, and JPT) separately and then taking the mean estimate across populations. Recombination estimates are provided in cM/Mb between each pair of SNPs. Further details are offered by The International HapMap Consortium (2005). The same procedure was applied, using LDhat, to obtain recombination rate estimates from the Perlegen data (for details, see Myers et al. 2005). The two data sets have complementary advantages: The HapMap data are derived from a larger sample, while the Perlegen data represent higher density SNPs ascertained using a more consistent protocol. These differences may explain a large part of the variation in magnitudes of the Ped/Pop statistic between the two genome scans.
Genome scans using the Ped/Pop statistic
By applying the Ped/Pop approach, we compared population-based recombination rates estimated from the HapMap and Perlegen data sets (Hinds et al. 2005; The International HapMap Consortium 2005) with those derived from the Rutgers pedigree genetic map (Matise et al. 2007) across the human genome to search for signals of recent selection. Since the estimates from the pedigree map are on a broader scale, we used the markers from this map to define the genetic intervals for which the two types of recombination estimate would be compared. Intervals were centered on each population marker owing to their greater density. Although the separation of markers in the pedigree map has a mean of ∼130 kb, its variance is substantial. We ensured that these intervals spanned at least 80 kb, to avoid the most unreliable estimates. Since the pedigree markers do not form a subset of the population markers, the intervals for population-based estimates were defined separately. These were chosen to span the population markers adjacent to the pedigree markers defining the corresponding pedigree-interval. Interpolation (to ensure corresponding intervals are of equal length) was not performed in order to avoid using estimates that relate to regions outside the given intervals; but only intervals that were at least 90% overlapping were compared as part of the genomic scan. Note that evaluating the Ped/Pop ratio for every population marker, using this scheme, assists in combining appropriate intervals (required to ensure intervals >80 kb), but often results in multiple Ped/Pop ratios with equal value at neighboring loci. However, since we consider only the value of the statistic (rather than its distribution) to detect candidate loci, this is not a cause for concern.
Simulation of null distribution for Ped/Pop
In our simulation of a realistic null distribution for the Ped/Pop statistic (Fig. 7; Supplemental Fig. S7), we adopt the model of Schaffner et al. (2005), who calibrated parameters of demography and recombination to produce a model consistent with a wide range of empirical measures relating to sequence variation and LD. We use the associated software, available from http://www.broad.mit.edu/∼sfs/cosi, to generate data sets reflecting 400-Mb regions of African, African-American, Asian, and European descent (achieved by combining two hundred 2-Mb regions). In attempting to recreate the Perlegen and HapMap data sets, we produced samples of 50 and 100 sequences, respectively, from the appropriate populations, retaining all SNPs for the analysis. We created a 400-Mb pseudo genetic map by first allocating markers on the map with between-marker distances corresponding to random draws from the between-marker distances of the actual genetic map, and then we gained pedigree-based estimates of recombination in the intervals of the map by varying the parameter-based recombination rates (from simulations) using the appropriate Poisson distribution (as described above). We then used LDhat to obtain population-based recombination rates from the data over the intervals stipulated by the pseudo genetic map. By repeating the scheme used in our empirical scan (described above), we obtained Ped/Pop values for the 400-Mb regions for each population sample. In order to more closely represent a full genome, we repeated the step of gaining pedigree-based estimates over the map (via draws from the Poisson distribution) seven times, giving a total genome of 2.8 Gb simulated under the Schaffner null for each population sample.
CLR statistic analysis
In order to investigate the robustness of the CLR statistic (Nielsen et al. 2005; Williamson et al. 2007), we simulated data to reflect Asian and European samples based on the Schaffner null model (Schaffner et al. 2005), since these were the populations dominating the selection candidate list produced in the Williamson et al. (2007) study (151/164 candidates). We produced 1000 data sets comprising 50 sequences of length 100 kb for each population sample, with all SNPs retained for the analysis. We also simulated a data set under the same parameter settings, for each population sample, but of length 10 Mb for use as a background frequency file for the “SweepFinder” package used in Williamson et al. (2007). The method implemented by the SweepFinder package (available from http://www.evolutionarygenomics.dk/pgs/programs.html), which calculates the CLR statistic, exploits the background frequency spectrum to test local regions for recent selection. We considered a 10-Mb region sufficient to generate a frequency spectrum closely matching the theoretical population frequency spectrum for the data of the relevant simulation settings. We then ran the SweepFinder software to infer the CLR statistic across the 100-kb regions (user-choices: gridsize = 50, folded = 1, inclusion of sites with frequency = 0).
The logistic selective sweep model
Kaplan et al. (1989) model a selective sweep dominated by a middle deterministic phase, where the frequency, p, of the selected allele, with selection coefficient 1 + s relative to the neutral allele, follows the logistic differential equation:
This model was simplified by Stephan et al. (1992) and Wiehe and Stephan (1993) so that the other two phases in the model by Kaplan et al. (1989) were ignored, such that the entire selective sweep could be modeled by Equation 1. This implies
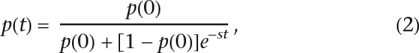 |
where t is the duration of the selective sweep, p(t) is the frequency of the selected allele at the end of the sweep, and p(0) is the frequency of the allele at the beginning of the sweep. From Equation 2, we can derive an equation for the time between given frequencies of the selective sweep:
where p(a) is the frequency of the selected variant at time t = a in the sweep and p(b) is the frequency at time t = b. We can use Equation 3 directly to calculate the expected time taken for a selected allele to go from one frequency to another during a sweep, according to the logistic model. For instance, considering an allele increasing in population frequency from 0.4 and 0.9 [so that p(a) = 0.4 and p(b) = 0.9] with selection coefficient s = 0.1, we find that t ≈ 26.
Acknowledgments
This work was supported by funding from the Biotechnology and Biological Sciences Research Council (UK). We thank Jamie O’Reilly, Ernest Turro, Will Astle, and three anonymous reviewers for their various helpful contributions. This article is dedicated to Jim O’Reilly, who passed away during the completion of this work. His memory will continue to act as a huge source of inspiration.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.067181.107.
References
- Bamshad M., Wooding S.P., Wooding S.P. Signatures of natural selection in the human genome. Nat. Rev. Genet. 2003;4:99–111. doi: 10.1038/nrg999. [DOI] [PubMed] [Google Scholar]
- Barreiro L.B., Laval G., Quach H., Patin E., Quintana-Murci L., Laval G., Quach H., Patin E., Quintana-Murci L., Quach H., Patin E., Quintana-Murci L., Patin E., Quintana-Murci L., Quintana-Murci L. Natural selection has driven population differentiation in modern humans. Nat. Genet. 2008;40:340–345. doi: 10.1038/ng.78. [DOI] [PubMed] [Google Scholar]
- Bersaglieri T., Sabeti P.C., Patterson N., Vanderploeg T., Schaffner S.E., Drake J.A., Rhodes M., Reich D.E., Hirschhorn J.N., Sabeti P.C., Patterson N., Vanderploeg T., Schaffner S.E., Drake J.A., Rhodes M., Reich D.E., Hirschhorn J.N., Patterson N., Vanderploeg T., Schaffner S.E., Drake J.A., Rhodes M., Reich D.E., Hirschhorn J.N., Vanderploeg T., Schaffner S.E., Drake J.A., Rhodes M., Reich D.E., Hirschhorn J.N., Schaffner S.E., Drake J.A., Rhodes M., Reich D.E., Hirschhorn J.N., Drake J.A., Rhodes M., Reich D.E., Hirschhorn J.N., Rhodes M., Reich D.E., Hirschhorn J.N., Reich D.E., Hirschhorn J.N., Hirschhorn J.N. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante C.D., Fledel-Alon A., Williamson S., Nielsen R., Hubisz M.T., Glanowski S., Tanenbaum D.M., White T.J., Sninsky J.J., Hernandez R.D., Fledel-Alon A., Williamson S., Nielsen R., Hubisz M.T., Glanowski S., Tanenbaum D.M., White T.J., Sninsky J.J., Hernandez R.D., Williamson S., Nielsen R., Hubisz M.T., Glanowski S., Tanenbaum D.M., White T.J., Sninsky J.J., Hernandez R.D., Nielsen R., Hubisz M.T., Glanowski S., Tanenbaum D.M., White T.J., Sninsky J.J., Hernandez R.D., Hubisz M.T., Glanowski S., Tanenbaum D.M., White T.J., Sninsky J.J., Hernandez R.D., Glanowski S., Tanenbaum D.M., White T.J., Sninsky J.J., Hernandez R.D., Tanenbaum D.M., White T.J., Sninsky J.J., Hernandez R.D., White T.J., Sninsky J.J., Hernandez R.D., Sninsky J.J., Hernandez R.D., Hernandez R.D., et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- Campbell R.B. Coalescent size versus coalescent time with strong selection. Bull. Math. Biol. 2007;69:2249–2259. doi: 10.1007/s11538-007-9218-9. [DOI] [PubMed] [Google Scholar]
- Carlson C.S., Thomas D.J., Eberle M.A., Swanson J.E., Livingston R.J., Rieder M.J., Nickerson D.A., Thomas D.J., Eberle M.A., Swanson J.E., Livingston R.J., Rieder M.J., Nickerson D.A., Eberle M.A., Swanson J.E., Livingston R.J., Rieder M.J., Nickerson D.A., Swanson J.E., Livingston R.J., Rieder M.J., Nickerson D.A., Livingston R.J., Rieder M.J., Nickerson D.A., Rieder M.J., Nickerson D.A., Nickerson D.A. Genomic regions exhibiting positive selection identified from dense genotype data. Genome Res. 2005;15:1553–1565. doi: 10.1101/gr.4326505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington M., Kissner T., Gerrard B., Ivanov S., O’Brien S.J., Dean M., Kissner T., Gerrard B., Ivanov S., O’Brien S.J., Dean M., Gerrard B., Ivanov S., O’Brien S.J., Dean M., Ivanov S., O’Brien S.J., Dean M., O’Brien S.J., Dean M., Dean M. Novel alleles of the chemokine-receptor gene CCR5. Am. J. Hum. Genet. 1997;61:1261–1267. doi: 10.1086/301645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Chimpanzee Sequencing and Analysis Consortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Eberle M.A., Rieder M.J., Kruglyak L., Nickerson D.A., Rieder M.J., Kruglyak L., Nickerson D.A., Kruglyak L., Nickerson D.A., Nickerson D.A. Allele frequency matching between SNPs reveals an excess of linkage disequilibrium in genic regions of the human genome. PLoS Genet. 2006;2:e142. doi: 10.1371/journal.pgen.0020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead P., Donnelly P., Donnelly P. Estimating recombination rates from population genetic data. Genetics. 2001;159:1299–1318. doi: 10.1093/genetics/159.3.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead P., Harding R.M., Schneider J.A., Myers S., Donnelly P., Harding R.M., Schneider J.A., Myers S., Donnelly P., Schneider J.A., Myers S., Donnelly P., Myers S., Donnelly P., Donnelly P. Application of coalescent methods to reveal fine-scale rate variation and recombination hotspots. Genetics. 2004;167:2067–2081. doi: 10.1534/genetics.103.021584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock A.M., Witonsky D.B., Gordon A.S., Eshel G., Pritchard J.K., Coop G., Di Rienzo A., Witonsky D.B., Gordon A.S., Eshel G., Pritchard J.K., Coop G., Di Rienzo A., Gordon A.S., Eshel G., Pritchard J.K., Coop G., Di Rienzo A., Eshel G., Pritchard J.K., Coop G., Di Rienzo A., Pritchard J.K., Coop G., Di Rienzo A., Coop G., Di Rienzo A., Di Rienzo A. Adaptations to climate in candidate genes for common metabolic disorders. PLoS Genet. 2008;15:e32. doi: 10.1371/journal.pgen.0040032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds D.A., Stuve L.L., Nilsen G.B., Halperin E., Eskin E., Ballinger D.G., Frazer K.A., Cox D.R., Stuve L.L., Nilsen G.B., Halperin E., Eskin E., Ballinger D.G., Frazer K.A., Cox D.R., Nilsen G.B., Halperin E., Eskin E., Ballinger D.G., Frazer K.A., Cox D.R., Halperin E., Eskin E., Ballinger D.G., Frazer K.A., Cox D.R., Eskin E., Ballinger D.G., Frazer K.A., Cox D.R., Ballinger D.G., Frazer K.A., Cox D.R., Frazer K.A., Cox D.R., Cox D.R. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- Hoggart C.J., Chadeau-Hyam M., Clark T.G., Lampariello R., Whittaker J.C., De Iorio M., Balding D.J., Chadeau-Hyam M., Clark T.G., Lampariello R., Whittaker J.C., De Iorio M., Balding D.J., Clark T.G., Lampariello R., Whittaker J.C., De Iorio M., Balding D.J., Lampariello R., Whittaker J.C., De Iorio M., Balding D.J., Whittaker J.C., De Iorio M., Balding D.J., De Iorio M., Balding D.J., Balding D.J. Sequence-level population simulations over large genomic regions. Genetics. 2007;177:1725–1731. doi: 10.1534/genetics.106.069088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys A.J., Neumann R., Neumann R. Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat. Genet. 2002;31:267–271. doi: 10.1038/ng910. [DOI] [PubMed] [Google Scholar]
- Jobling M.A., Hurles M.E., Tyler-Smith C., Hurles M.E., Tyler-Smith C., Tyler-Smith C. Human evolutionary genetics: Origins, peoples & disease. Garland Sciences; Oxford: 2004. [Google Scholar]
- Kaplan N.L., Hudson R.R., Langley C.H., Hudson R.R., Langley C.H., Langley C.H. The “hitchhiking effect” revisited. Genetics. 1989;123:887–899. doi: 10.1093/genetics/123.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley J.L., Madeoy J., Calhoun J.C., Swanson W., Akey J.M., Madeoy J., Calhoun J.C., Swanson W., Akey J.M., Calhoun J.C., Swanson W., Akey J.M., Swanson W., Akey J.M., Akey J.M. Genomic signatures of positive selection in humans and the limits of the outlier approaches. Genome Res. 2006;16:980–989. doi: 10.1101/gr.5157306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A., Thorleifsson G., Stefansson H., Masson G., Helgason A., Gudbjartsson D.F., Jonsdottir G.M., Gudjonsson S.A., Sverrisson S., Thorlacius T., Thorleifsson G., Stefansson H., Masson G., Helgason A., Gudbjartsson D.F., Jonsdottir G.M., Gudjonsson S.A., Sverrisson S., Thorlacius T., Stefansson H., Masson G., Helgason A., Gudbjartsson D.F., Jonsdottir G.M., Gudjonsson S.A., Sverrisson S., Thorlacius T., Masson G., Helgason A., Gudbjartsson D.F., Jonsdottir G.M., Gudjonsson S.A., Sverrisson S., Thorlacius T., Helgason A., Gudbjartsson D.F., Jonsdottir G.M., Gudjonsson S.A., Sverrisson S., Thorlacius T., Gudbjartsson D.F., Jonsdottir G.M., Gudjonsson S.A., Sverrisson S., Thorlacius T., Jonsdottir G.M., Gudjonsson S.A., Sverrisson S., Thorlacius T., Gudjonsson S.A., Sverrisson S., Thorlacius T., Sverrisson S., Thorlacius T., Thorlacius T., et al. Sequence variants in the RNF212 gene associate with genome-wide recombination rate. Science. 2008;7:1398–1401. doi: 10.1126/science.1152422. [DOI] [PubMed] [Google Scholar]
- Lewontin R.C., Krakauer J., Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–195. doi: 10.1093/genetics/74.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Stephens M., Stephens M. Modelling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics. 2003;165:2213–2233. doi: 10.1093/genetics/165.4.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise T.C., Chen F., Chen W., De La Vega F.M., Hansen M., He C., Hyland F.C., Kennedy G.C., Kong X., Murray S.S., Chen F., Chen W., De La Vega F.M., Hansen M., He C., Hyland F.C., Kennedy G.C., Kong X., Murray S.S., Chen W., De La Vega F.M., Hansen M., He C., Hyland F.C., Kennedy G.C., Kong X., Murray S.S., De La Vega F.M., Hansen M., He C., Hyland F.C., Kennedy G.C., Kong X., Murray S.S., Hansen M., He C., Hyland F.C., Kennedy G.C., Kong X., Murray S.S., He C., Hyland F.C., Kennedy G.C., Kong X., Murray S.S., Hyland F.C., Kennedy G.C., Kong X., Murray S.S., Kennedy G.C., Kong X., Murray S.S., Kong X., Murray S.S., Murray S.S., et al. A second-generation combined linkage physical map of the human genome. Genome Res. 2007;17:1783–1786. doi: 10.1101/gr.7156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G. The structure of linkage disequilibrium around a selective sweep. Genetics. 2007;175:1395–1406. doi: 10.1534/genetics.106.062828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G., Spencer C.C., Spencer C.C. Scanning the human genome for positive selection. Curr. Opin. Genet. Dev. 2006;16:624–629. doi: 10.1016/j.gde.2006.09.004. [DOI] [PubMed] [Google Scholar]
- McVean G.A., Myers S.R., Hunt S., Deloukas P., Bentley D.R., Donnelly P., Myers S.R., Hunt S., Deloukas P., Bentley D.R., Donnelly P., Hunt S., Deloukas P., Bentley D.R., Donnelly P., Deloukas P., Bentley D.R., Donnelly P., Bentley D.R., Donnelly P., Donnelly P. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- Myers S., Bottolo L., Freeman C., McVean G., Donnelly P., Bottolo L., Freeman C., McVean G., Donnelly P., Freeman C., McVean G., Donnelly P., McVean G., Donnelly P., Donnelly P. A fine-scale map of recombination rates and recombination hotspots in the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- Myers S., Spencer C.C., Auton A., Bottolo L., Freeman C., Donnelly P., McVean G., Spencer C.C., Auton A., Bottolo L., Freeman C., Donnelly P., McVean G., Auton A., Bottolo L., Freeman C., Donnelly P., McVean G., Bottolo L., Freeman C., Donnelly P., McVean G., Freeman C., Donnelly P., McVean G., Donnelly P., McVean G., McVean G. The distribution and causes of meiotic recombination in the human genome. Biochem. Soc. Trans. 2006;34:526–530. doi: 10.1042/BST0340526. [DOI] [PubMed] [Google Scholar]
- Nielsen R. Statistical tests of selective neutrality in the age of genomics. Heredity. 2001;86:641–647. doi: 10.1046/j.1365-2540.2001.00895.x. [DOI] [PubMed] [Google Scholar]
- Nielsen R., Williamson S., Kim Y., Hubisz M.J., Clark A.G., Bustamante C., Williamson S., Kim Y., Hubisz M.J., Clark A.G., Bustamante C., Kim Y., Hubisz M.J., Clark A.G., Bustamante C., Hubisz M.J., Clark A.G., Bustamante C., Clark A.G., Bustamante C., Bustamante C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings P.S., Hermisson J., Hermisson J. Soft sweeps II: Molecular population genetics of adaptation from recurrent mutation or migration. Mol. Biol. Evol. 2006a;23:1076–1084. doi: 10.1093/molbev/msj117. [DOI] [PubMed] [Google Scholar]
- Pennings P.S., Hermisson J., Hermisson J. Soft Sweeps III: The signature of selection from recurrent mutation. PLoS Genet. 2006b;15:e186. doi: 10.1371/journal.pgen.0020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M., Coop G., Wall J.D., Coop G., Wall J.D., Wall J.D. The signature of positive selection on standing genetic variation. Evolution Int. J. Org. Evolution. 2005;59:2312–2323. [PubMed] [Google Scholar]
- Reed F.A., Tishkoff S.A., Tishkoff S.A. Positive selection can create false hotspots of recombination. Genetics. 2006;172:2011–2014. doi: 10.1534/genetics.105.052183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti P.C., Reich D.E., Higgins J.M., Levine H.Z., Richter D.J., Schaffner S.F., Gabriel S.B., Platko J.V., Patterson N.J., McDonald N.J., Reich D.E., Higgins J.M., Levine H.Z., Richter D.J., Schaffner S.F., Gabriel S.B., Platko J.V., Patterson N.J., McDonald N.J., Higgins J.M., Levine H.Z., Richter D.J., Schaffner S.F., Gabriel S.B., Platko J.V., Patterson N.J., McDonald N.J., Levine H.Z., Richter D.J., Schaffner S.F., Gabriel S.B., Platko J.V., Patterson N.J., McDonald N.J., Richter D.J., Schaffner S.F., Gabriel S.B., Platko J.V., Patterson N.J., McDonald N.J., Schaffner S.F., Gabriel S.B., Platko J.V., Patterson N.J., McDonald N.J., Gabriel S.B., Platko J.V., Patterson N.J., McDonald N.J., Platko J.V., Patterson N.J., McDonald N.J., Patterson N.J., McDonald N.J., McDonald N.J., et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- Sabeti P.C., Walsh E., Schaffner S.F., Varilly P., Fry B., Hutcheson H.B., Cullen M., Mikkelsen T.S., Roy J., Patterson N., Walsh E., Schaffner S.F., Varilly P., Fry B., Hutcheson H.B., Cullen M., Mikkelsen T.S., Roy J., Patterson N., Schaffner S.F., Varilly P., Fry B., Hutcheson H.B., Cullen M., Mikkelsen T.S., Roy J., Patterson N., Varilly P., Fry B., Hutcheson H.B., Cullen M., Mikkelsen T.S., Roy J., Patterson N., Fry B., Hutcheson H.B., Cullen M., Mikkelsen T.S., Roy J., Patterson N., Hutcheson H.B., Cullen M., Mikkelsen T.S., Roy J., Patterson N., Cullen M., Mikkelsen T.S., Roy J., Patterson N., Mikkelsen T.S., Roy J., Patterson N., Roy J., Patterson N., Patterson N., et al. The case for selection at CCR5-Δ32. PLoS Biol. 2005;3:e378. doi: 10.1371/journal.pbio.0030378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti P.C., Schaffner S.F., Fry B., Lohmueller J., Varilly P., Shamovsky O., Palma A., Mikkelsen T.S., Altshuler D., Lander E.S., Schaffner S.F., Fry B., Lohmueller J., Varilly P., Shamovsky O., Palma A., Mikkelsen T.S., Altshuler D., Lander E.S., Fry B., Lohmueller J., Varilly P., Shamovsky O., Palma A., Mikkelsen T.S., Altshuler D., Lander E.S., Lohmueller J., Varilly P., Shamovsky O., Palma A., Mikkelsen T.S., Altshuler D., Lander E.S., Varilly P., Shamovsky O., Palma A., Mikkelsen T.S., Altshuler D., Lander E.S., Shamovsky O., Palma A., Mikkelsen T.S., Altshuler D., Lander E.S., Palma A., Mikkelsen T.S., Altshuler D., Lander E.S., Mikkelsen T.S., Altshuler D., Lander E.S., Altshuler D., Lander E.S., Lander E.S. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- Sabeti P.C., Varilly P., Fry B., Lohmueller J., Hostetter E., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R., Varilly P., Fry B., Lohmueller J., Hostetter E., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R., Fry B., Lohmueller J., Hostetter E., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R., Lohmueller J., Hostetter E., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R., Hostetter E., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R., Xie X., Byrne E.H., McCarroll S.A., Gaudet R., Byrne E.H., McCarroll S.A., Gaudet R., McCarroll S.A., Gaudet R., Gaudet R., et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;18:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner S.F., Foo C., Gabriel S., Reich D., Daly M.J., Altshuler D., Foo C., Gabriel S., Reich D., Daly M.J., Altshuler D., Gabriel S., Reich D., Daly M.J., Altshuler D., Reich D., Daly M.J., Altshuler D., Daly M.J., Altshuler D., Altshuler D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliekelman P., Garner C., Slatkin M., Garner C., Slatkin M., Slatkin M. Natural selection and resistance to HIV. Nature. 2001;411:545–546. doi: 10.1038/35079176. [DOI] [PubMed] [Google Scholar]
- Smith N.G., Fearnhead P., Fearnhead P. A comparison of three estimators of the population-scaled recombination rate: Accuracy and robustness. Genetics. 2005;171:2051–2062. doi: 10.1534/genetics.104.036293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed T.P., Zhao H., Zhao H. Chromosome maps. In: Balding D.J., et al., editors. Handbook of statistical genetics, 3d ed. John Wiley and Sons; Hoboken, NJ: 2007. pp. 3–39. [Google Scholar]
- Spencer C.C., Coop G., Coop G. SelSim: A program to simulate population genetic data with natural selection and recombination. Bioinformatics. 2004;20:3673–3675. doi: 10.1093/bioinformatics/bth417. [DOI] [PubMed] [Google Scholar]
- Stephan W., Wiehe T., Lenz M.W., Wiehe T., Lenz M.W., Lenz M.W. The effect of strongly selected substitutions on neutral polymorphism: Analytical results from diffusion theory. Theor. Popul. Biol. 1992;42:237–254. [Google Scholar]
- Stumpf M.P., McVean G.A., McVean G.A. Estimating recombination rates from population-genetic data. Nat. Rev. Genet. 2003;4:959–968. doi: 10.1038/nrg1227. [DOI] [PubMed] [Google Scholar]
- Tang K., Thornton K.R., Stoneking M., Thornton K.R., Stoneking M., Stoneking M. A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 2007;5:e171. doi: 10.1371/journal.pbio.0050171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff S.A., Reed F.A., Ranciaro A., Voight B.F., Babbitt C.C., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M., Reed F.A., Ranciaro A., Voight B.F., Babbitt C.C., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M., Ranciaro A., Voight B.F., Babbitt C.C., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M., Voight B.F., Babbitt C.C., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M., Babbitt C.C., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M., Powell K., Mortensen H.M., Hirbo J.B., Osman M., Mortensen H.M., Hirbo J.B., Osman M., Hirbo J.B., Osman M., Osman M., et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 2007;39:31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight B.F., Kudaravalli S., Wen X., Pritchard J.K., Kudaravalli S., Wen X., Pritchard J.K., Wen X., Pritchard J.K., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir B.S., Cardon L.R., Anderson A.D., Nielsen D.M., Hill W.G., Cardon L.R., Anderson A.D., Nielsen D.M., Hill W.G., Anderson A.D., Nielsen D.M., Hill W.G., Nielsen D.M., Hill W.G., Hill W.G. Measures of human population structure show heterogeneity among genomic regions. Genome Res. 2005;15:1468–1476. doi: 10.1101/gr.4398405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiehe T., Stephan W., Stephan W. Analysis of genetic hitchhiking model and its applications to DNA Drosophila melanogaster. Mol. Biol. Evol. 1993;10:842–854. doi: 10.1093/oxfordjournals.molbev.a040046. [DOI] [PubMed] [Google Scholar]
- Williamson S.H., Hubisz M.J., Clark A.G., Paysuer B.A., Bustamante C.D., Nielsen R., Hubisz M.J., Clark A.G., Paysuer B.A., Bustamante C.D., Nielsen R., Clark A.G., Paysuer B.A., Bustamante C.D., Nielsen R., Paysuer B.A., Bustamante C.D., Nielsen R., Bustamante C.D., Nielsen R., Nielsen R. Localising recent adaptive evolution in the human genome. PLoS Genet. 2007;3:e90. doi: 10.1371/journal.pgen.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winckler W., Myers S.R., Richter D.J., Onofrio R.C., McDonald G.J., Bontrop R.E., McVean G.A., Gabriel S.B., Reich D., Donnelly P., Myers S.R., Richter D.J., Onofrio R.C., McDonald G.J., Bontrop R.E., McVean G.A., Gabriel S.B., Reich D., Donnelly P., Richter D.J., Onofrio R.C., McDonald G.J., Bontrop R.E., McVean G.A., Gabriel S.B., Reich D., Donnelly P., Onofrio R.C., McDonald G.J., Bontrop R.E., McVean G.A., Gabriel S.B., Reich D., Donnelly P., McDonald G.J., Bontrop R.E., McVean G.A., Gabriel S.B., Reich D., Donnelly P., Bontrop R.E., McVean G.A., Gabriel S.B., Reich D., Donnelly P., McVean G.A., Gabriel S.B., Reich D., Donnelly P., Gabriel S.B., Reich D., Donnelly P., Reich D., Donnelly P., Donnelly P., et al. Comparison of fine-scale recombination rates in humans and chimpanzees. Science. 2005;308:107–111. doi: 10.1126/science.1105322. [DOI] [PubMed] [Google Scholar]
- Yauk C.L., Boise P.R., Jeffreys A.J., Boise P.R., Jeffreys A.J., Jeffreys A.J. High-resolution sperm typing of meiotic recombination in the mouse MHC Eβ gene. EMBO J. 2003;22:1389–1397. doi: 10.1093/emboj/cdg136. [DOI] [PMC free article] [PubMed] [Google Scholar]