

Abstract
Accurately defining the coding potential of an organism, i.e., all protein-encoding open reading frames (ORFs) or “ORFeome,” is a prerequisite to fully understand its biology. ORFeome annotation involves iterative computational predictions from genome sequences combined with experimental verifications. Here we reexamine a set of Saccharomyces cerevisiae “orphan” ORFs recently removed from the original ORFeome annotation due to lack of conservation across evolutionarily related yeast species. We show that many orphan ORFs produce detectable transcripts and/or translated products in various functional genomics and proteomics experiments. By combining a naïve Bayes model that predicts the likelihood of an ORF to encode a functional product with experimental verification of strand-specific transcripts, we argue that orphan ORFs should still remain candidates for functional ORFs. In support of this model, interstrain intraspecies genome sequence variation is lower across orphan ORFs than in intergenic regions, indicating that orphan ORFs endure functional constraints and resist deleterious mutations. We conclude that ORFs should be evaluated based on multiple levels of evidence and not be removed from ORFeome annotation solely based on low sequence conservation in other species. Rather, such ORFs might be important for micro-evolutionary divergence between species.
Comparative genomics, involving homology searching of genome sequences between evolutionarily related species, is a powerful tool for predicting functional regions in a genome sequence without prior biological knowledge. To date, complete genome sequences are available for more than 500 different organisms across all three domains of life (Liolios et al. 2006). Comparative genomics of bacteria, yeast, worm, fly, and human have led to extensive revision of complete sets of predicted protein-encoding open reading frames (ORFs), or “ORFeomes” (McClelland et al. 2000; Brachat et al. 2003; Cliften et al. 2003; Kellis et al. 2003; Stein et al. 2003; Clamp et al. 2007; Clark et al. 2007). Removal from earlier versions of predicted ORFeomes of ORFs that are poorly or not conserved in other species (“orphan ORFs”) is a critical revision proposed by these comparative genomic studies. The principle underlying removal of orphan ORFs is that selective constraints on functional DNA sequences should prevent deleterious mutations from occurring (Hardison 2003).
However, lack of evolutionary conservation does not guarantee lack of functional significance. It may be imprudent to eliminate putative ORFs from predicted ORFeomes solely based on lack of cross-species conservation. Different species, no matter how evolutionarily close, might express distinct ORF products. In support of this possibility, the pilot Encyclopedia of DNA Elements (ENCODE) project on 1% of the human genome has revealed that experimentally identified functional elements are not necessarily evolutionary constrained (Birney et al. 2007). In addition, although evolutionary conservation implies functionality for the product of a predicted ORF, experimental validation is required to demonstrate its biological significance. Therefore, cautious experimental reinvestigation of the functionality of predicted ORFs is needed to improve the accuracy of genome annotation.
To this end we set out to examine potential functionality of orphan ORFs in Saccharomyces cerevisiae based on available experimental evidence. Three independent comparative genomic analyses (Brachat et al. 2003; Cliften et al. 2003; Kellis et al. 2003) have predicted 648 annotated ORFs as “spurious” or “false,” representing 10% of originally annotated ORFs. Notably, 10 out of these 648 orphan ORFs have since been validated as functional by small-scale experiments. For example, although YDR504C lacks clear orthologs in other yeast species, its deletion causes lethality upon exposure to high temperature while in stationary phase (Martinez et al. 2004). Given the time-consuming efforts of traditional “one-gene-at-a-time” inquiries, many predicted ORFs have not been individually characterized. However, as the first sequenced eukaryotic organism, S. cerevisiae has been used intensively for functional genomics and proteomics studies, providing valuable functional evidence that allow further evaluation of coding potential of the orphan ORFs.
Using currently available functional genomics and proteomics data sets, we collate functional evidence for a significant portion of S. cerevisiae orphan ORFs, finding that many orphan ORFs produce detectable transcripts and/or translated products. Using a naïve Bayes model, we predict the likelihood that any S. cerevisiae ORF encodes a functional product and show that the number of orphan ORFs with potential functional significance is higher than expected by chance. Notably, we provide experimental verification for strand-specific transcription of many orphan ORFs. Finally, we report that interstrain intraspecies genome sequence variation is lower across orphan ORFs than in intergenic regions. Altogether our results demonstrate that orphan ORFs should not be excluded from current ORFeome annotation simply because they fail to show interspecies sequence conservation. We suggest that orphan ORFs should be included in future genome-wide experimental studies to reveal their bona fide identity either as functional ORFs or as randomly occurring misannotated ORFs.
Results
Evidence for biological significance of S. cerevisiae orphan ORFs
The genome annotation of S. cerevisiae has undergone continuous modification through computational and experimental efforts since the original release in 1996 (Goffeau et al. 1996; Fisk et al. 2006). Three independent comparative genomic analyses compared the conservation of DNA or predicted protein sequences among several ascomycete species (Brachat et al. 2003; Cliften et al. 2003; Kellis et al. 2003), recommending that 402, 513, and 495 ORFs, respectively, be removed from the S. cerevisiae predicted ORFeome because their putative counterparts in other yeast species accumulate stop codons and frame-shift mutations (Fig. 1A). The union of these three comparative analyses is a set of 648 orphan ORFs called “spurious” or “false” in these studies (Fig. 1A).
Figure 1.

Experimental evidence for S. cerevisiae orphan ORFs. (A) Percentages indicate proportions of orphan ORFs detected at least in one of 13 functional genomics and proteomics data sets (Table 1). Note that ORFs rejected by all three comparative genomic studies analyzed here (Brachat et al. 2003; Cliften et al. 2003; Kellis et al. 2003) show similar percentages. (B) Supporting experimental evidence for each of 648 ORFs observed as orphan by three comparative genomic studies (Brachat et al. 2003; Cliften et al. 2003; Kellis et al. 2003). Complete lists of ORFs and supporting experimental evidence are in Supplemental Table 2. Columns are ordered from the ORF with most evidence (ORF X1; left) to the one with the least evidence (ORF X648; right). Data sets were grouped together by type of experimental approach, transcriptional on top and translational at the bottom. In total, there are 477 orphan ORFs with transcriptional evidence, 180 with translational evidence, and 145 with both transcriptional and translational evidence.
High-throughput functional genomics and proteomics approaches have recently accelerated functional characterization of predicted ORFs. Several of these genome-wide approaches, such as gene-expression profiling or in vivo characterization of protein complexes, have detected transcripts or translated products of orphan ORFs. For example, in a proteome-wide purification of yeast protein complexes (Krogan et al. 2006), 85 proteins identified by mass spectrometry were encoded by orphan ORFs.
To provide a systematic reanalysis of S. cerevisiae orphan ORFs, we collected 13 large-scale studies (Table 1) informing on either transcription or translation of orphan ORFs. The transcriptome studies included tiling arrays (David et al. 2006), high-density Affymetrix chip analysis (Holstege et al. 1998), SAGE analysis (Velculescu et al. 1997), and cDNA sequencing (Miura et al. 2006). Because many (69%) of the orphan ORFs overlap with another annotated ORF, we only included transcriptome studies able to detect strand-specific transcripts. Protein–protein interaction studies included proteome-scale yeast two-hybrid screens (Uetz et al. 2000; Ito et al. 2001) and affinity pull-downs of tagged proteins followed by mass spectrometry (Gavin et al. 2002, 2006; Ho et al. 2002; Krogan et al. 2006). For yeast two-hybrid studies, we considered an ORF being translated only if its product was involved in a protein–protein interaction as a prey. Protein expression studies included global surveys of protein abundance (Ghaemmaghami et al. 2003) and subcellular localization (Kumar et al. 2002; Huh et al. 2003).
Table 1.
Thirteen functional genomics and proteomics data sets integrated in our analysis

Out of the 648 orphan ORFs, most (79%) have been detected in at least one of these data sets. The proportion of orphan ORFs detected was nearly the same for ORFs rejected by each of the three comparative genomics analyses independently (80% for Brachat, 79% for Cliften, and 79% for Kellis) and for the 276 orphan ORFs discarded by all three (79%) (Fig. 1A). Among the 648 orphan ORFs, many were detected by more than one approach. In total, 145 orphan ORFs (22%) were both detected as transcripts and translated products (Fig. 1B). A similar distribution of functional evidence was observed for the orphan ORFs rejected by all three comparative genomic analyses (Supplemental Fig. 1).
Evaluating biological significance of S. cerevisiae ORFs by a naïve Bayes approach
High-throughput approaches have inherently limited coverage (not all ORFs are detectable) and precision (detection of some ORFs might be artifactual). Therefore information from large-scale data sets needs to be accepted cautiously. We chose a naïve Bayes model to quantify the observations reported above, because this approach can integrate dissimilar types of data sets into a common probabilistic framework with maximal coverage and precision (Jansen et al. 2003; Yu et al. 2004). By use of such an integration scheme, evidence (i.e., features) from several data types can be accumulated to estimate with increasing confidence the likelihood that an ORF encodes a functional product.
As with any machine learning algorithm, naïve Bayes models need a training set of gold standard positives (GSPs) and negatives (GSNs). The Saccharomyces Genome Database (SGD), the arbiter of genome annotation for budding yeasts, has categorized all S. cerevisiae ORFs into three major groups based on conservation across species and on available experimental characterization: “verified” (4449 ORFs), “uncharacterized” (1333 ORFs), and “dubious” (823 ORFs) (Fisk et al. 2006). Both verified ORFs and uncharacterized ORFs are conserved across species. Verified ORFs have clear small-scale experimental evidence for the existence of functional ORF products, but uncharacterized ORFs do not. Dubious ORFs are thought not to encode a functional product due to (1) lack of conservation across species, and/or (2) absence of any small-scale experiment demonstrating detectable mRNA or protein production or phenotypic effects. We used all 4449 verified ORFs as the GSPs and all 823 dubious ORFs as the GSNs. Although ideally the GSNs should be depleted of functional ORFs, this cannot exactly be true for the dubious set. However, the dubious set is likely enriched with nonfunctional ORFs. It is common practice to use an “enriched” set of negatives in training data sets (Miller et al. 2005; Xia et al. 2006).
We calculated the ratio of the fraction of GSPs present in each of the 13 functional genomics and proteomics data sets divided by the fraction of GSNs present in each data set, which measures the confidence levels (Supplemental Table 1). The product of these ratios of the 13 data sets for each ORF is defined as the likelihood ratio (LR) of an ORF, i.e., the likelihood of each ORF to encode a functional product (see Methods). We used the base 10 logarithmic form of LR (LLR) as final prediction scores (Supplemental Table 2). Out of the large-scale studies integrated, several did measure similar biological features of ORFs and ORF products. However, we treated all 13 data sets as independent features, due to the low correlation between them (Supplemental Tables 3, 4).
To evaluate the performance of the naïve Bayes model, we used threefold cross-validation (see Methods). After randomly dividing both the GSPs and GSNs into three separate equal sets, we used two of the three sets as the training set to calculate LLRs and the remaining set as the test set to identify positives and negatives. The true-positive rate (TP rate: fraction of GSPs that are predicted to be functional) and the false-positive rate (FP rate: fraction of GSNs that are predicted to be functional) were calculated at different LLR cutoffs. The resulting couplets (TP rate–FP rate) were used to plot a receiver operating characteristic (ROC) curve. We ran this process three times so that each of the three sets was a test set and the remaining two constituted the training set. Each ROC curve looked similar (Supplemental Fig. 2), which validated the overall quality of our training set. A final ROC curve was plotted by using potential LLR cutoffs from all three training subsets and their associated TP rate and FP rate based on the predictions from the complete training set (Fig. 2A). The significant deviation of the final ROC curve from the 45° random ROC line indicates that our model has substantial predictive value (area under ROC curve = 0.982). To assess the contribution of each data set to the final prediction scores, we successively omitted one data set and repeated the training and cross-validation procedures. We plotted ROC curves for all procedures (Supplemental Fig. 3) and observed little difference when excluding any single data set. Thus it seems that no single data set dominates the prediction.
Figure 2.

Evaluating functionality of S. cerevisiae ORFs. (A) ROC curve (blue) for naïve Bayes predictions based on 13 functional genomics and proteomics data sets. The diagonal (black dotted line) is the expected ROC curve for random, where the TP rate equals the FP rate. The two LLR cutoffs highlighted on the curve were used later as thresholds for categorizing orphan ORFs. (B) All 6718 S. cerevisiae ORFs were divided into 20 bins by decreasing LLR. Each bin has similar numbers of ORFs. The false-positive rates associated with the minimum LLR in each bin are listed. Distributions of verified ORFs, orphan dubious ORFs, “other” dubious ORFs, and all other ORFs in each bin are shown. Orphan dubious ORFs tend to have a higher LLR than ORFs classified as dubious for other reasons.
We divided all 6718 S. cerevisiae ORFs into 20 bins ranked by decreasing LLR, with each bin containing similar numbers of ORFs. Verified ORFs localized mostly in the higher LLR bins (92.5% of all verified ORFs distributed between bin 1 and bin 15), while dubious ORFs localized in lower LLR bins (only 4.98% of dubious ORFs distributed between bin 1 and bin 15) (Fig. 2B). Such segregation between verified ORFs and dubious ORFs was expected, given that the ORFs used in the training as GSPs (verified ORFs) are bound to have a higher LLR than the ones used in the training as GSNs (dubious ORFs). An unanticipated result of the naïve Bayes predictions is that orphan dubious ORFs have overall higher LLR (P < 10−15 by Mann-Whitney U test) (Fig. 2B) than ORFs classified as dubious for reasons other than strict lack of interspecies sequence conservation (e.g., a mutant phenotype described for the ORF could be ascribed to mutation of an overlapping well-characterized ORF) (Fisk et al. 2006). This suggests that orphan dubious ORFs might be more likely to encode functional products than “other” dubious ORFs.
For an ORF to be considered “most-likely” functional in our naïve Bayes predictions, its posterior odds (the product of the prior odds and the likelihood ratio) has to be larger than 1 (see Methods). We can estimate that the prior odds for any given ORF to be most-likely functional is ∼5.4 (4449 GSPs divided by 823 GSNs). Hence, we used LLR = log10(1/5.4) = −0.7 (FP rate = 0.07) as the cutoff for an ORF to be most-likely functional (bins 1–15). Among the 648 orphan ORFs, 54 ORFs with LLR ≥ −0.7 were thus assigned to a set of most-likely functional orphan ORFs. Although the percentage of verified ORFs decreased significantly from bin 16 to bin 20 compared with the first 15 bins (Fig. 2B), there were still 3.4% and 2.5% of verified ORFs (152 and 111 ORFs) in bins 16 and 17, respectively. We classified the 199 orphan ORFs in bins 16 and 17, with an LLR between −0.7 (FP rate = 0.07) and −3.1 (FP rate = 0.32), as “moderately-likely” to encode a functional product. The remaining 395 orphan ORFs distributed between bins 18 and 20 were called “least-likely” functional ORFs. Detectability limitations in the large-scale data sets integrated in our predictions may have biased against these least-likely ORFs. Integration of new lines of experimental evidence in the future could still potentially identify promising functional ORF candidates among the least-likely ORFs.
Experimental evidence for expression of S. cerevisiae orphan ORFs
We next experimentally measured mRNA expression for orphan ORFs using reverse transcription–polymerase chain reaction (RT-PCR) (Fig. 3A). Strand specificity was needed to ensure that the transcripts detected were transcribed from the predicted DNA strand and to exclude artifacts caused from read-through transcription on the opposite strand (Craggs et al. 2001).
Figure 3.

Two-step strand-specific RT-PCR. (A) Schematic diagram of the strand-specific RT-PCR procedure. (B) Electrophoretic analysis of strand-specific RT-PCR products. Reverse ORF-specific primers (OSPR), with sequences complementary to the ORF-coding strand, were used for first-strand cDNA synthesis. Second-step PCR amplifications used a pair of forward (OSPF) and reverse ORF-specific primers (OSPR). As controls, the first step of RT-PCR was performed without reverse transcriptase for detecting contamination by genomic DNA, or without the OSPR primer for detecting residual reverse transcriptase activity in second-step PCR reactions. Two intron-containing verified ORFs, YER133W (genomic DNA length: 1464 bp; coding sequence length: 939 bp) and YBR078W (genomic DNA length: 1737 bp; coding sequence length: 1407 bp), were used to test the strand specificity. An extra control for these two verified ORFs was a standard PCR action using yeast genomic DNA as template and the same pair of ORF-specific primers. The observed difference in the length of PCR products amplified from genomic DNA versus poly(A) mRNA manifested the strand specificity. Strand-specific RT-PCR results of 201 nonoverlapping orphan ORFs were analyzed on 1% agarose E-gel (Invitrogen). Of the reactions 53% (105 ORFs) gave rise to visible RT-PCR products of the expected sizes. Three orphan ORFs, YJL199C (327 bp), YJR108W (372 bp), and YDR344C (444 bp), are shown as examples of successful RT-PCR reactions. (C) Comparison of the average LLR between nonoverlapping ORFs detected and undetected by strand-specific RT-PCR. Error bars, SEM.
We tested strand specificity on two verified S. cerevisiae ORFs that both contain introns: YER133W (GLC7) and YBR078W (ECM33) (see Methods). Given the presence of introns in these ORFs, the sense-strand transcripts should be appreciably shorter in length than the antisense-strand transcripts. Spliced transcripts of the expected sizes were obtained in reactions where strand-specific primer was added for cDNA synthesis (Fig. 3B). No RT-PCR products were obtained in reactions without RT, demonstrating absence of contaminating genomic DNA in the poly(A) mRNA template preparation. No RT-PCR products were observed in the absence of cDNA primer for first-strand cDNA synthesis, demonstrating that the second step of standard PCR amplification contained no active reverse transcriptase for the synthesis of incorrect strand cDNA from antisense strand–specific primer. The identities of RT-PCR products were confirmed by sequencing.
Thereafter we applied our strand-specific RT-PCR to 201 orphan ORFs that do not overlap any other annotated ORF. The requirement for nonoverlap further reduces the false-positive rate, because it is less likely that there would be any transcription from the incorrect strand. Among 201 nonoverlapping orphan ORFs tested under conditions of growth on rich media, RT-PCR products of expected size were obtained for 105 ORFs (Supplemental Table 2). Although the available supporting experimental evidence for these 105 ORFs is not strikingly different from the ORFs whose transcripts were not detected by strand-specific RT-PCR (Supplemental Fig. 4), the detected ORFs have a significantly higher average LLR (−3.4 ± 0.2) than the ones undetected by RT-PCR (−3.8 ± 0.2, P = 0.03 by Mann-Whitney U test) (Fig. 3C), demonstrating the validity and robustness of our predictions for positives. In particular, YJL199C, a dubious ORF, has the highest LLR among 201 tested ORFs and was detected by RT-PCR. YJL199C was recently predicted to encode a metabolic protein based on large-scale protein–protein interaction studies (Samanta and Liang 2003).
Notably, out of 49 orphan ORFs tested that had not been detected by any of the 13 data sets (Table 1), 29 were expressed (Supplemental Table 2), among which YPR096C was recently found to encode a ribosome-interacting protein (Fleischer et al. 2006) and YOR235W was shown through a genome-wide phenotypic analysis to be involved in DNA recombination events (Alvaro et al. 2007). Therefore, we suggest that more experimentation is needed before rejecting ORFs from the S. cerevisiae ORFeome annotation.
Interstrain intraspecies sequence conservation for S. cerevisiae orphan ORFs
The available experimental evidence from large-scale data sets, combined with our experimental support for many orphan ORFs, implies that lack of interspecies conservation does not necessarily dispel the bona fide functionality of an ORF. Functional orphan ORFs may have a relaxed selective constraint due to their dispensable roles in other species and may therefore rapidly lose sequence similarity even in closely related species (Schmid and Aquadro 2001). However, select species–specific functions may stringently constrain sequence divergence of functional orphan ORFs within species (Domazet-Loso and Tautz 2003). Therefore, we examined the intraspecies conservation of orphan ORFs in S. cerevisiae, using single nucleotide polymorphism (SNP) information from genome resequencing of multiple strains of S. cerevisiae by the Saccharomyces Genome Resequencing Project (SGRP) (http://www.sanger.ac.uk/Teams/Team71/durbin/sgrp/index.shtml). Among the 37 currently available strain sequences, four (SK1, W303, Y55, and DBVPG6765) have been sequenced at twofold coverage or higher. We used the SNP data from these four genomes to assess nucleotide variation in different genomic regions across S. cerevisiae strains. We compared nucleotide divergence among three genomic features: orphan ORFs, nonorphan ORFs, and intergenic regions, considering only the regions that do not overlap with any other annotated ORF (see Methods). Across the four strains analyzed, orphan ORFs showed higher nucleotide divergence (7.0 ± 0.4 SNPs per kb) than did nonorphan ORFs (3.7 ± 0.1 SNPs per kb, P < 10−5 by Mann-Whitney U test), but less than intergenic regions (15.5 ± 0.2 SNPs per kb, P < 10−15 by Mann-Whitney U test) (Fig. 4A). Such intermediate nucleotide divergence for orphan ORFs suggests that at least a portion of them are subject to significant intraspecies evolutionary constraints. Such “interstrain intraspecies” conservation of orphan ORFs indicates potential functionality of an ORF in addition to experimental evidence.
Figure 4.

Interstrain intraspecies sequence conservation for orphan ORFs. (A) Distribution of nucleotide divergence in different genomic features. We binned three types of genomic features, (1) non-orphan ORFs (red curve), (2) orphan ORFs predicted by three comparative genomic analyses (blue curve) (Brachat et al. 2003; Cliften et al. 2003; Kellis et al. 2003), and (3) intergenic regions (green curve), using a window of an average three SNPs per kb across four S. cerevisiae strains. Each dot represents the fraction of genomic features in each bin. Numbers on the X-axis represent the maximum number of SNPs per kb in each bin. For instance the first bin collects the genomic regions that have between zero and three SNPs per kb in four strains. The inset zooms in on the 0–21 SNPs per kb range with SEM displayed. (B) Comparison of nucleotide divergence among three predicted categories of orphan ORFs based on their LLRs. Error bars, SEM in each category. (C) Comparison of the percentage of ORFs among the three predicted categories of orphan ORFs that have reading frames preserved across four S. cerevisiae strains.
Among the 648 orphan ORFs, the most-likely functional ones displayed a significantly lower nucleotide divergence (3.8 ± 0.7 SNPs per kb) than both moderately-likely (6.4 ± 0.8 SNPs per kb, P = 0.016 by Mann-Whitney U test) and least-likely ORFs (7.7 ± 0.6 SNPs per kb, P = 0.005 by Mann-Whitney U test) (Fig. 4B). Although the moderately-likely category does have a lower nucleotide divergence than least-likely category, the difference is not significant (P > 0.05 by Mann-Whitney U test). Because different types of SNPs, such as synonymous or nonsynonymous substitutions, could have distinct effects on an ORF product, we applied another test to compare sequence conservation among the three groups, measuring the percentage of ORFs with preserved reading frames (absence of stop codons or frame-shift mutations) across all four S. cerevisiae strains. A decreasing trend was observed from most-likely to least-likely ORFs (Fig. 4C), with significant differences among the three categories (P = 0.03 by χ2 test). The coexistence of high interstrain intraspecies conservation with high likelihood of functionality demonstrates that some orphan ORFs face functional constraints that protect them from deleterious intraspecies mutations.
In summary, analysis of nucleotide variation in multiple S. cerevisiae strains, combined with multiple lines of experimental evidence, suggest that reevaluation of the functionality of all ORFs, especially orphan ORFs, is warranted.
Discussion
We report here that many interspecies nonconserved ORFs or orphan ORFs predicted by comparative genomic analyses in S. cerevisiae show evidence of transcription or translation, as reported in various functional genomics or proteomics data sets. We used a naïve Bayes probabilistic integration of a heterogeneous set of large-scale data sets to predict the likelihood that a predicted ORF encodes a functional product. Threefold cross-validation demonstrated high performance for this approach, which revealed that orphan ORFs are more likely functional than are ORFs classified as dubious for reasons other than strict lack of sequence conservation across species. Independent strand-specific RT-PCR confirmed that many orphan ORFs are indeed expressed. Although presence of transcripts is not sufficient by itself to conclude that an ORF encodes a functional product, the correspondence between our RT-PCR results and naïve Bayes prediction scores demonstrated both the potential functionality of orphan ORFs and the robustness of our prediction method. Confirming that orphan ORFs could be functional, many show signs of interstrain intraspecies negative selection, such as lower nucleotide divergence than intergenic regions and retaining an intact reading frame in multiple S. cerevisiae strains.
Collectively our findings argue that the likelihood that an ORF encodes a functional product is best evaluated by combining multiple lines of experimental and evolutionary evidence (Snyder and Gerstein 2003). The potential functionality of orphan ORFs in S. cerevisiae suggests that experimentally verified functional sequences are not always conserved across species. Such nonconserved functional sequences might be responsible for species-specific phenotypic differences, making S. cerevisiae “cerevisiae” and not some other species in the Saccharomyces genus. An alternative explanation is that there are some functional elements evolving neutrally and conferring no specific benefit to the organism (Birney et al. 2007). Either way, experimental investigation has an irreplaceable role in determining biologically relevant DNA sequences. Comparative genomics has demonstrated analytic power in predicting functional regions before availability of any experimental information (Hardison 2003). When experimental information does become available (mainly from high-throughput functional genomics and proteomics analyses), then its integration should revise the genome annotation accordingly. The naïve Bayes model implemented here can be readily applied to all organisms.
Although we provide confidence scores about the likelihood of a predicted ORF to encode a functional product, comprehensive functional characterization of an ORF needs more concrete evidence from genetics, cell biology, and biochemistry than simple evidence of transcription or translation. The functional genomics or proteomics data sets used in our naïve Bayes predictions only investigated a few growth conditions, generally growth on rich media, limiting investigation of functions unique to the development and physiology of S. cerevisiae. Given the limited functional information obtained so far under laboratory conditions about uncharacterized ORFs (Pena-Castillo and Hughes 2007), perhaps what is needed are studies of yeast cells outside the laboratory. Upon such a shift, data sets generated under diverse conditions will become available, and our approach will then be available to aid precise and powerful annotation of genomes.
Methods
Large-scale data sets analysis
We collected 13 published functional genomics and proteomics data sets of S. cerevisiae, summarized in Table 1 with references to the data sources. Only ORFs identified by the same primary SGDID in the publication and in the January 2007 version of SGD annotation were included. We assigned “presence” or “absence” of transcript or translated product of every ORF in each data set. For protein complexes characterization data sets (Gavin et al. 2002, 2006; Ho et al. 2002; Krogan et al. 2006) all proteins that were identified as peptides were considered “present,” independent of further filtration by the investigators. For high-throughput yeast two-hybrid (Uetz et al. 2000; Ito et al. 2001), only proteins identified as preys were considered present. Only protein–protein interactions classified as “core” by Ito et al. (2001) were included. Transcripts identified by SAGE (Velculescu et al. 1997) and assigned to “class 1” by the investigators were considered present; all others, absent. We divided the Affymetrix Genechip data (Holstege et al. 1998) into two groups: intensity of expression strictly positive but less than or equal to 1, and intensity strictly more than 1. These two groups were treated separately in the naïve Bayes model. The normalized intensity of expression per probe (David et al. 2006) was averaged, and the percentage of probes whose intensity was higher than this average was considered as the intensity of expression of each ORF. We then extracted four groups (undetected, intensity strictly positive but less than 0.4, intensity strictly more than or equal to 0.4 but less than 0.8, and intensity strictly more than or equal to 0.8) that were treated separately in the naïve Bayes model. The remaining data sets were not reprocessed.
The naïve Bayes model
If the numbers of positives are known among the total number of ORFs, the “prior” odds of finding a positive are
The “posterior” odds are the odds of finding a positive after considering N different feature data sets with values f1 ... fN:
The likelihood ratio LR is defined as
According to Bayes rule, the posterior odds can be expressed as
If the N features are conditionally independent, LR can be simplified to
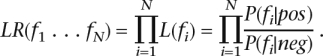 |
LR can be computed from contingency tables relating positive and negative examples with the N features (we binned the feature values f1 . . . fN into discrete intervals). Since Oprior is a fixed value, Opost is determined by LR. We used log-likelihood ratio (log10 LR or LLR) as the final prediction score. The higher the LLR of a certain ORF, the more likely it is a positive, i.e., a functional ORF.
Threefold cross-validation
We divided the whole training set into three subsets randomly. We then trained the model with two subsets and tested its performance on the third subset. We repeated this step three times so that each subset was used once to test the performance. We calculated the ROC curve with the predictions for the whole training set by combining the results from the three repeated tests.
Strand-specific RT-PCR
S. cerevisiae strain S288C was grown in yeast extract-peptone-dextrose (YPD) medium at 30°C to mid-exponential phase. Yeast cells were then harvested and used for total RNA isolation with an RNeasy kit (Qiagen). Poly(A) RNA was subsequently enriched by Oligotex mRNA kit (Qiagen). Before RT-PCR experiments, Poly(A) RNA was subjected to DNA-free DNase treatment (Ambion) to eliminate genomic DNA contamination. Genomic DNA was extracted from yeast culture by the DNeasy blood and tissue kit (Qiagen). We modified a strand-specific RT-PCR method previously described (Craggs et al. 2001), using the GeneAmp thermostable rTth reverse transcriptase RNA PCR kit (Applied Biosystems). DNase-treated poly(A) RNA sample (25 ng) was denatured for 5 min at 70°C with 2 μL of 10× rTth reverse transcriptase buffer and 1 μL of 10 μM reverse ORF-specific primer complementary to the ORF-coding strand (OSPR). While the template and the primer were still incubating at 70°C, a preheated reaction mixture was added consisting of 2 μL of 10 mM MnCl2 solution, 1.6 μL of 10 mM dNTP mix, and 2.5U of rTth polymerase. The temperature was lowered for 2 min to 55°C for annealing and then raised for 30 min to 70°C for the first-strand cDNA synthesis. After the cDNA synthesis, 20 μL of prewarmed 1× chelating buffer was added to chelate Mn2+ followed by heating the mixture for 30 min at 98°C to inactivate the reverse transcriptase activity of rTth. Second-step PCR reactions were performed in a 50-μL reaction volume using one-tenth of the synthesized first-strand cDNA as template, forward ORF-specific primer (OSPF) and OSPR as primers, and one unit of High Fidelity Platinum Taq polymerase (Invitrogen). The OSPR complementary to the ORF-coding strand was used in both first-strand cDNA synthesis and second-step PCR amplification. The OSPF complementary to the opposite strand was used only in the second-step PCR amplification. Both OSPR and OSPF were designed using the OSP Program (Hillier and Green 1991). The OSPR starts from the last nucleotide of the termination codon, while the OSPF starts from A of the ATG initiation codon. Primers used for RT-PCR of 201 nonoverlapping orphan ORFs are listed in Supplemental Table 5.
Interstrain intraspecies conservation analysis
SNP information from the four strains SK1, Y55, DBVPG6765, and W303 were extracted from the website of the Sanger Institute Saccharomyces Genome Resequencing Project (http://www.sanger.ac.uk/Teams/Team71/durbin/) on September 18, 2007 (R. Durbin and E. Louis, pers. comm.). The preassembly SNPs were taken into account only when their quality was “confirmed.” They were mapped to the ORFeome of the reference strain S288C as annotated by SGD on January 2007, as well as to intergenic regions that are annotated as “not feature” (ftp://genome-ftp.stanford.edu/pub/yeast/data_download/sequence/genomic_sequence/intergenic/NotFeature.fasta.gz). The nucleotide divergence of each ORF was then computed by averaging the number of SNPs per kb found in each of the four strains, counting insertions and deletions as one event independently of their length. For overlapping ORFs, only the regions unique to the ORFs themselves were considered for counting SNPs. To be considered as a preserved reading frame in our analysis, the ORF had to show neither stop codons nor frame-shift mutations in any of the four strains. The reading frame of an ORF was not considered preserved if the ORF had an insertion or deletion (indel) longer or equal to 20 bp, no matter whether the indel caused a frame-shift or not.
Acknowledgments
We thank R. Durbin and E. Louis for providing SNP information and F. Roth (HMS) for helpful discussions. We thank the members of the Vidal Lab and the Center for Cancer Systems Biology (CCSB) for their scientific and technical support, especially M. Boxem, K. Venkatesan, M. Yildirim, K. Salehi-Ashtiani, M. Dreze, S. Milstein, and C. Fraughton. This work was supported by an Ellison Foundation grant awarded to M.V. and by Institute Sponsored Research funds from the Dana-Farber Cancer Institute Strategic Initiative awarded to M.V. and CCSB.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.076661.108.
References
- Alvaro D., Lisby M., Rothstein R., Lisby M., Rothstein R., Rothstein R. Genome-wide analysis of Rad52 foci reveals diverse mechanisms impacting recombination. PLoS Genet. 2007;3:e228. doi: 10.1371/journal.pgen.0030228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E., Stamatoyannopoulos J.A., Dutta A., Guigo R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., Stamatoyannopoulos J.A., Dutta A., Guigo R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., Dutta A., Guigo R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., Guigo R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., Snyder M., Dermitzakis E.T., Thurman R.E., Dermitzakis E.T., Thurman R.E., Thurman R.E., et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachat S., Dietrich F., Voegeli S., Zhang Z., Stuart L., Lerch A., Gates K., Gaffney T., Philippsen P., Dietrich F., Voegeli S., Zhang Z., Stuart L., Lerch A., Gates K., Gaffney T., Philippsen P., Voegeli S., Zhang Z., Stuart L., Lerch A., Gates K., Gaffney T., Philippsen P., Zhang Z., Stuart L., Lerch A., Gates K., Gaffney T., Philippsen P., Stuart L., Lerch A., Gates K., Gaffney T., Philippsen P., Lerch A., Gates K., Gaffney T., Philippsen P., Gates K., Gaffney T., Philippsen P., Gaffney T., Philippsen P., Philippsen P. Reinvestigation of the Saccharomyces cerevisiae genome annotation by comparison to the genome of a related fungus: Ashbya gossypii. Genome Biol. 2003;4:R45. doi: 10.1186/gb-2003-4-7-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clamp M., Fry B., Kamal M., Xie X., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Fry B., Kamal M., Xie X., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Kamal M., Xie X., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Xie X., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Cuff J., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Lin M.F., Kellis M., Lindblad-Toh K., Lander E.S., Kellis M., Lindblad-Toh K., Lander E.S., Lindblad-Toh K., Lander E.S., Lander E.S. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. 2007;104:19428–19433. doi: 10.1073/pnas.0709013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A.G., Eisen M.B., Smith D.R., Bergman C.M., Oliver B., Markow T.A., Kaufman T.C., Kellis M., Gelbart W., Iyer V.N., Eisen M.B., Smith D.R., Bergman C.M., Oliver B., Markow T.A., Kaufman T.C., Kellis M., Gelbart W., Iyer V.N., Smith D.R., Bergman C.M., Oliver B., Markow T.A., Kaufman T.C., Kellis M., Gelbart W., Iyer V.N., Bergman C.M., Oliver B., Markow T.A., Kaufman T.C., Kellis M., Gelbart W., Iyer V.N., Oliver B., Markow T.A., Kaufman T.C., Kellis M., Gelbart W., Iyer V.N., Markow T.A., Kaufman T.C., Kellis M., Gelbart W., Iyer V.N., Kaufman T.C., Kellis M., Gelbart W., Iyer V.N., Kellis M., Gelbart W., Iyer V.N., Gelbart W., Iyer V.N., Iyer V.N., et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Cliften P., Sudarsanam P., Desikan A., Fulton L., Fulton B., Majors J., Waterston R., Cohen B.A., Johnston M., Sudarsanam P., Desikan A., Fulton L., Fulton B., Majors J., Waterston R., Cohen B.A., Johnston M., Desikan A., Fulton L., Fulton B., Majors J., Waterston R., Cohen B.A., Johnston M., Fulton L., Fulton B., Majors J., Waterston R., Cohen B.A., Johnston M., Fulton B., Majors J., Waterston R., Cohen B.A., Johnston M., Majors J., Waterston R., Cohen B.A., Johnston M., Waterston R., Cohen B.A., Johnston M., Cohen B.A., Johnston M., Johnston M. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- Craggs J.K., Ball J.K., Thomson B.J., Irving W.L., Grabowska A.M., Ball J.K., Thomson B.J., Irving W.L., Grabowska A.M., Thomson B.J., Irving W.L., Grabowska A.M., Irving W.L., Grabowska A.M., Grabowska A.M. Development of a strand-specific RT-PCR based assay to detect the replicative form of hepatitis C virus RNA. J. Virol. Methods. 2001;94:111–120. doi: 10.1016/s0166-0934(01)00281-6. [DOI] [PubMed] [Google Scholar]
- David L., Huber W., Granovskaia M., Toedling J., Palm C.J., Bofkin L., Jones T., Davis R.W., Steinmetz L.M., Huber W., Granovskaia M., Toedling J., Palm C.J., Bofkin L., Jones T., Davis R.W., Steinmetz L.M., Granovskaia M., Toedling J., Palm C.J., Bofkin L., Jones T., Davis R.W., Steinmetz L.M., Toedling J., Palm C.J., Bofkin L., Jones T., Davis R.W., Steinmetz L.M., Palm C.J., Bofkin L., Jones T., Davis R.W., Steinmetz L.M., Bofkin L., Jones T., Davis R.W., Steinmetz L.M., Jones T., Davis R.W., Steinmetz L.M., Davis R.W., Steinmetz L.M., Steinmetz L.M. A high-resolution map of transcription in the yeast genome. Proc. Natl. Acad. Sci. 2006;103:5320–5325. doi: 10.1073/pnas.0601091103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domazet-Loso T., Tautz D., Tautz D. An evolutionary analysis of orphan genes in Drosophila. Genome Res. 2003;13:2213–2219. doi: 10.1101/gr.1311003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk D.G., Ball C.A., Dolinski K., Engel S.R., Hong E.L., Issel-Tarver L., Schwartz K., Sethuraman A., Botstein D., Cherry J.M., Ball C.A., Dolinski K., Engel S.R., Hong E.L., Issel-Tarver L., Schwartz K., Sethuraman A., Botstein D., Cherry J.M., Dolinski K., Engel S.R., Hong E.L., Issel-Tarver L., Schwartz K., Sethuraman A., Botstein D., Cherry J.M., Engel S.R., Hong E.L., Issel-Tarver L., Schwartz K., Sethuraman A., Botstein D., Cherry J.M., Hong E.L., Issel-Tarver L., Schwartz K., Sethuraman A., Botstein D., Cherry J.M., Issel-Tarver L., Schwartz K., Sethuraman A., Botstein D., Cherry J.M., Schwartz K., Sethuraman A., Botstein D., Cherry J.M., Sethuraman A., Botstein D., Cherry J.M., Botstein D., Cherry J.M., Cherry J.M., et al. Saccharomyces cerevisiae S288C genome annotation: A working hypothesis. Yeast. 2006;23:857–865. doi: 10.1002/yea.1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischer T.C., Weaver C.M., McAfee K.J., Jennings J.L., Link A.J., Weaver C.M., McAfee K.J., Jennings J.L., Link A.J., McAfee K.J., Jennings J.L., Link A.J., Jennings J.L., Link A.J., Link A.J. Systematic identification and functional screens of uncharacterized proteins associated with eukaryotic ribosomal complexes. Genes & Dev. 2006;20:1294–1307. doi: 10.1101/gad.1422006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A.-C., Bosche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J.M., Michon A.-M., Cruciat C.-M., Bosche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J.M., Michon A.-M., Cruciat C.-M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J.M., Michon A.-M., Cruciat C.-M., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J.M., Michon A.-M., Cruciat C.-M., Marzioch M., Bauer A., Schultz J., Rick J.M., Michon A.-M., Cruciat C.-M., Bauer A., Schultz J., Rick J.M., Michon A.-M., Cruciat C.-M., Schultz J., Rick J.M., Michon A.-M., Cruciat C.-M., Rick J.M., Michon A.-M., Cruciat C.-M., Michon A.-M., Cruciat C.-M., Cruciat C.-M., et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Gavin A.-C., Aloy P., Grandi P., Krause R., Boesche M., Marzioch M., Rau C., Jensen L.J., Bastuck S., Dumpelfeld B., Aloy P., Grandi P., Krause R., Boesche M., Marzioch M., Rau C., Jensen L.J., Bastuck S., Dumpelfeld B., Grandi P., Krause R., Boesche M., Marzioch M., Rau C., Jensen L.J., Bastuck S., Dumpelfeld B., Krause R., Boesche M., Marzioch M., Rau C., Jensen L.J., Bastuck S., Dumpelfeld B., Boesche M., Marzioch M., Rau C., Jensen L.J., Bastuck S., Dumpelfeld B., Marzioch M., Rau C., Jensen L.J., Bastuck S., Dumpelfeld B., Rau C., Jensen L.J., Bastuck S., Dumpelfeld B., Jensen L.J., Bastuck S., Dumpelfeld B., Bastuck S., Dumpelfeld B., Dumpelfeld B., et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Dephoure N., O’Shea E.K., Weissman J.S., O’Shea E.K., Weissman J.S., Weissman J.S. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Goffeau A., Barrell B.G., Bussey H., Davis R.W., Dujon B., Feldmann H., Galibert F., Hoheisel J.D., Jacq C., Johnston M., Barrell B.G., Bussey H., Davis R.W., Dujon B., Feldmann H., Galibert F., Hoheisel J.D., Jacq C., Johnston M., Bussey H., Davis R.W., Dujon B., Feldmann H., Galibert F., Hoheisel J.D., Jacq C., Johnston M., Davis R.W., Dujon B., Feldmann H., Galibert F., Hoheisel J.D., Jacq C., Johnston M., Dujon B., Feldmann H., Galibert F., Hoheisel J.D., Jacq C., Johnston M., Feldmann H., Galibert F., Hoheisel J.D., Jacq C., Johnston M., Galibert F., Hoheisel J.D., Jacq C., Johnston M., Hoheisel J.D., Jacq C., Johnston M., Jacq C., Johnston M., Johnston M., et al. Life with 6000 genes. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Hardison R.C. Comparative genomics. PLoS Biol. 2003;1:e58. doi: 10.1371/journal.pbio.0000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier L., Green P., Green P. OSP: A computer program for choosing PCR and DNA sequencing primers. PCR Methods Appl. 1991;1:124–128. doi: 10.1101/gr.1.2.124. [DOI] [PubMed] [Google Scholar]
- Ho Y., Gruhler A., Heilbut A., Bader G.D., Moore L., Adams S.L., Millar A., Taylor P., Bennett K., Boutilier K., Gruhler A., Heilbut A., Bader G.D., Moore L., Adams S.L., Millar A., Taylor P., Bennett K., Boutilier K., Heilbut A., Bader G.D., Moore L., Adams S.L., Millar A., Taylor P., Bennett K., Boutilier K., Bader G.D., Moore L., Adams S.L., Millar A., Taylor P., Bennett K., Boutilier K., Moore L., Adams S.L., Millar A., Taylor P., Bennett K., Boutilier K., Adams S.L., Millar A., Taylor P., Bennett K., Boutilier K., Millar A., Taylor P., Bennett K., Boutilier K., Taylor P., Bennett K., Boutilier K., Bennett K., Boutilier K., Boutilier K., et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Holstege F.C.P., Jennings E.G., Wyrick J.J., Lee T.I., Hengartner C.J., Green M.R., Golub T.R., Lander E.S., Young R.A., Jennings E.G., Wyrick J.J., Lee T.I., Hengartner C.J., Green M.R., Golub T.R., Lander E.S., Young R.A., Wyrick J.J., Lee T.I., Hengartner C.J., Green M.R., Golub T.R., Lander E.S., Young R.A., Lee T.I., Hengartner C.J., Green M.R., Golub T.R., Lander E.S., Young R.A., Hengartner C.J., Green M.R., Golub T.R., Lander E.S., Young R.A., Green M.R., Golub T.R., Lander E.S., Young R.A., Golub T.R., Lander E.S., Young R.A., Lander E.S., Young R.A., Young R.A. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- Huh W.K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O'Shea E.K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O'Shea E.K., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O'Shea E.K., Carroll A.S., Howson R.W., Weissman J.S., O'Shea E.K., Howson R.W., Weissman J.S., O'Shea E.K., Weissman J.S., O'Shea E.K., O'Shea E.K. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Ito T., Chiba T., Ozawa R., Yoshida M., Hattori M., Sakaki Y., Chiba T., Ozawa R., Yoshida M., Hattori M., Sakaki Y., Ozawa R., Yoshida M., Hattori M., Sakaki Y., Yoshida M., Hattori M., Sakaki Y., Hattori M., Sakaki Y., Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R., Yu H., Greenbaum D., Kluger Y., Krogan N.J., Chung S., Emili A., Snyder M., Greenblatt J.F., Gerstein M., Yu H., Greenbaum D., Kluger Y., Krogan N.J., Chung S., Emili A., Snyder M., Greenblatt J.F., Gerstein M., Greenbaum D., Kluger Y., Krogan N.J., Chung S., Emili A., Snyder M., Greenblatt J.F., Gerstein M., Kluger Y., Krogan N.J., Chung S., Emili A., Snyder M., Greenblatt J.F., Gerstein M., Krogan N.J., Chung S., Emili A., Snyder M., Greenblatt J.F., Gerstein M., Chung S., Emili A., Snyder M., Greenblatt J.F., Gerstein M., Emili A., Snyder M., Greenblatt J.F., Gerstein M., Snyder M., Greenblatt J.F., Gerstein M., Greenblatt J.F., Gerstein M., Gerstein M. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003;302:449–453. doi: 10.1126/science.1087361. [DOI] [PubMed] [Google Scholar]
- Kellis M., Patterson N., Endrizzi M., Birren B., Lander E.S., Patterson N., Endrizzi M., Birren B., Lander E.S., Endrizzi M., Birren B., Lander E.S., Birren B., Lander E.S., Lander E.S. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Krogan N.J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A.P., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A.P., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A.P., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A.P., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A.P., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A.P., Li J., Pu S., Datta N., Tikuisis A.P., Pu S., Datta N., Tikuisis A.P., Datta N., Tikuisis A.P., Tikuisis A.P., et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Kumar A., Agarwal S., Heyman J.A., Matson S., Heidtman M., Piccirillo S., Umansky L., Drawid A., Jansen R., Liu Y., Agarwal S., Heyman J.A., Matson S., Heidtman M., Piccirillo S., Umansky L., Drawid A., Jansen R., Liu Y., Heyman J.A., Matson S., Heidtman M., Piccirillo S., Umansky L., Drawid A., Jansen R., Liu Y., Matson S., Heidtman M., Piccirillo S., Umansky L., Drawid A., Jansen R., Liu Y., Heidtman M., Piccirillo S., Umansky L., Drawid A., Jansen R., Liu Y., Piccirillo S., Umansky L., Drawid A., Jansen R., Liu Y., Umansky L., Drawid A., Jansen R., Liu Y., Drawid A., Jansen R., Liu Y., Jansen R., Liu Y., Liu Y., et al. Subcellular localization of the yeast proteome. Genes & Dev. 2002;16:707–719. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liolios K., Tavernarakis N., Hugenholtz P., Kyrpides N.C., Tavernarakis N., Hugenholtz P., Kyrpides N.C., Hugenholtz P., Kyrpides N.C., Kyrpides N.C. The Genomes On Line Database (GOLD) v.2: A monitor of genome projects worldwide. Nucleic Acids Res. 2006;34:D332–D334. doi: 10.1093/nar/gkj145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez M.J., Roy S., Archuletta A.B., Wentzell P.D., Anna-Arriola S.S., Rodriguez A.L., Aragon A.D., Quinones G.A., Allen C., Werner-Washburne M., Roy S., Archuletta A.B., Wentzell P.D., Anna-Arriola S.S., Rodriguez A.L., Aragon A.D., Quinones G.A., Allen C., Werner-Washburne M., Archuletta A.B., Wentzell P.D., Anna-Arriola S.S., Rodriguez A.L., Aragon A.D., Quinones G.A., Allen C., Werner-Washburne M., Wentzell P.D., Anna-Arriola S.S., Rodriguez A.L., Aragon A.D., Quinones G.A., Allen C., Werner-Washburne M., Anna-Arriola S.S., Rodriguez A.L., Aragon A.D., Quinones G.A., Allen C., Werner-Washburne M., Rodriguez A.L., Aragon A.D., Quinones G.A., Allen C., Werner-Washburne M., Aragon A.D., Quinones G.A., Allen C., Werner-Washburne M., Quinones G.A., Allen C., Werner-Washburne M., Allen C., Werner-Washburne M., Werner-Washburne M. Genomic analysis of stationary-phase and exit in Saccharomyces cerevisiae: Gene expression and identification of novel essential genes. Mol. Biol. Cell. 2004;15:5295–5305. doi: 10.1091/mbc.E03-11-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland M., Florea L., Sanderson K., Clifton S.W., Parkhill J., Churcher C., Dougan G., Wilson R.K., Miller W., Florea L., Sanderson K., Clifton S.W., Parkhill J., Churcher C., Dougan G., Wilson R.K., Miller W., Sanderson K., Clifton S.W., Parkhill J., Churcher C., Dougan G., Wilson R.K., Miller W., Clifton S.W., Parkhill J., Churcher C., Dougan G., Wilson R.K., Miller W., Parkhill J., Churcher C., Dougan G., Wilson R.K., Miller W., Churcher C., Dougan G., Wilson R.K., Miller W., Dougan G., Wilson R.K., Miller W., Wilson R.K., Miller W., Miller W. Comparison of the Escherichia coli K-12 genome with sampled genomes of a Klebsiella pneumoniae and three Salmonella enterica serovars, Typhimurium, Typhi and Paratyphi. Nucleic Acids Res. 2000;28:4974–4986. doi: 10.1093/nar/28.24.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.P., Lo R.S., Ben-Hur A., Desmarais C., Stagljar I., Noble W.S., Fields S., Lo R.S., Ben-Hur A., Desmarais C., Stagljar I., Noble W.S., Fields S., Ben-Hur A., Desmarais C., Stagljar I., Noble W.S., Fields S., Desmarais C., Stagljar I., Noble W.S., Fields S., Stagljar I., Noble W.S., Fields S., Noble W.S., Fields S., Fields S. Large-scale identification of yeast integral membrane protein interactions. Proc. Natl. Acad. Sci. 2005;102:12123–12128. doi: 10.1073/pnas.0505482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura F., Kawaguchi N., Sese J., Toyoda A., Hattori M., Morishita S., Ito T., Kawaguchi N., Sese J., Toyoda A., Hattori M., Morishita S., Ito T., Sese J., Toyoda A., Hattori M., Morishita S., Ito T., Toyoda A., Hattori M., Morishita S., Ito T., Hattori M., Morishita S., Ito T., Morishita S., Ito T., Ito T. A large-scale full-length cDNA analysis to explore the budding yeast transcriptome. Proc. Natl. Acad. Sci. 2006;103:17846–17851. doi: 10.1073/pnas.0605645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Castillo L., Hughes T.R., Hughes T.R. Why are there still over 1000 uncharacterized yeast genes? Genetics. 2007;176:7–14. doi: 10.1534/genetics.107.074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta M.P., Liang S., Liang S. Predicting protein functions from redundancies in large-scale protein interaction networks. Proc. Natl. Acad. Sci. 2003;100:12579–12583. doi: 10.1073/pnas.2132527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid K.J., Aquadro C.F., Aquadro C.F. The evolutionary analysis of “orphans” from the Drosophila genome identifies rapidly diverging and incorrectly annotated genes. Genetics. 2001;159:589–598. doi: 10.1093/genetics/159.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M., Gerstein M., Gerstein M. Defining genes in the genomics era. Science. 2003;300:258–260. doi: 10.1126/science.1084354. [DOI] [PubMed] [Google Scholar]
- Stein L.D., Bao Z., Blasiar D., Blumenthal T., Brent M.R., Chen N., Chinwalla A., Clarke L., Clee C., Coghlan A., Bao Z., Blasiar D., Blumenthal T., Brent M.R., Chen N., Chinwalla A., Clarke L., Clee C., Coghlan A., Blasiar D., Blumenthal T., Brent M.R., Chen N., Chinwalla A., Clarke L., Clee C., Coghlan A., Blumenthal T., Brent M.R., Chen N., Chinwalla A., Clarke L., Clee C., Coghlan A., Brent M.R., Chen N., Chinwalla A., Clarke L., Clee C., Coghlan A., Chen N., Chinwalla A., Clarke L., Clee C., Coghlan A., Chinwalla A., Clarke L., Clee C., Coghlan A., Clarke L., Clee C., Coghlan A., Clee C., Coghlan A., Coghlan A., et al. The genome sequence of Caenorhabditis briggsae: A platform for comparative genomics. PLoS Biol. 2003;1:e45. doi: 10.1371/journal.pbio.0000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P., Giot L., Cagney G., Mansfield T.A., Judson R.S., Knight J.R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Giot L., Cagney G., Mansfield T.A., Judson R.S., Knight J.R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Cagney G., Mansfield T.A., Judson R.S., Knight J.R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Mansfield T.A., Judson R.S., Knight J.R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Judson R.S., Knight J.R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Knight J.R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Lockshon D., Narayan V., Srinivasan M., Pochart P., Narayan V., Srinivasan M., Pochart P., Srinivasan M., Pochart P., Pochart P., et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Velculescu V.E., Zhang L., Zhou W., Vogelstein J., Basrai M.A., Bassett D.E., Hieter P., Vogelstein B., Kinzler K.W., Zhang L., Zhou W., Vogelstein J., Basrai M.A., Bassett D.E., Hieter P., Vogelstein B., Kinzler K.W., Zhou W., Vogelstein J., Basrai M.A., Bassett D.E., Hieter P., Vogelstein B., Kinzler K.W., Vogelstein J., Basrai M.A., Bassett D.E., Hieter P., Vogelstein B., Kinzler K.W., Basrai M.A., Bassett D.E., Hieter P., Vogelstein B., Kinzler K.W., Bassett D.E., Hieter P., Vogelstein B., Kinzler K.W., Hieter P., Vogelstein B., Kinzler K.W., Vogelstein B., Kinzler K.W., Kinzler K.W. Characterization of the yeast transcriptome. Cell. 1997;88:243–251. doi: 10.1016/s0092-8674(00)81845-0. [DOI] [PubMed] [Google Scholar]
- Xia Y., Lu L.J., Gerstein M., Lu L.J., Gerstein M., Gerstein M. Integrated prediction of the helical membrane protein interactome in yeast. J. Mol. Biol. 2006;357:339–349. doi: 10.1016/j.jmb.2005.12.067. [DOI] [PubMed] [Google Scholar]
- Yu H., Luscombe N.M., Lu H.X., Zhu X., Xia Y., Han J.-D.J., Bertin N., Chung S., Vidal M., Gerstein M., Luscombe N.M., Lu H.X., Zhu X., Xia Y., Han J.-D.J., Bertin N., Chung S., Vidal M., Gerstein M., Lu H.X., Zhu X., Xia Y., Han J.-D.J., Bertin N., Chung S., Vidal M., Gerstein M., Zhu X., Xia Y., Han J.-D.J., Bertin N., Chung S., Vidal M., Gerstein M., Xia Y., Han J.-D.J., Bertin N., Chung S., Vidal M., Gerstein M., Han J.-D.J., Bertin N., Chung S., Vidal M., Gerstein M., Bertin N., Chung S., Vidal M., Gerstein M., Chung S., Vidal M., Gerstein M., Vidal M., Gerstein M., Gerstein M. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 2004;14:1107–1118. doi: 10.1101/gr.1774904. [DOI] [PMC free article] [PubMed] [Google Scholar]