

Abstract
Histone modifications are major epigenetic factors regulating gene expression. They play important roles in maintaining stem cell pluripotency and in cancer pathogenesis. Different modifications may combine to form complex “histone codes.” Recent high-throughput technologies, such as “ChIP-chip” and “ChIP-seq,” have generated high-resolution maps for many histone modifications on the human genome. Here we use these maps to build a Bayesian network to infer causal and combinatorial relationships among histone modifications and gene expression. A pilot network derived by the same method among polycomb group (PcG) genes and H3K27 trimethylation is accurately supported by current literature. Our unbiased network model among histone modifications is also well supported by cross-validation results. It not only confirmed already known relationships, such as those of H3K27me3 to gene silencing, H3K4me3 to gene activation and the effect of bivalent modification of both H3K4me3 and H3K27me3, but also identified many other relationships that may predict new epigenetic interactions important in epigenetic gene regulation. Our automated inference method, which is potentially applicable to other ChIP-chip or ChIP-seq data analyses, provides a much-needed guide to deciphering the complex histone codes.
Histone methylation is one of the major types of chromatin modifications that are responsible for epigenetic regulation of gene expression. Modifications usually occur on the lysine residues at the N terminus of histones. Although different modifications are broadly associated with activation or repression of gene expression, their relationship to one another and their combinatorial function remain mysteries under intensive investigation (Berger 2007). Barski et al. (2007) have performed chromatin immunoprecipitation (ChIP) followed by high-throughput sequencing (ChIP-seq) in human T cells, using antibodies against 20 human histone lysine and arginine methylations, as well as histone variant H2A.Z, RNA polymerase II (Pol II), and the insulator binding protein CTCF, to map the genomic locations of these modifications and DNA/chromatin binding factors. This study not only confirmed the known associations of different modifications with gene expression, and discovered novel ones, but also provided an important resource for sorting out the logical relationships among these modifications. The binding sites are mapped at the whole genome level with single-nucleosome resolutions, providing more than enough data points and resolution to infer causal relationships among the modifications. Under such circumstances, robust Bayesian networks can be built to reveal the causal relationships.
The basic principle of a Bayesian network is to derive dependency among variables through examining the conditional probability and joint conditional probability distributions of different events. The final result is visualized in a directed acyclic graph (a graph without loops), where an edge from a source to a target node indicates that the occurrence of the target node depends on that of the source node (Needham et al. 2006). Under certain assumptions, edges in a Bayesian network can correspond to causal relationships. Here we used the WinMine package to derive statistical inference models (Chickering 2002), because it contains an improved algorithm to distinguish compelled versus reversible edges. Compelled edges correspond to causal influences, whereas reversible edges are not necessarily causal but might be merely correlated (Chickering 1995). We first tested the validity of the algorithms by applying them to a smaller-scale “ChIP-chip” (microarray after ChIP) data set (Boyer et al. 2006) where the causal relationships among nodes have been clearly demonstrated. We then applied the algorithms to the larger-scale “ChIP-seq” (sequencing after ChIP) data for 20 histone methylations and three other factors (Barski et al. 2007). The Bayesian network derived agrees with the clustering results among genes and histone modifications, as well as current literature about them. Some of the relationships have been tested previously in mammalian embryonic stem (ES) cells, fruitfly, or other organisms, supporting the validity of our model. Other relationships inferred from our model have not yet been tested experimentally and represent potentially new causal and/or combinatorial relationships. Such relationships provide a blueprint for mapping the complex “histone code.”
Results
Bayesian network to reconstruct causal relationships among polycomb complexes
To illustrate how Bayesian network can be used to derive causal relationships beyond simple correlations, we carried out a proof-of-concept analysis on a smaller data set of the same type. It has been found that trimethylation of lysine 27 on histone 3 (H3K27me3) at a gene in stem cells is dependent on polycomb repressive complex 2 (PRC2) binding. PRC2 comprises of the core proteins EED, SUZ12, and a methyl transferase EZH2 that directly catalyze the H3K27me3. Polycomb repressive complex 1 (PRC1) can then recognize the H3K27me3 and is recruited to the gene regulatory region and serves to stabilize H3K27me3 (Sparmann and van Lohuizen 2006). Based on these findings, we should expect a dependency of H3K27me3 modification on PRC2 binding and that PRC1 binding should depend on H3K27me3 modification. Boyer et al. (2006) have performed ChIP in mouse ES cells, followed by microarray analysis (ChIP-chip) to determine the genome-wide H3K27me3 modification sites and the binding sites of EED, SUZ12, and two PRC1 components RNF2 and PHC1. We focused on the simple network between EED, SUZ12, RNF2 binding, and H3K27me3 modification, using a data set provided by the authors for ∼20,000 genes with binding events discretized to 1 (binding) and 0 (no binding) values. A simple Venn diagram reveals the four factors share many targeting genes (Fig. 1A). Measuring the correlations among the four binding and modification events demonstrates that these events are tightly correlated, except for H3K27me3 (pairwise correlations can be found in Supplemental Table 1). However, none can indicate any causal relationships or dependency among these events. With Bayesian network analysis, we found that, as expected, the H3K27me3 modification is dependent on the binding of both EED and SUZ12, and that RNF2 binding is dependent on H3K27me3 modification as well as binding of EED and SUZ12 (Fig. 1B). It should be noted that only H3K27me3 has been demonstrated to directly bind PRC1, but an examination of the conditional probability distribution table for the RNF2 node indicates the binding of RNF2 is dependent on all three factors together and that no single factor or even pair of factors is predictive of RNF2 binding to the same gene (Fig. 1B). This is a new insight into the mechanism of PRC1 recruitment. The network structures are unchanged whether we based our analysis on the 1 kb upstream of and downstream from transcription start site (TSS), or 8 kb upstream of to 2 kb downstream from TSS. Furthermore, the relationships among the four factors do not change if ChIP data for another PRC1 component, PHC1, are included. Only three new edges connect to PHC1, from H3K27me3, EED, and RNF2. These suggest the network inferred is robust when subjected to minor variations. Unfortunately, when Chickering’s algorithm was applied to find the compelled edges in the network, none of the edges can be determined as causal relationships. This could be due to the fact that only a small number (five) of factors have been examined. If H3K27me3 is dependent on one additional factor that is independent of EED or SUZ12, all but EED → SUZ12 can be claimed as compelled edges or causal relationships (Supplemental Fig. 1). This is possible given that 51% of H3K27me3 signal has no overlap to EED or SUZ12 (Fig. 1A), and its profile is not highly correlated to those of the others (Supplemental Table 1).
Figure 1.

Inferring causal relationships of H3K27 trimethylation and binding of polycomb complexes. (A) Venn diagram visualizing the targeting genes shared by double, triple, or quadruple combinations among SUZ12, EED, RNF2, and H3K27me3. (B) Bayesian network inferred among the four factors. A directed edge denotes the occurrence of the target node is dependent on that of the source node, or that the occurrence of the source node is predictive of the target node. The probability distributions giving rise to the dependency for each node are given next to the node. For example, the conditional probability of RNF2 binding given the occurrence of H3K27me3, EED, and/or SUZ12 is listed below the RNF2 node.
Having ensured that a Bayesian network detects known and novel causal relationship on a smaller data set of the same type, we proceed to build one among all the 20 histone modifications and three other DNA binding factors. Compared with the Boyer’s data set, Barski’s data set is generated by direct Illumina sequencing (formerly Solexa sequencing) rather than microarray after ChIP. This latest technology (a.k.a. ChIP-seq) has been shown to have higher precision than ChIP-chip. The 30–50-nucleotide-(nt) long sequence reads of the ChIP DNA fragments are mapped onto the genome to determine the number of times a certain interval of genomic DNA is precipitated and detected. The output of the ChIP-seq experiments is digital sequence counts per interval of genomic DNA.
Two major groups of histone modifications relate to transcription activation and repression
Different histone modifications have been broadly categorized into either activating or repressing modifications for transcription of protein coding genes and are mainly associated with modifications in promoter regions of the genes (Martin and Zhang 2005). We also found that the histone modifications segregated into two large clusters, when their ChIP-seq counts within 1 kb upstream of and downstream from the TSS (TSS ± 1kb) of genes were analyzed by hierarchical clustering (see Methods) (Fig. 2A,B). The two clusters apparently correspond to transcription activating (Group A) and transcription repressing (Group R) binding/modifications, because (1) Group A contains binding of Pol II and H2A.Z that indicate active transcription, as well as known transcription activating modifications, such as H3K4 mono-, di-, and trimethylations, while Group R contains well-known transcription repressing modifications, H3K27me3, H3K9me3, and H3K20me3; and (2) protein-coding genes also segregate into two major clusters that are of high (Cluster H) and low (Cluster L) expression levels in T cells (Fig. 2B). Cluster H has high counts for Group A binding/modification and low counts for Group R, whereas Cluster L is the opposite. If genes are sorted by their expression levels in T cells, it is obvious that the genes having low expression level have more repressive marks and fewer activating marks (Fig. 2C, top), and the other way around for those having high expression level (Fig. 2C, middle). Other, less-well-understood modifications, such as H3K27me1, H3K9me1, H2BK5me1, and H4K20me1 modifications are included in Group A and H3K79me1, H3K79me2, H3K79me3, H3K20me3, and H4R3me2, etc. in Group R (Fig. 2B), suggesting that these modifications might be also associated with transcription activation and repression, respectively.
Figure 2.

Two major groups of genes regulated by histone modifications. (A) Based on the histone modification counts within 1 kb upstream of and downstream from the transcription start site (TSS), two major groups of modifications can be detected as two major clusters above the heat plot. One group contains the known transcription activating binding/modifications, such as Pol II, H2A.Z, H3K4 mono-, di-, and trimethylations, while the other contains the known transcription repressing modifications, such as H3K27me3, H3K9me3, H3K79me3, and H4K20me3. (B) When the expression levels in T cells are compared, genes in the group with activating modifications (Cluster H) and those in the other group with repressive modifications (Cluster L) are, respectively, 1.14 and 0.78 times of the average log2 transformed expression levels of all genes measured on the microarray (both P < 1.62 × 10−12). An asterisk above a bar indicates that the group’s mean value is not significantly different from the average, otherwise the difference is significant. (C) Genes with measured expression levels are arranged from top to bottom according to increasing expression levels in T cells, as indicated by the gradient arrow.
Consistent with Cluster H and L being transcriptionally activated and repressed genes, respectively, Cluster H is enriched in house-keeping genes and T-cell-specific genes, whereas Cluster L is enriched in tissue-specific genes and pathways for alternative cell fates (neuron, keratinocyte, muscle, skeletal, and epithelial cells), as indicated by gene ontology (GO) and KEGG pathway annotations (Table 1; Supplemental Tables 2, 3). Interestingly, genes with low expression levels have much more homogeneous modification profiles and clearer bound or unbound signals compared with highly expressed genes. This is especially evident when comparing the major subclusters in Clusters H and L (Fig. 2A, Clusters 3,10), or when sorting genes according to expression levels (Fig. 2C). The implication is that histone methylation might dominate in transcription repression, whereas transcription factors or other mechanisms might be more important to transcription activation. In agreement with this hypothesis, highly expressed genes nearly always have high Pol II binding rather than any particular histone modification, whereas those with the lowest expression levels are associated with H3K27me3 modification (Fig. 2C). Furthermore, our Bayesian network model reveals that H3K4me3 and other activating modifications influence gene expression indirectly through Pol II, whereas the H3K27me3 modification directly suppresses gene expression in addition to inhibiting Pol II binding (see below).
Table 1.
GO terms and KEGG pathways enriched in Cluster H and L

Only annotations that are enriched >1.5-fold over the average and with P < 10E–8 for GO or P < 10E–5 for KEGG are listed here. Full lists and more details are provided in Supplemental Tables 2 and 3.
Notably, two gene subclusters within Cluster H, Clusters 1 and 6, have lower than average expression levels. They both contain H3K27me3 modifications in addition to transcription activating modifications, suggesting that transcription repression function of H3K27me3 is dominant over the activating modifications (also see below). In ES cells, bivalent modifications (i.e., both transcription activating and transcription repressing) have been proposed to hold genes in a temporary silent state, “poised” for rapid activation upon removal of H3K27me3 (Berger 2007). In T cells, bivalent modifications are associated with genes with very diverse functions, including thromboxane receptor activity, ephrin receptor signaling, Rho protein signaling, and so on (Supplemental Tables 2, 3, Cluster 1, no enriched function annotations were found in Cluster 6). It is not clear whether these functions are poised for activation in T cells. In contrast to Clusters 1 and 6, subcluster 8 within Cluster L not only lacks the major transcription activating modifications (except H3K27me1, H3K4me1, H2BK5me1 and H4K20me1) but also the repressive modifications H3K27me3, H3K79me3, H3K9me3, and H4K20me3. Subcluster 8 has average expression level (Fig. 2B), and low, but not the lowest, level of Pol II binding (Fig. 2A). This subcluster may correspond to background or default transcription, and so not enriched for many function categories (Supplemental Tables 2, 3).
Bayesian network to infer causal relationships among histone modifications and gene expression
The binary division of the modifications does not imply that a single modification is sufficient to cause transcription activation or repression or that any of the modifications are necessary for gene expression regulation. Accumulating evidence suggests that the regulation of gene expression by histone modifications is not as simple as an on-off switch, but involves complex combinatorial effects, some times referred to as the “histone code” (Berger 2007). However, only the relationships of few modifications and how they affect gene expression have been solved so far. Here, we try to build a more comprehensive and unbiased model by inferring the causal relationships among various histone modifications, chromatin binding events and gene expression in a “gene-centric” Bayesian network, where high, medium, or low binding of a protein or occurrence of a modification (revealed by each ChIP) to each gene’s regulatory region is treated as an observable event.
To derive such a model, we first summed up the sequence counts within 1 kb upstream of and downstream from each gene’s TSS (TSS ± 1kb) so that one gene has one count for a single histone modification or binding factor. We then discretized the sequence counts for each of the 17,757 genes to three levels, low, medium, and high, by an unsupervised learning method, the k-means clustering algorithm (see Methods) (Fig. 3A; Supplemental Table 4). To incorporate the gene expression data, only the 12,078 genes that have both expression measurements and ChIP data are used to infer Bayesian networks. To extract the relationships to gene expression, an additional node “gene expression” was introduced, where each gene’s expression level is discretized into low, medium and high based on its expression level in the T cells (see Methods) (Fig. 3A; Table 2). Alternatively, we also discretized a gene expression value relative to the gene’s overall expression distribution among 79 human tissues (see Methods) (Su et al. 2004). Although the two different discretization methods give rise to very different classification of gene expressions (Table 2; Supplemental Table 5), the Bayesian network models generated are exactly the same. Removing one to six modifications or factors weakly associated with gene expression also does not perturb the rest of the network (data not shown). Both of the above results support the robustness of the model in addition to the cross-validation results (see below). We also explored other k values in k-means clustering for data discretization. At k = 5, the k-means algorithm can no longer generate clustering result for H3K79me2 ChIP data set. At k = 3, the network is the most robust and performs the best according to the cross-validation (see below) results compared with k = 2 or 4 levels (Supplemental Fig. 2). We therefore presented the Bayesian network based on the k = 3 results.
Figure 3.

Causal relationships among histone modifications and gene expression. (A) Flowchart of a Bayesian network construction using sequence counts within TSS ± 1kb. See text for details. (B) The coverage and accuracy of models derived from sequence counts within TSS ± 600bp, TSS ± 1kb, and TSS ± 2kb. For each N (an integer from one to 10) nine out of 10 group combinations, the models’ accuracy and coverage are calculated generating a curve for each sequence range used to construct the models. We performed random grouping 100 times, and hence, the coverage and accuracy at each N is the average of 100 trials. The vertical and horizontal bars on the curve denote the standard deviations of accuracy and coverage at each point. (C) The common Bayesian network (see text for details) consisted of only compelled edges agreed by all 100 trials. The model is based on the sequence counts in TSS ± 1kb. The edge colors indicate the correlations (measured by Pearson correlation coefficient [PCC]) among the various modification/binding factors; nodes are colored by their correlation to gene expression. Colors are scaled as shown in the color legend. The edge directions have the same meanings as in Fig. 1B. (D) The causal relationships in the Bayesian network model are not expected by shuffled sequence counts among genes for each ChIP. Comparing to that of the real data, when the sequence counts are shuffled among genes, each Bayesian network contains zero compelled edge. Each point on a curve represents the average results of 100 tests or 100 simulations, with the vertical bars on the curve denoting the standard deviations at each point.
Table 2.
Boundaries used to define low, medium and high gene expression levels

Within T-cell levels are based on all genes’ expression levels in T cells, and the cross-tissue expression levels are based on a gene’s expression level in T cells compared with those in other tissues.
aCross-tissue expression level boundaries for each gene are listed in Supplemental Table 5.
To ensure the robustness of the Bayesian network generated, we randomly partitioned the genes into 10 nonoverlapping groups. We then used each nine-group combination among the 10 groups (i.e., leave one group out) to train a Bayesian network (Fig. 3A). In order to generate testable predictions for causal relationships, all the reversible edges were removed from each inferred network, and only the compelled edges were kept. The Bayesian networks generated by each of the nine-group combinations were then compared with the common network agreed by N combinations, where N is an integer from 1 to 10. As there is no positive training data set, the overlap to the common network serves as a surrogate for robustness measurement. We define the accuracy as the percentage of edges identified in each network that are also found in the common network, and the coverage as the percentage of edges in the common network that are identified by a particular network model. We performed random grouping 100 times and repeated the cross-validation described above on each trial of 10 random groups to derive average and standard deviation of cross-validation accuracy and coverage (Fig. 3A,B). We found that the boundary around TSS can affect the robustness of the network models. When the models’ coverage is plotted against their accuracy, it is clear that the models derived from TSS ± 1kb regions has the highest area under the curve (AUC) over those derived from TSS ± 600bp or TSS ± 2kb regions (Fig. 3B). We therefore adopted the overlapping network closest to the upper right corner of the coverage ∼ accuracy plot. This is the common network agreed by seven of the 10 models derived from sequence counts within TSS ± 1kb (Fig. 3B). Among the 100 trials of obtaining best 10-fold cross-validated common network, 32 compelled edges are common to all 100 trials, which in total inferred 37 compelled edges. These 32 inferred causal relationships connect 22 out of the 24 binding/modification events examined by ChIP (Fig. 3C). In contrast, if the ChIP sequence counts were permuted among different genes, each network model contained on average only three reversible edges and no compelled edge (Fig. 3D), indicating the large network size and the overlap among the different models could not be due to random chance (empirical P < 0.001) (see Methods).
To visualize the modes or signs of action (i.e., activation or suppression) among modifications and gene expressions, the edges in the network are colored according to the level of correlations between the two nodes linked by the edge, and the nodes are colored by their correlation to gene expression (see Methods) (Fig. 3C). Pearson correlation coefficient (PCC) between the two vectors of sequence counts for the 17,757 genes identified by the two ChIPs (nodes) is used to measure the correlation between a pair of ChIPs. PCC between a vector of ChIP sequence counts for the 12,078 genes (only the genes that have expression measurements) and another vector of gene expression values of the genes is used to measure the correlation between a ChIP and gene expression. If we consider the anti-correlation between the two modifications together with the conditional dependency as an inhibitory effect, and correlation plus conditional dependency as activating effect, H3K27me3 is predicted to be the strongest and the only direct inhibitory factor causal to gene expression, whereas H3K4me3 has the strongest activating effect but indirectly through Pol II (Fig. 3C).
The network is hierarchical with approximately four levels. As expected, the gene expression node is at the bottom of the hierarchy with one direct activating effect from Pol II and one direct suppressing effect from H3K27me3 (Fig. 3C). H4K20me3, a hallmark of heterochromatin (Schotta et al. 2004; Talasz et al. 2005), a transcription repressing modification (H3K27me3), and three transcription activating modifications (H3K4me1, H3K4me2, and H3K9me1) are at the top of the hierarchy.
A chain of three nodes negatively associated with gene expression, H3K9me3 → H3K9me2 → H3K79me2, is downstream of H3K27me3 and H4K20me3 (Fig. 3C). Three other nodes, H3K27me2, H4R3me2, and H3K36me3, are also negatively correlated with gene expression, with H4R3me2 dependent on H3K27me2 and H3K4me3, and H3K36me3 on H3K4me3 only (Fig. 3C). The rest of the nodes, including all the monomethylations, H3K4me3, Pol II, H2A.Z, and CTCF, are all directly or indirectly associated and positively correlated with gene expression. Among them, a chain of causal relationships formed among four monomethylations, H3K4me1 → H2BK5me1 → H3K36me1 → H3K79me1, seems to be significantly longer than expected (P = 0.044 assuming normal distribution) (see Methods) (Fig. 3C). The biological meaning of this observation is currently unknown.
The modifications or binding events, such as H2A.Z and CTCF binding at the bottom of the cascade, are not predicted to be causal to gene expression (Fig. 3C).
Existing experimental support for the inferred relationships
Since epigenetic modifications reflect the gene expression status in a particular tissue and state, the same gene is likely to be modified differently in distinct tissues or conditions. Even the methyltransferase or demethylase complexes catalyzing the modifications might be different. However, the relationship of each modification to gene expression status, and the relationships among various modifications present in T cells might reflect their interaction patterns in general toward forming the histone code and therefore might be the same or similar in different tissues or under different conditions.
Ohm et al. (2007) have observed that cancer stem cells possess two additional repressive modifications, H3K9me2 and H3K9me3, besides the H3K27me3 seen in normal ES cells and proposed that these might lead to heritable gene suppression in tumor cells. In agreement with their observation, we found that H3K27me3, together with H4K20me3, a modification involved in DNA repair (Sanders et al. 2004), is predicted to be causal to H3K9me3, which may in turn lead to H3K9me2 (Fig. 3C). It is therefore interesting to test if H4K20me3 formation during DNA repair synergizes with H3K27me3 to create H3K9me3 and H3K9me2 and facilitate the transition from normal to cancer stem cells. Current literature already points to such a possibility: In Tetrahymena, H3K27me3 has been demonstrated to regulate H3K9 methylations (Liu et al. 2007); H4K20 methylation has been suggested to further stabilize polycomb complex binding in addition to H3K27me3 (Schwartz and Pirrotta 2007).
Consistent with the many results with polycomb gene mutations (Schuettengruber et al. 2007; Schwartz and Pirrotta 2007), H3K4me3 and H3K27me3 assume central roles in the network, with the highest out degrees (number of edges pointing away from the node) (Fig. 3C). H3K4me3 is known as a strong transcription activating modification (Bernstein et al. 2002; Kim et al. 2005; Barski et al. 2007; Berger 2007) that may serve as a “memory” mark to reinforce future histone modifications and transcription on the marked genes (Ng et al. 2003; Martin and Zhang 2005). Our model puts H3K4me3 downstream of H3K4me1 and H3K4me2, suggesting a directional equilibrium among mono-, di-, and trimethyl H3K4. It is also downstream of H3K9me1 and upstream of CTCF, H2A.Z, Pol II, H3K36me3, and H4R3me2. The causal effect of the trxG complex (responsible for catalyzing H3K4me3) to Pol II binding has been demonstrated by direct genetic experiments (Schuettengruber et al. 2007; Schwartz and Pirrotta 2007). For example, in polytene chromosomes of fruitflies mutant for the trxG gene kis, the level of elongating Pol II decreases dramatically (Srinivasan et al. 2005). Mutations in the mouse trx counterpart Mll1 affect Pol II binding level and distribution on the Hoxa9 gene promoter (Milne et al. 2005)
Our model also predicts that H3K4me3 might inhibit the formation of H3K36me3 and H4R3me2 (Fig. 3C). The inhibition of H3K36me3 by H3K4me3 is supported by various experimental results. H3K4me3 and H3K36me3 seem to have mutually exclusive localizations, H3K4me3 peaking at the 5′ end of genes, H3K36me3 at the 3′ end, and tail gradually into each other’s territory (Li et al. 2007). H3K4me3 is known to recruit either a transcription activating complex, which leads to recruitment of the SAGA complex, transcription initiation, and elongation, or transcription repression complexes such as Sin3-Hdac1 and JMJD2A (Berger 2007). JMJD2A is a demethylase for H3K36me3 (Huang et al. 2006; Tsukada et al. 2006; Berger 2007). Thus, the inferred causal effect of H3K4me3 to H3K36me3 might correspond to the biochemical relationships of H3K4me3 → JMJD2A → H3K36me3.
H3K27me3 strongly suppresses expression (Boyer et al. 2006; Lee et al. 2006; Roh et al. 2006). Our model predicts that, in addition to being inhibitory to Pol II and gene expression, H3K27me3 also inhibits H2BK5me1. In ES cells, developmental genes or genes involved in cell differentiation are bivalently modified with both H3K4me3 and H3K27me3 (Szutorisz et al. 2005; Bernstein et al. 2006). This bivalent modification is believed to repress the expression of developmental genes in ES cells but makes them poised for rapid activation upon removal of H3K27me3 (Berger 2007). Our model supports this postulate through the epistatic relationship of the two modifications: H3K27me3 clearly has a dominant role over H3K4me3 by inhibiting both Pol II and its ultimate target gene expression (Fig. 3C), thus the end result of the bivalent modification is repression instead of activation. Mechanistically, the inhibition of Pol II and gene expression by H3K27me3 might be due to a reduced DNA accessibility, caused by the polycomb complexes (Fitzgerald and Bender 2001), or a block to RNA synthesis by Pol II (Dellino et al. 2004). It is still controversial about which of the two mechanisms is correct, because the former lacks strong in vivo evidence and the latter has only been shown on genes that are prebound by Pol II or “pre-set” genes (Schwartz and Pirrotta 2007). Our model indicates that both of these mechanisms might be at work in vivo. In our model the H3K27me3 → Pol II relationship is consistent with the first mechanism, where Pol II binding is prohibited by H3K27me3. The second scenario of gene expression inhibition by H3K27me3 in the presence of prebound Pol II may correspond to the direct H3K27me3 → gene expression relationship in our model. In ES cells, the bivalently modified genes probably need the latter mechanism to achieve a dominant “off” state, while maintaining an open chromatin structure (Bernstein et al. 2007).
Consistent with the histone codes hypothesis, many modifications are predicted to be the result of combinatorial upstream modification events. For example, Pol II binding is predicted to be the combinatorial result of H3K4me3 and H3K27me3 modifications (Fig. 3C). This is supported by the antagonistic effects between the trxG and PcG complexes (which catalyze H3K4me3 and H3K27me3, respectively) on Pol II binding and transcription activity (Schuettengruber et al. 2007; Schwartz and Pirrotta 2007). The detailed joint conditional probabilities for each node can be found in Supplemental Table 6.
Although some nodes, such as the insulator CTCF (Meneghini et al. 2003) and anti-silencer H2A.Z binding, are strongly correlated with gene expression, our model inferred that CTCF and H2A.Z are influenced by H3K4me3 synergistically with Pol II or H3K4me2 and that they do not directly influence gene expression (Fig. 3C). This is consistent with the fact that both are enriched at the insulator sites that limit the spread of gene activation (Bruce et al. 2005; Barski et al. 2007).
In summary, the causal relationships among histone modifications revealed by our Bayesian network model indicate that various histone modifications form a hierarchical cascade, and in combination regulate gene expression. The causal relationships in many cases reflect sequential events during dynamic chromatin remodeling and gene regulation. As expected, H3K4me3 and H3K27me3 are the strongest transcription activating and repressing modifications.
Discussion
Although many other modifications are strongly correlated or anti-correlated with gene expression, they are not directly causal to gene expression regulation and correlate more weakly to gene expressions than H3K4me3 and H3K27me3 do. The other modifications might stabilize these major two modifications (as most are dependent on the two modification “hubs”), be by-products of transcription, or be required for other processes, such as epigenetic inheritance and higher-order organization of chromosomes.
Due to the strict requirement of acyclic structure in the Bayesian networks, bidirectional interactions are bound to be missed in at least one direction. For example, there is evidence that initial Pol II binding to a TSS can recruit the trxG complex (Ng et al. 2003; Martin and Zhang 2005), which then keeps the chromatin structure in a transcription active mode to allow for more Pol II binding to occur (Schuettengruber et al. 2007). In our model, we only identified the H3K4me3 → Pol II direction, probably because this direction can be detected at higher frequency than the initial Pol II binding step. Similarly the initial Pol II binding is also able to recruit a demethylase to H3K27me3 (Smith et al. 2008). The positive feedback loops formed by the bidirectional interactions might reinforce a signal of transcription action or repression and thus create “memory” on the genes.
The new relationships predicted by our model may also be very important for solving the histone code. For example, a rarely studied modification, H2BK5me1, is predicted to have a central role in relaying information from H3K27me3 and H3K4me1 to H3K36me1, H3R2me2, and H4K20me1 (Fig. 3C). It should be noted that even the direct causal relationships predicted in the Bayesian network model may not correspond to direct biochemical interactions. They are instead more likely to correspond to epistatic genetic relationships. Genetic mutant evidence is therefore important in validating the predicted relationships.
Although the accuracy of the model can only be experimentally validated after a significant number of the inferred causal relationships have been tested, individual or a small number of the relationships can be independently subjected to experimental examination. To facilitate experimental testing, we expanded the network model with known methyltransferases and demethylases that potentially govern the state of methylation on a specific histone site (Supplemental Fig. 3; Supplemental Table 7). This provides a mapping between the model and possible ways of perturbing the network and testing the model. For example, knowing that trxG genes create H3K4me3 and JARID1B and C destroy it, one might perturb H3K4me3 by knocking out/down either its methyltransferase (trxG genes) or demethylase (JARID1B and C) and then examine the binding of Pol II to the promoter of genes. According to the model, we would expect that down-regulating trxG proteins will decrease Pol II binding, whereas down-regulating JARID1B and C will increase Pol II binding on genes that have H3K4me3 at their promoter regions, such as the HOX genes. These results have already been demonstrated in kis mutant Drosophila melanogaster and in Mll1 mutant mouse cell lines (Milne et al. 2005; Srinivasan et al. 2005). Other inferred causal interactions can be tested similarly, when specific methyltransferase and demethylase have been found. To test a predicted synergistic effect, such as H4K20me3 and H3K27me3 together lead to H3K9me3, one can examine if H3K9me3 is formed at the promoter regions of genes that normally have only H3K27me3 when H4K20me3 is increased by overexpression of its methyltransferase WHSC1 (also known as MMSET) (Marango et al. 2008). However, knocking down/out an H4K20me3 demethylase might be more convincing, when such an enzyme is identified. It should also be noted that tissue and state specificities of the modification enzymes and redundancies among them may complicate the experimental results.
Since some of the relationships we found in T cells are the same as those regulating stem cell pluripotency and differentiation as well as cancer pathogenesis, the model we inferred from T cells may also provide clues for gene regulation mechanisms in these processes.
From the methodology perspective, our pipeline of integrating various ChIP and gene expression data to infer causal relationships among chromatin-associated factors/modifications and gene expression adds a powerful and much-needed tool for analyzing the ever-increasing ChIP-chip and ChIP-seq data.
Methods
Data sets
The PcG and H3K27me3 ChIP-chip data were obtained from Boyer et al. (2006). The 20 histone modification and three other factor binding ChIP-seq data were obtained from Barski et al. (2007). RefSeq version 35 downloaded on March 10, 2005 was used to determine the genomic coordinate of the sequences in the ChIP-seq data. Gene expression data containing the expression intensity of ∼15,000 genes in 79 human tissues measured by DNA microarray was obtained from Su et al. (2004). GO annotations were downloaded from ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/ on September 1, 2007. KEGG pathway annotations were obtained from ftp://ftp.genome.jp/pub/kegg/xml/organisms/hsa/ on April 25, 2007.
Bayesian network inference
We used the WinMine package (http://research.microsoft.com/~dmax/winmine/tooldoc.htm) to calculate joint conditional probability and build the preliminary potential Bayesian networks. Bayesian network structure is searched by the following algorithms (Heckerman et al. 1995): First define a function S(xi|πi) where xi represents the current node and πi represents all the parental nodes of χi. The function is a custom-defined evaluation function that is only related to the current node and its parental nodes. It can take many forms. The most basic utilizes conditional probability. When searching for structures in an acyclic direct graph, a weight is defined for edge xi to xj as w(xi,xj) = logS(xi|xj) − logS(xi|Φ), where Φ is a node set containing no edges. Because the weights w(xi,xj) and w(xj,xi) are different, the directionality of an edge can thus be determined; that is, the one maximizing 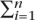 w(χi|πi) will be adopted. Thus for a certain network structure, the sum of the total weight of all its edges can be calculated as s(χi|πi) = w(χi|πi) − s(χi|Φ). Because s(χi|Φ) is a constant, searching for the best network structure maximizes the value for w(χi|πi). However, this is a NP-hard problem that can be only approximated by a heuristic search method. It uses a greedy algorithm to search the maximal value for w(χi|πi). The algorithm starts with an initial state corresponding to a network model containing no edge. It does not require an input order of the nodes for searching, and the network generated is not dependent on the order of nodes on the input list. As for all greedy algorithms, the maximal value might be trapped at a local maximum. We then implemented an algorithm of compelled edge identification as previously described (Chickering 1995) to find causal relationships.
w(χi|πi) will be adopted. Thus for a certain network structure, the sum of the total weight of all its edges can be calculated as s(χi|πi) = w(χi|πi) − s(χi|Φ). Because s(χi|Φ) is a constant, searching for the best network structure maximizes the value for w(χi|πi). However, this is a NP-hard problem that can be only approximated by a heuristic search method. It uses a greedy algorithm to search the maximal value for w(χi|πi). The algorithm starts with an initial state corresponding to a network model containing no edge. It does not require an input order of the nodes for searching, and the network generated is not dependent on the order of nodes on the input list. As for all greedy algorithms, the maximal value might be trapped at a local maximum. We then implemented an algorithm of compelled edge identification as previously described (Chickering 1995) to find causal relationships.
GO term and KEGG pathway enrichment
GO terms were first filtered as described previously (Xia et al. 2006). Then, GO term and KEGG pathway enrichment was determined by Fisher exact test followed by Benjamini-Hochberg correction (Benjamini and Hochberg 1995) for multiple hypothesis testing on all the GO terms tested in each gene set.
Allocating sequence counts to gene regulatory regions
Barski et al. (2007) have provided the ChIP results as sequence counts per 200 bp or 400 bp (only for Pol II and H2A.Z) intervals. Counts in 400-bp intervals were first equally divided into two contiguous 200-bp intervals. Only when more than half of an interval is within a certain regulatory region, e.g., TSS ± 1kb, we allocate the sequence counts in the interval to the regulatory region. Sequence counts in all the intervals thus determined are then summed up for each gene.
Clustering genes using histone modification profiles
We first filtered for genes that have nonzero counts for at least three ChIPs, then used a hierarchical clustering algorithm implemented in Cluster 3.0 (Eisen et al. 1998) to group the histone modifications and genes. Sequence counts within TSS ± 1kb for each gene were first adjusted by log transformation, median centering genes, normalizing genes, median centering samples, and normalizing samples. Then, hierarchical uncentered correlation and centroid linkage were used for clustering in both gene and sample dimensions or only for the samples if genes are sorted by their expression levels. The clustering results were visualized in JavaTreeView 1.0.12 (Saldanha 2004).
Discretization of ChIP signal intensities and gene expression values
Using k-means clustering algorithm implemented in Cluster 3.0 with k = 3 and 100 repeats, a ChIP signal intensity on each gene was categorized into low, medium, or high based on the sequence counts for the ChIP within the regulatory regions of 17,757 genes. The T-cell expression levels of 12,849 genes that have been measured on DNA microarray are similarly discretized based on either genes expression intensity within T cells or across tissues. When the input gene expression levels for each gene among 79 tissues are used for k-means clustering (tissue-wise comparison), the discretization considers tissue-specific expression for the same gene, which usually has the same promoter in different tissues. When the T-cell expression levels of all genes are used as the input gene expression levels (gene-wise comparison), the discretization compares genes of different promoters and assumes a more general role of histone modification in gene expression regulation. Discretization using other k values (from 2, 4, and 5) was similarly performed.
Randomizing sequence counts among genes for a ChIP
To examine if the Bayesian network derived can be expected randomly, we randomly assigned the low, medium, and high discretized values for each ChIP among the 12,078 genes, keeping the same value distribution as the original ChIP data. We generated 100 such randomized data sets. For each data set, we generated Bayesian networks exactly the same way as for the real data. The overlaps of networks generated by 10 different nine out of 10 group combinations were also examined. The results of the 100 simulations were compared to the network generated using the real data.
Generating random networks to evaluate the significance of a path length
One thousand networks of the same in and out degree distributions as the one shown in Figure 3C were randomly constructed using an algorithm described by Milo et al. (2002). Then the longest single directional chains of mono-, di-, and trimethylations were searched in each network to determine the P values of obtaining a chain of mono-, di-, and trimethylations as long as or longer than those found in the real model.
Acknowledgments
We thank Dr. Nicholas Baker at Albert Einstein College of Medicine for editing the manuscript and Dr. Yi Zhang at University of North Carolina for helpful suggestions. This work was supported by grants from the China National Science Foundation (grant nos. 30588001 and 30620120433), Chinese Ministry of Science and Technology (grant no. 2006CB910700), and funds from the Chinese Academy of Sciences to J.-D.J.H.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.073080.107.
References
- Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K., Wang Z., Wei G., Chepelev I., Zhao K., Wei G., Chepelev I., Zhao K., Chepelev I., Zhao K., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y., Hochberg Y.1995Controlling the false discovery rate—A practical and powerful approach to multiple testing J. R. Stat. Soc. Ser. B Methodolog. 57289 -300 [Google Scholar]
- Berger S.L. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Bernstein B.E., Humphrey E.L., Erlich R.L., Schneider R., Bouman P., Liu J.S., Kouzarides T., Schreiber S.L., Humphrey E.L., Erlich R.L., Schneider R., Bouman P., Liu J.S., Kouzarides T., Schreiber S.L., Erlich R.L., Schneider R., Bouman P., Liu J.S., Kouzarides T., Schreiber S.L., Schneider R., Bouman P., Liu J.S., Kouzarides T., Schreiber S.L., Bouman P., Liu J.S., Kouzarides T., Schreiber S.L., Liu J.S., Kouzarides T., Schreiber S.L., Kouzarides T., Schreiber S.L., Schreiber S.L. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. 2002;99:8695–8700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Fry B., Meissner A., Wernig M., Plath K., Meissner A., Wernig M., Plath K., Wernig M., Plath K., Plath K., et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bernstein B.E., Meissner A., Lander E.S., Meissner A., Lander E.S., Lander E.S. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Boyer L.A., Plath K., Zeitlinger J., Brambrink T., Medeiros L.A., Lee T.I., Levine S.S., Wernig M., Tajonar A., Ray M.K., Plath K., Zeitlinger J., Brambrink T., Medeiros L.A., Lee T.I., Levine S.S., Wernig M., Tajonar A., Ray M.K., Zeitlinger J., Brambrink T., Medeiros L.A., Lee T.I., Levine S.S., Wernig M., Tajonar A., Ray M.K., Brambrink T., Medeiros L.A., Lee T.I., Levine S.S., Wernig M., Tajonar A., Ray M.K., Medeiros L.A., Lee T.I., Levine S.S., Wernig M., Tajonar A., Ray M.K., Lee T.I., Levine S.S., Wernig M., Tajonar A., Ray M.K., Levine S.S., Wernig M., Tajonar A., Ray M.K., Wernig M., Tajonar A., Ray M.K., Tajonar A., Ray M.K., Ray M.K., et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- Bruce K., Myers F.A., Mantouvalou E., Lefevre P., Greaves I., Bonifer C., Tremethick D.J., Thorne A.W., Crane-Robinson C., Myers F.A., Mantouvalou E., Lefevre P., Greaves I., Bonifer C., Tremethick D.J., Thorne A.W., Crane-Robinson C., Mantouvalou E., Lefevre P., Greaves I., Bonifer C., Tremethick D.J., Thorne A.W., Crane-Robinson C., Lefevre P., Greaves I., Bonifer C., Tremethick D.J., Thorne A.W., Crane-Robinson C., Greaves I., Bonifer C., Tremethick D.J., Thorne A.W., Crane-Robinson C., Bonifer C., Tremethick D.J., Thorne A.W., Crane-Robinson C., Tremethick D.J., Thorne A.W., Crane-Robinson C., Thorne A.W., Crane-Robinson C., Crane-Robinson C. The replacement histone H2A.Z in a hyperacetylated form is a feature of active genes in the chicken. Nucleic Acids Res. 2005;33:5633–5639. doi: 10.1093/nar/gki874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chickering D.M. A transformational characterization of equivalent bayesian network structures. In: Besnard P., Hanks S., Hanks S., editors. Proceedings of 11th Conference on Uncertainty in Artificial Intelligence. Morgan Kaufmann Publishers; San Mateo, CA: 1995. pp. 87–98. [Google Scholar]
- Chickering D.M. Microsoft Research Technical Report MSR-TR-2002-103. Microsoft; Redmond, WA: 2002. The WinMine toolkit. [Google Scholar]
- Dellino G.I., Schwartz Y.B., Farkas G., McCabe D., Elgin S.C., Pirrotta V., Schwartz Y.B., Farkas G., McCabe D., Elgin S.C., Pirrotta V., Farkas G., McCabe D., Elgin S.C., Pirrotta V., McCabe D., Elgin S.C., Pirrotta V., Elgin S.C., Pirrotta V., Pirrotta V. Polycomb silencing blocks transcription initiation. Mol. Cell. 2004;13:887–893. doi: 10.1016/s1097-2765(04)00128-5. [DOI] [PubMed] [Google Scholar]
- Eisen M.B., Spellman P.T., Brown P.O., Botstein D., Spellman P.T., Brown P.O., Botstein D., Brown P.O., Botstein D., Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald D.P., Bender W., Bender W. Polycomb group repression reduces DNA accessibility. Mol. Cell. Biol. 2001;21:6585–6597. doi: 10.1128/MCB.21.19.6585-6597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckerman D., Geiger D., Chickering D.M., Geiger D., Chickering D.M., Chickering D.M. Learning Bayesian networks: The combination of knowledge and statistical data. Mach. Learn. 1995;20:197–243. [Google Scholar]
- Huang Y., Fang J., Bedford M.T., Zhang Y., Xu R.M., Fang J., Bedford M.T., Zhang Y., Xu R.M., Bedford M.T., Zhang Y., Xu R.M., Zhang Y., Xu R.M., Xu R.M. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- Kim T.H., Barrera L.O., Zheng M., Qu C., Singer M.A., Richmond T.A., Wu Y., Green R.D., Ren B., Barrera L.O., Zheng M., Qu C., Singer M.A., Richmond T.A., Wu Y., Green R.D., Ren B., Zheng M., Qu C., Singer M.A., Richmond T.A., Wu Y., Green R.D., Ren B., Qu C., Singer M.A., Richmond T.A., Wu Y., Green R.D., Ren B., Singer M.A., Richmond T.A., Wu Y., Green R.D., Ren B., Richmond T.A., Wu Y., Green R.D., Ren B., Wu Y., Green R.D., Ren B., Green R.D., Ren B., Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.I., Jenner R.G., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K., Jenner R.G., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K., Chevalier B., Johnstone S.E., Cole M.F., Isono K., Johnstone S.E., Cole M.F., Isono K., Cole M.F., Isono K., Isono K., et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Carey M., Workman J.L., Carey M., Workman J.L., Workman J.L. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Liu Y., Taverna S.D., Muratore T.L., Shabanowitz J., Hunt D.F., Allis C.D., Taverna S.D., Muratore T.L., Shabanowitz J., Hunt D.F., Allis C.D., Muratore T.L., Shabanowitz J., Hunt D.F., Allis C.D., Shabanowitz J., Hunt D.F., Allis C.D., Hunt D.F., Allis C.D., Allis C.D. RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes & Dev. 2007;21:1530–1545. doi: 10.1101/gad.1544207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marango J., Shimoyama M., Nishio H., Meyer J.A., Min D.J., Sirulnik A., Martinez-Martinez Y., Chesi M., Bergsagel P.L., Zhou M.M., Shimoyama M., Nishio H., Meyer J.A., Min D.J., Sirulnik A., Martinez-Martinez Y., Chesi M., Bergsagel P.L., Zhou M.M., Nishio H., Meyer J.A., Min D.J., Sirulnik A., Martinez-Martinez Y., Chesi M., Bergsagel P.L., Zhou M.M., Meyer J.A., Min D.J., Sirulnik A., Martinez-Martinez Y., Chesi M., Bergsagel P.L., Zhou M.M., Min D.J., Sirulnik A., Martinez-Martinez Y., Chesi M., Bergsagel P.L., Zhou M.M., Sirulnik A., Martinez-Martinez Y., Chesi M., Bergsagel P.L., Zhou M.M., Martinez-Martinez Y., Chesi M., Bergsagel P.L., Zhou M.M., Chesi M., Bergsagel P.L., Zhou M.M., Bergsagel P.L., Zhou M.M., Zhou M.M., et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111:3145–3154. doi: 10.1182/blood-2007-06-092122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C., Zhang Y., Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- Meneghini M.D., Wu M., Madhani H.D., Wu M., Madhani H.D., Madhani H.D. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell. 2003;112:725–736. doi: 10.1016/s0092-8674(03)00123-5. [DOI] [PubMed] [Google Scholar]
- Milne T.A., Dou Y., Martin M.E., Brock H.W., Roeder R.G., Hess J.L., Dou Y., Martin M.E., Brock H.W., Roeder R.G., Hess J.L., Martin M.E., Brock H.W., Roeder R.G., Hess J.L., Brock H.W., Roeder R.G., Hess J.L., Roeder R.G., Hess J.L., Hess J.L. MLL associates specifically with a subset of transcriptionally active target genes. Proc. Natl. Acad. Sci. 2005;102:14765–14770. doi: 10.1073/pnas.0503630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U., Itzkovitz S., Kashtan N., Chklovskii D., Alon U., Kashtan N., Chklovskii D., Alon U., Chklovskii D., Alon U., Alon U. Network motifs: Simple building blocks of complex networks. Science. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- Needham C.J., Bradford J.R., Bulpitt A.J., Westhead D.R., Bradford J.R., Bulpitt A.J., Westhead D.R., Bulpitt A.J., Westhead D.R., Westhead D.R. Inference in Bayesian networks. Nat. Biotechnol. 2006;24:51–53. doi: 10.1038/nbt0106-51. [DOI] [PubMed] [Google Scholar]
- Ng H.H., Robert F., Young R.A., Struhl K., Robert F., Young R.A., Struhl K., Young R.A., Struhl K., Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Ohm J.E., McGarvey K.M., Yu X., Cheng L., Schuebel K.E., Cope L., Mohammad H.P., Chen W., Daniel V.C., Yu W., McGarvey K.M., Yu X., Cheng L., Schuebel K.E., Cope L., Mohammad H.P., Chen W., Daniel V.C., Yu W., Yu X., Cheng L., Schuebel K.E., Cope L., Mohammad H.P., Chen W., Daniel V.C., Yu W., Cheng L., Schuebel K.E., Cope L., Mohammad H.P., Chen W., Daniel V.C., Yu W., Schuebel K.E., Cope L., Mohammad H.P., Chen W., Daniel V.C., Yu W., Cope L., Mohammad H.P., Chen W., Daniel V.C., Yu W., Mohammad H.P., Chen W., Daniel V.C., Yu W., Chen W., Daniel V.C., Yu W., Daniel V.C., Yu W., Yu W., et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh T.Y., Cuddapah S., Cui K., Zhao K., Cuddapah S., Cui K., Zhao K., Cui K., Zhao K., Zhao K. The genomic landscape of histone modifications in human T cells. Proc. Natl. Acad. Sci. 2006;103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha A.J. Java Treeview—Extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Sanders S.L., Portoso M., Mata J., Bahler J., Allshire R.C., Kouzarides T., Portoso M., Mata J., Bahler J., Allshire R.C., Kouzarides T., Mata J., Bahler J., Allshire R.C., Kouzarides T., Bahler J., Allshire R.C., Kouzarides T., Allshire R.C., Kouzarides T., Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Schotta G., Lachner M., Sarma K., Ebert A., Sengupta R., Reuter G., Reinberg D., Jenuwein T., Lachner M., Sarma K., Ebert A., Sengupta R., Reuter G., Reinberg D., Jenuwein T., Sarma K., Ebert A., Sengupta R., Reuter G., Reinberg D., Jenuwein T., Ebert A., Sengupta R., Reuter G., Reinberg D., Jenuwein T., Sengupta R., Reuter G., Reinberg D., Jenuwein T., Reuter G., Reinberg D., Jenuwein T., Reinberg D., Jenuwein T., Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes & Dev. 2004;18:1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B., Chourrout D., Vervoort M., Leblanc B., Cavalli G., Chourrout D., Vervoort M., Leblanc B., Cavalli G., Vervoort M., Leblanc B., Cavalli G., Leblanc B., Cavalli G., Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Schwartz Y.B., Pirrotta V., Pirrotta V. Polycomb silencing mechanisms and the management of genomic programs. Nat. Rev. Genet. 2007;8:9–22. doi: 10.1038/nrg1981. [DOI] [PubMed] [Google Scholar]
- Smith E.R., Lee M.G., Winter B., Droz N.M., Eissenberg J.C., Shiekhattar R., Shilatifard A., Lee M.G., Winter B., Droz N.M., Eissenberg J.C., Shiekhattar R., Shilatifard A., Winter B., Droz N.M., Eissenberg J.C., Shiekhattar R., Shilatifard A., Droz N.M., Eissenberg J.C., Shiekhattar R., Shilatifard A., Eissenberg J.C., Shiekhattar R., Shilatifard A., Shiekhattar R., Shilatifard A., Shilatifard A. Drosophila UTX is a histone H3 Lys27 demethylase that colocalizes with the elongating form of RNA polymerase II. Mol. Cell. Biol. 2008;28:1041–1046. doi: 10.1128/MCB.01504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann A., van Lohuizen M., van Lohuizen M. Polycomb silencers control cell fate, development, and cancer. Nat. Rev. Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- Srinivasan S., Armstrong J.A., Deuring R., Dahlsveen I.K., McNeill H., Tamkun J.W., Armstrong J.A., Deuring R., Dahlsveen I.K., McNeill H., Tamkun J.W., Deuring R., Dahlsveen I.K., McNeill H., Tamkun J.W., Dahlsveen I.K., McNeill H., Tamkun J.W., McNeill H., Tamkun J.W., Tamkun J.W. The Drosophila trithorax group protein Kismet facilitates an early step in transcriptional elongation by RNA Polymerase II. Development. 2005;132:1623–1635. doi: 10.1242/dev.01713. [DOI] [PubMed] [Google Scholar]
- Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Zhang J., Soden R., Hayakawa M., Kreiman G., Soden R., Hayakawa M., Kreiman G., Hayakawa M., Kreiman G., Kreiman G., et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szutorisz H., Canzonetta C., Georgiou A., Chow C.M., Tora L., Dillon N., Canzonetta C., Georgiou A., Chow C.M., Tora L., Dillon N., Georgiou A., Chow C.M., Tora L., Dillon N., Chow C.M., Tora L., Dillon N., Tora L., Dillon N., Dillon N. Formation of an active tissue-specific chromatin domain initiated by epigenetic marking at the embryonic stem cell stage. Mol. Cell. Biol. 2005;25:1804–1820. doi: 10.1128/MCB.25.5.1804-1820.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talasz H., Lindner H.H., Sarg B., Helliger W., Lindner H.H., Sarg B., Helliger W., Sarg B., Helliger W., Helliger W. Histone H4-lysine 20 monomethylation is increased in promoter and coding regions of active genes and correlates with hyperacetylation. J. Biol. Chem. 2005;280:38814–38822. doi: 10.1074/jbc.M505563200. [DOI] [PubMed] [Google Scholar]
- Tsukada Y., Fang J., Erdjument-Bromage H., Warren M.E., Borchers C.H., Tempst P., Zhang Y., Fang J., Erdjument-Bromage H., Warren M.E., Borchers C.H., Tempst P., Zhang Y., Erdjument-Bromage H., Warren M.E., Borchers C.H., Tempst P., Zhang Y., Warren M.E., Borchers C.H., Tempst P., Zhang Y., Borchers C.H., Tempst P., Zhang Y., Tempst P., Zhang Y., Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Xia K., Xue H., Dong D., Zhu S., Wang J., Zhang Q., Hou L., Chen H., Tao R., Huang Z., Xue H., Dong D., Zhu S., Wang J., Zhang Q., Hou L., Chen H., Tao R., Huang Z., Dong D., Zhu S., Wang J., Zhang Q., Hou L., Chen H., Tao R., Huang Z., Zhu S., Wang J., Zhang Q., Hou L., Chen H., Tao R., Huang Z., Wang J., Zhang Q., Hou L., Chen H., Tao R., Huang Z., Zhang Q., Hou L., Chen H., Tao R., Huang Z., Hou L., Chen H., Tao R., Huang Z., Chen H., Tao R., Huang Z., Tao R., Huang Z., Huang Z., et al. Identification of the proliferation/differentiation switch in the cellular network of multicellular organisms. PLoS Comput. Biol. 2006;2:e145. doi: 10.1371/journal.pcbi.0020145. [DOI] [PMC free article] [PubMed] [Google Scholar]