

Abstract
G protein-coupled receptors (GPCRs) interact directly with heterotrimeric G proteins to transduce physiological signals. Early studies of this interaction concluded that GPCRs (R) and G proteins (G) collide with each other randomly after receptor activation and that R-G complexes are transient. More recent studies have suggested that inactive R and G are preassembled (precoupled) as stable R-G complexes. Here we examine the stability of complexes formed between cyan fluorescent protein-labeled α2A-adrenoreceptors (C-α2ARs) and G proteins in cells using fluorescence recovery after photobleaching (FRAP). Labeled G proteins diffused in the plasma membrane with equal mobility in the absence and presence of immobile C-α2ARs. Immobile C-α2ARs activated labeled G proteins, demonstrating functional coupling without stable physical association. In contrast, a stable R-G interaction was detected when G proteins were deprived of nucleotides and C-α2ARs were active, as predicted by the ternary complex model. Overexpression of regulator of G protein signaling 4 (RGS4) accelerated the onset of effector activation but did not detectably alter the interaction between C-α2ARs and G proteins. We conclude that at most a small fraction of C-α2ARs and G proteins exist as R-G complexes at any moment.—Qin, K., Sethi, P. R., Lambert, N. A. Abundance and stability of complexes containing inactive G protein-coupled receptors and G proteins.
Keywords: FRAP, heterotrimers, precoupling, collision
G protein-coupled receptors (GPCRs) and heterotrimeric G proteins interact directly with each other to carry out many physiological signaling events. Although the importance of the receptor-G protein (R-G) interaction for signaling is clear, the dynamics of R-G complex association and dissociation are not completely understood. A large body of evidence is consistent with a “collision coupling” model in which both R and G are freely mobile in the plasma membrane and interact with each other transiently (1). In this model, receptors bind ligand, interact with Gαβγ-GDP to catalyze GDP release, and dissociate from G proteins immediately after Gα binds to GTP. The ability of a single active receptor to activate multiple G proteins in this manner is thought to be necessary for the high sensitivity of phototransduction (2, 3). However, the results of several studies (4,5,6,7) suggest that R-G complexes can be assembled before ligand binding and may remain assembled during signaling. The theoretical advantages of signaling by “precoupled” R-G complexes are that precoupling provides a potential mechanism for specificity if only appropriate R-G complexes are formed and that precoupling eliminates the time required for an active receptor to diffuse and bind to an appropriate G protein.
The defining difference between these two models is the stability (and thus the abundance) of inactive R-G complexes. At one extreme (collision coupling), R and G remain associated no longer than any two randomly interacting proteins, and a ligand is likely to encounter a receptor that is not bound to a G protein. At the other extreme (precoupling), R and G form a stable complex that persists for periods that are significant when compared with the intervals between ligand-binding events, and a ligand is thus more likely to encounter a receptor that is already bound to a G protein than if there were no precoupling. Few techniques are available to assess the stability of protein-protein interactions in living cells, thus little information is available regarding the stability and abundance of inactive R-G complexes under physiological conditions. Here we address this question using a technique based on fluorescence recovery after photobleaching (FRAP). We show that immobilization of α2A-adrenoreceptors (α2ARs) in the plasma membrane has no detectable effect on the mobility of the G protein heterotrimers that are activated by these receptors. These results set an upper limit on the steady-state abundance of α2AR-G protein complexes.
MATERIALS AND METHODS
Plasmid DNA constructs
C-α2AR subunits consisted of (starting at the amino terminus) a cleavable signal peptide from human growth hormone, enhanced (E) cyan fluorescent protein (CFP), and the human α2ARs. Gα-V subunits incorporated the yellow fluorescent protein venus between amino acids 91 and 92 and a C to G mutation at the −4 position to render them insensitive to pertussis toxin (PTX) -mediated ADP ribosylation. Venus-labeled Gβγ dimers were constructed as described previously (8) and in the text and were fluorescent by virtue of bimolecular fluorescence complementation (9). All constructs were made using an adaptation of the QuikChange (Stratagene, La Jolla, CA, USA) mutagenesis protocol, were expressed from pcDNA3.1 (Invitrogen, Carlsbad, CA, USA), and were verified by automated sequencing. YN-Gγ2 and YC-Gβ1 were generously provided by Steve Ikeda and Huanmian Chen (National Institute on Alcohol Abuse and Alcoholism, Rockville, MD, USA). Kir3.x plasmids were generously provided by Lily Y. Jan (University of California, San Francisco, CA, USA).
Cell culture and transfection
Human embryonic kidney (HEK) 293 cells and Chinese hamster ovary (CHO) -K1 cells (American Type Culture Collection, Manassas, VA, USA) were propagated in plastic flasks and on polylysine-coated glass coverslips according to the supplier’s protocol. Cells were transfected using polyethyleneimine (HEK 293) or Effectene (Qiagen, Valencia, CA; CHO-K1) and were used for experiments 12–24 h later. For most experiments, C-α2ARs and G protein subunits were transfected at a 3:1:1:1 ratio, and in all cases complementary unlabeled G protein subunits were cotransfected with labeled subunits. For experiments involving inwardly rectifying potassium (GIRK) channel activation, cells were also transfected with Kir3.1 and Kir3.2d, and PTX (100 ng/ml; List Biologicals, Campbell, CA, USA) was added to the culture medium immediately after transfection.
Cross-linking and permeabilization
For avidin-mediated cross-linking, cells were rinsed 3 times in buffer containing 150 mM NaCl, 2.5 mM KCl, 10 mM HEPES, 12 mM glucose, 0.5 mM CaCl2, and 0.5 mM MgCl2 (pH 8.0) and incubated at room temperature for 15 min in 0.5 mg/ml NHS-sulfo-LC-LC-biotin (Pierce, Rockford, IL, USA). Cells were washed an additional 3 times and incubated for 15 min in 0.1 mg/ml avidin. In some control experiments, cells were biotinylated but were not exposed to avidin. All experiments were performed within 1 h of avidin exposure. For antibody-mediated cross-linking (used for permeabilization experiments), cells were rinsed 3 times in buffer containing 140 mM potassium gluconate, 5 mM KCl, 10 mM HEPES, 1 mM EGTA, 0.3 mM CaCl2, and 1 mM MgCl2 (pH 7.2) and incubated at room temperature for 5 min in a 1:200 dilution of polyclonal rabbit anti-GFP IgG (Invitrogen A11122). Cells were washed and incubated for 5 min in a 1:1000 dilution of goat anti-rabbit antibody (Invitrogen B2770). Cells were permeabilized with 1000 U/ml α-hemolysin (Sigma, St. Louis, MO, USA; H9395) in the presence of 5 mM KCN and incubated for at least 10 min before imaging. As described in the text, the permeabilization buffer contained either rauwolscine (10 μM), norepinephrine (NE; 100 μM), or NE plus 0.3 mM GTPγS.
FRAP and imaging
Coverslips were transferred to the stage of a Leica (Wetzlar, Germany) SP2 scanning confocal microscope and imaged using an ×63, 1.4 NA objective. Cells were excited using 458 nm (for CFP) or 512 nm (for venus) laser lines. For translocation experiments, cells were perfused and exposed to NE as described below. For FRAP experiments, low intensity illumination was used during a control (prebleach) period, after which a 4 μm segment of the plasma membrane edge was irreversibly photobleached by increasing the laser intensity to 100%. Recovery of fluorescence into the bleached segment of plasma membrane was monitored for 3 min using low intensity illumination. Average pixel intensity in the bleached region was corrected for photobleaching during low intensity illumination, normalized, and plotted vs. time. For Fig. 6, curves were fitted or simulated using the empirical equation F(t) = Fi + Fm {1−[w2(w2+4πDt)−1]0.5}, where Fi is the fluorescence intensity immediately after photobleaching, Fm is the intensity recovered, w is the width of the bleached plasma membrane, and D is the effective one-dimensional diffusion coefficient (10). Fluorescence recovery depended on the size of the bleached area, thus recovery reflected lateral diffusion of labeled proteins.
Figure 1.

Immobile C-α2ARs do not restrict the mobility of Gα-V subunits. A) Exemplary images of an avidin-cross-linked cell expressing C-α2ARs and Gαi3-V during a FRAP experiment; inset in top left panel indicates position of remaining panels. Images are shown before (prebleach), immediately after (10 s), and 3 min after (180 s) photobleaching of a 4 μm segment of plasma membrane. C-α2AR fluorescence fails to recover into the bleached region, whereas Gαi3-V fluorescence recovers completely. B) Mean CFP intensity vs. time is plotted for avidin-cross-linked cells expressing C-α2ARs or the control protein C-TM, unlabeled Gβ1 and Gγ2 subunits, and Gαi3-V; in all FRAP plots, gray lines indicate mean ± se, and photobleaching occurred at 10 s. C) CFP fluorescence recovery at 180 s is plotted for biotinylated control cells expressing C-α2ARs (n=7), avidin-cross-linked cells expressing C-α2ARs (n=8), and avidin-cross-linked cells expressing C-TM (n=19); error bars represent se. D) Mean venus intensity vs. time is plotted for avidin-cross-linked cells expressing C-α2ARs or the control protein C-TM and Gαi3-V. E) Venus fluorescence recovery at 180 s is plotted for avidin-cross-linked cells expressing C-α2ARs or C-TM and Gαi3-V (n=11 and 11, respectively), Gαi1-V (n=9 and 8), or GαoA-V (n=12 and 10).
Figure 2.

C-α2ARs are present in excess of Gαi3-V subunits. Venus intensity [arbitrary units (a.u.)] in a plasma membrane region of interest is plotted against CFP intensity in the same region for individual cells expressing C-TM-V (n=25), C-TM and Gαi3-V (n=20), or C-α2ARs and Gαi3-V (n=25). A linear least-squares fit (slope=0.81; r=0.97) to the C-TM-V values is shown superimposed.
Figure 3.

Immobile C-α2ARs activate but do not restrict the mobility of heterotrimers containing Gβ1γ2/11-V dimers. A) Mean venus intensity vs. time is plotted for avidin-cross-linked cells expressing C-α2ARs (n=11) or the control protein C-TM (n=11) together with unlabeled GαoA subunits and Gβ1γ2/11-V dimers. B) Normalized ratio of plasma membrane to intracellular Gβ1γ2/11-V fluorescence is plotted against time for cells expressing either C-TM (n=12) or C-α2ARs (n=9) together with unlabeled GαoA and Gβ1γ2/11-V dimers; translocation of Gβ1γ2/11-V from the plasma membrane to the cell interior indicates heterotrimer activation and dissociation. NE (100 μM) was applied where indicated by horizontal bar.
Figure 4.

Agonist-activated immobile C-α2ARs restrict the mobility of Gαi3-V subunits in permeabilized, nucleotide-deprived cells. A) Mean venus intensity vs. time is plotted for antibody-cross-linked cells expressing C-α2ARs, unlabeled Gβ1 and Gγ2 subunits, and Gαi3-V. Cells were permeabilized with α-hemolysin and exposed to the antagonist rauwolscine (10 μM; n=12), NE (100 μM; n=22), or NE and 0.3 mM GTPγS (n=20), a poorly hydrolyzable analog of GTP. B) CFP and venus fluorescence recovery at 180 s is plotted for the same cells as A; *P < 0.001.
Figure 5.

RGS4 overexpression accelerates GIRK channel activation but does not induce stable complexes between C-α2ARs and GαoA-V-containing heterotrimers. A) Mean venus intensity vs. time is plotted for avidin-cross-linked cells expressing unlabeled Gβ1 and Gγ2 subunits, GαoA-V, and either the control protein C-TM (n=9), C-α2ARs alone (n=9), or C-α2ARs together with RGS4 (n=9). B) Exemplary traces from cells expressing C-α2ARs, unlabeled Gβ1 and Gγ2 subunits, GαoA-V, and GIRK channel subunits with or without RGS4; NE (100 μM) was applied where indicated by the horizontal bar. C) Grouped data from experiments identical to those shown in B showing the mean monoexponential GIRK activation time constant (τ) for control cells (n=16) and cells expressing RGS4 (n=16; *P<0.001).
Figure 6.

Limits of detection of FRAP for binding. Fluorescence recovery curves (taken from Fig. 3) are replotted, and the control trace (C-TM) is fitted to an empirical equation yielding a diffusion coefficient (D=0.11 μm2/s; see Materials and Methods). The fitted curve is taken to represent the mobility of heterotrimers without binding (bound fraction=0). Additional curves are simulated for transient binding of G proteins to C-α2ARs with bound fractions of 0.2 and 0.5 (see Discussion).
Electrophysiology
Whole-cell voltage-clamp recordings were made using standard procedures from transfected, PTX-treated cells on the stage of an inverted fluorescence microscope. Cells were held at a membrane potential of −60 mV and each second were stepped to −100 mV for 0.2 s and then ramped from −100 to 0 mV at a rate of 0.18 mV/ms. Patch electrodes (4–5 MΩ) were filled with a solution containing 140 mM K-gluconate, 5 mM KCl, 0.2 mM EGTA, 10 mM HEPES, 3 mM MgATP, and 0.3 mM Na2GTP (pH 7.2, ∼295 mOsm/kg H2O). Cells were perfused with a solution containing 122.5 or 150 mM NaCl, 30 mM or 5 mM KCl, 10 mM HEPES, 10 mM glucose, 1.5 mM CaCl2, and 2.5 mM MgCl2 (pH 7.2, ∼320 mOsm/kg H2O). Solution changes were made using a multiport attachment and perfusion capillary positioned directly in front of the cell under study.
RESULTS
Immobile C-α2ARs do not change Gα-V mobility
To determine the stability of protein-protein interactions using FRAP, one member of a complex is immobilized and the mobility of a potential binding partner is measured. If a protein-protein complex is highly stable, then immobilizing one binding partner will greatly decrease the mobility of the other. To test the stability of R-G complexes, we measured the mobility of heterotrimeric G proteins after immobilization of α2ARs fused to ECFP (C) at their amino terminus (C-α2ARs). We focused on α2ARs in this study because these receptors have been the subject of numerous biochemical and biophysical investigations of R-G precoupling (4,5,6, 11). Fusing ECFP to the extracellular amino terminus of α2ARs provided a means of measuring the mobility of this receptor using FRAP as well as a means of immobilizing this receptor (using avidin- or antibody-mediated cross-linking; see Materials and Methods) without altering the intracellular portions of the receptor that interact with G proteins. C-α2ARs expressed in HEK 293 cells by transient transfection were trafficked to the cell surface (Fig. 1A) and were functional with respect to signaling (see below). These receptors activate PTX-sensitive G proteins of the Gαi/o family. Therefore, in cells expressing C-α2ARs, we coexpressed Gαi or Gαo subunits with the yellow fluorescent protein venus (V) fused in the loop between helices αA and αB in the helical domain (Gα-V). Previous studies have shown that insertion of fluorescent proteins at this site does not prevent functional activation by GPCRs, and resonance energy transfer (RET) studies of precoupling have used identical fusion proteins (4, 6, 11). Unlabeled Gβ1 and Gγ2 subunits were also expressed in these experiments to provide exogenous Gβγ dimers. As expected, Gα-V subunits were trafficked to the plasma membrane (Fig. 1A) and were functional, as heterotrimers containing these subunits could be activated by C-α2ARs (see below).
To determine if Gα-V-containing heterotrimers formed stable precoupled R-G complexes with C-α2ARs, we compared the mobility of these heterotrimers in cells expressing either immobile C-α2ARs or an immobile control protein consisting of an extracellular ECFP and a single transmembrane domain (C-TM). Cell surface biotinylation followed by exposure to soluble avidin cross-links and immobilizes proteins that are exposed to the extracellular environment (8). Accordingly, both C-α2ARs and the C-TM control protein were completely immobile after avidin-mediated cross-linking, as indicated by the nearly complete absence of fluorescence recovery 180 s after photobleaching (Fig. 1A, B). Since heterotrimeric G proteins are attached to the inner leaflet of the plasma membrane by lipid anchors and are not exposed to the extracellular space, they are not directly immobilized by avidin-mediated cross-linking. Therefore, any change in their mobility after avidin-mediated cross-linking can be attributed to an interaction with an immobilized protein. However, Gα-V subunits (and presumbably Gα-V-containing heterotrimers) remained equally mobile after cross-linking in cells expressing C-TM or C-α2ARs (Fig. 1A, D). Similar results were obtained for Gαi3-V, Gαi1-V, and GαoA-V, suggesting that none of these isoforms formed highly stable R-G complexes with C-α2ARs (Fig. 1E). Similar results were obtained when cells also expressed GIRK channels (see Materials and Methods). Fractional fluorescence recovery of GαoA-V was 0.94 ± 0.03 (n=11) in cells expressing C-α2ARs and 0.87 ± 0.02 (n=12) in cells expressing C-α2ARs and GIRK channels (P>0.05). Gα-V mobility was not changed by avidin-mediated cross-linking itself, as fluorescence recovery was indistinguishable in control and cross-linked cells (data not shown). This result suggests that Gα-V subunits and heterotrimers do not bind to endogenous proteins (such as endogenous GPCRs) that are immobilized by avidin-mediated cross-linking.
If Gα-V subunits were greatly outnumbered by endogenous Gα subunits in these experiments, then precoupling to endogenous G proteins might have prevented precoupling to Gα-V-containing G proteins by competition. To assess the relative abundance of endogenous and expressed Gα subunits, we measured activation of GIRK channels by C-α2ARs and endogenous G proteins with and without expression of Gα-V subunits. Expression of exogenous Gα subunits can prevent Gβγ signaling (including GIRK channel activation) by “buffering” free Gβγ dimers if the exogenous Gα subunits are sufficiently abundant (12). Activation of GIRK channels by endogenous G proteins (550±96 pA; n=10) was greatly decreased by expression of either Gαi3-V (69±37 pA; n=9; P<0.001) or GαoA-V (139±72 pA; n=9; P<0.005). These experiments suggest that under the conditions of our experiments expressed Gα-V subunits represented a substantial fraction of the total population of Gα subunits.
In order for this type of assay to detect a protein interaction, the immobile protein (in this case C-α2ARs) must be sufficiently abundant to inhibit the mobility of a substantial fraction of the potential binding partners (in this case Gα-V-containing heterotrimers). We therefore estimated the Gα-V:C-α2AR stoichiometry in these experiments by comparing the V:C fluorescence intensity ratio in cells expressing C-α2AR and Gα-V to those expressing a control fusion protein consisting of ECFP, a transmembrane domain and venus (C-TM-V). The V:C intensity ratio was obtained for C-TM-V by linear regression (Fig. 2) and was found to be 0.81 (n=25). In cells expressing C-α2AR and Gαi3-V, the average V:C fluorescence intensity ratio was 0.25 ± 0.03 (n=20), and all of the values for individual cells fell below 0.81 (Fig. 2), suggesting that under these conditions C-α2ARs outnumbered Gαi3-V subunits ∼3-fold. Similar V:C ratios were obtained for cells coexpressing C-TM and Gαi3-V (0.21±0.03; n=25) and C-α2AR and GαoA-V (0.74±0.11; n=26). These results suggest that C-α2ARs were present in sufficient numbers to interact with a significant fraction of Gα-V-containing heterotrimers.
Immobile C-α2ARs do not change Gβγ-V mobility
Several studies (4, 6, 11, 13) have detected either basal or agonist-induced RET between GPCRs and G proteins when the latter were labeled at the amino terminus of either the Gβ1 or Gγ2 subunit. Therefore, we repeated our FRAP experiments with Gβγ dimers that were labeled by bimolecular fluorescence complementation (9). Complementary fragments of venus were fused to the amino terminus of Gβ1 and Gγ2, and the resulting Gβ1γ2-V dimers were expressed together with unlabeled Gαi1 subunits. As was the case with labeled Gα subunits, the mobility of Gβ1γ2-V was the same in the absence and presence of immobile C-α2ARs; fractional recovery of Gβ1γ2-V fluorescence at 180 s was 0.89 ± 0.04 in control cells expressing C-TM (n=11) and 0.84 ± 0.04 in cells expressing C-α2ARs (P=0.49; n=13). To verify that we were measuring the mobility of heterotrimers rather than free Gβγ dimers and that these heterotrimers could functionally interact with C-α2ARs, we repeated this experiment with a chimera (Gγ2/11) consisting of Gγ2 with six amino acids at the extreme carboxyl-terminus replaced with those found in Gγ11. Gβ1γ2/11-V dimers are attached to the plasma membrane by a farnesyl group as opposed to the geranylgeranyl group found on Gβ1γ2-V dimers, and this relatively weak attachment allows them to dissociate from the plasma membrane after dissociation from Gα (14, 15). Gβ1γ2/11-V dimers were coexpressed with unlabeled GαoA subunits, and their mobility was unaffected by the presence of immobile C-α2ARs (Fig. 3A). However, activation of C-α2ARs by the agonist NE (30 μM) induced reversible translocation of Gβ1γ2/11-V fluorescence from the plasma membrane to the cell interior (Fig. 3B). This result indicates that C-α2ARs were functional and present at a level sufficient to activate a substantial fraction of Gβ1γ2/11-V-containing heterotrimers and that Gβ1γ2/11-V dimers were part of functional heterotrimers. Thus, although C-α2ARs and Gβ1γ2/11-V-containing heterotrimers were functionally coupled, they were not physically associated to the extent that Gβ1γ2/11-V mobility was detectably inhibited by immobilized C-α2ARs.
Immobile C-α2ARs decrease Gα-V mobility after nucleotide depletion and agonist activation
We next attempted to produce a stable interaction between C-α2ARs and G proteins by depriving the latter of guanine nucleotides and applying an agonist. The ternary complex model of GPCR signaling predicts that agonist-bound GPCRs bind to nucleotide-free (empty) G proteins with a high affinity and that G protein binding reciprocally stablizes an active receptor state with a high affinity for agonist (16). Guanine nucleotides were depleted by permeablizing cells in nucleotide-free buffer with staphylococcal α-hemolysin (α-toxin), which forms 2 nm pores in the plasma membrane, and by adding 5 mM KCN to inhibit ATP (and GTP) synthesis. In preliminary experiments, we found that avidin-mediated cross-linking interferes with α-toxin permeabilization; therefore, C-α2ARs were selectively immobilized in these experiments by sequential incubation with a polyclonal anti-GFP antibody and an appropriate secondary antibody (see Materials and Methods). Under these conditions, Gαi3-V mobility was significantly reduced by application of NE (100 μM; P<0.001; Fig. 4A, B) but not by application of the antagonist rauwolscine (10 μM). The effect of NE was reversed by loading cells with the poorly hydrolyzable GTP analog GTPγS (0.3 mM; Fig. 4A, B). There was no significant difference in C-α2AR mobility in these three conditions (P>0.05; Fig. 4B), and NE had no effect on Gαi3-V mobility in unpermeabilized cells (P>0.05; n=9). These results are consistent with many previous experimental results documenting high affinity ternary (ligand-R-G) complexes and show that our FRAP assay is capable of detecting C-α2AR:Gαi3-V complexes.
Regulator of G protein signaling (RGS) 4 does not induce stable C-α2AR:GαoA-V complexes
Finally, we investigated the possibility that RGS proteins might facilitate a physical interaction between C-α2ARs and G proteins. RGS proteins interact directly or indirectly with GPCRs (17,18,19) and inactive G proteins (20) and could thus serve as scaffolds for R-G complexes. This type of scaffolding has been suggested as a possible mechanism whereby RGS proteins accelerate the activation of GIRK channels (21, 22), but little evidence is available to support or refute this suggestion. To test this possibility, we overexpressed RGS4 in CHO-K1 cells that also expressed C-α2ARs and PTX-resistant GαoA-V. CHO-K1 cells were used for these experiments because previous work demonstrating acceleration of GIRK activation by RGS proteins used these cells (23). As was the case in our previous experiments, immobile C-α2ARs had no detectable effect on the mobility of GαoA-V (Fig. 5A). In addition, RGS4 overexpression did not produce a significant decrease in GαoA-V mobility, as would be expected if this protein induced the formation of stable C-α2AR:GαoA-V complexes (Fig. 5A). To document the functionality of GαoA-V-containing heterotrimers and the physiological function of RGS4, we reconstituted activation of GIRK channels in cells treated with PTX to inactivate endogenous G proteins. The onset of GIRK currents was accelerated in cells that expressed RGS4 (Fig. 5B,C), as previously reported (23, 24). These results show that C-α2ARs and GαoA-V-containing heterotrimers were functionally coupled, that RGS4 could interact with GαoA-V, and that RGS4 could accelerate activation of GIRK channels without inducing the formation of detectable R-G complexes.
DISCUSSION
Many biochemical studies, beginning with those of Tolkovsky and Levitzki (1), support the conclusion that activated GPCRs act catalytically by colliding with freely diffusing G proteins in the plasma membrane (25). However, other results are difficult to reconcile with unrestricted exchange of G proteins between GPCRs and can be explained if a fraction of GPCRs and G proteins are either directly precoupled or compartmentalized in a membrane subdomain (5, 26,27,28,29).
The interaction between GPCRs and G proteins has recently been examined in live cells using RET techniques (4, 6, 7, 11, 13, 30). RET between two labeled proteins occurs if these proteins are within ∼10 nm of each other and are properly oriented. Several studies (4, 6, 13, 30) have detected basal RET signals between labeled GPCRs and G proteins that persist after receptor activation, again consistent with the idea that a fraction of these proteins exist either as precoupled R-G complexes or within a restricted membrane domain. In contrast, a similar study detected only agonist-induced RET and concluded that GPCRs and G proteins associate by random collision after activation (11). This discrepancy highlights one of the inherent limitations of RET-based techniques. The absolute magnitude of RET depends on several factors that can not be measured or predicted and thus provides little information regarding the fraction of binding partners that are associated. Since random interactions between membrane-associated proteins can generate significant RET signals (31, 32), the choice of negative controls becomes critical for distinguishing random interactions from specific interactions. In addition, RET techniques measure signals from populations of proteins and do not provide direct information regarding the kinetics of protein complex association and dissociation. Complex association and dissociation will produce changes in RET, but these can not readily be distinguished from changes in orientation of one or both of the binding partners. Because of these limitations, previous studies have not provided estimates of the rates of R-G association and dissociation or the fraction of GPCRs and G proteins that are associated at rest. Thus, it is unclear if GPCRs and G proteins form highly stable R-G complexes that remain intact between signaling events or alternatively if these proteins associate transiently and spend little time as a precoupled complex.
This uncertainty prompted us to examine the question of R-G precoupling using a technique with different limitations. Our FRAP experiments suggest that α2ARs and the G proteins that they activate do not form highly stable R-G complexes under basal conditions. This conclusion is based on the observation that the lateral mobility of labeled Gα-V subunits or Gβγ-V dimers is the same in the absence and presence of immobile C-α2ARs when the latter are present in excess. C-α2ARs, Gα-V subunits, and Gβγ-V dimers were functionally coupled with each other as indicated by Gβγ-V translocation and GIRK channel activation. Immobile C-α2ARs did restrict the mobility of Gα-V-containing heterotrimers when agonist was present and guanine nucleotides were absent, as predicted by the ternary complex model (16, 33). This observation indicates that our FRAP assay would have been capable of detecting a stable interaction between these molecules under these conditions had one existed. An RGS protein that accelerated signaling did not detectably stabilize the interaction between C-α2ARs and G proteins. Our results support the conclusions of Hein et al. (11) who did not detect basal RET between labeled α2ARs and G proteins and showed that G protein abundance could become rate limiting for agonist-induced RET at low levels of expression. The absence of stable precoupling is also consistent with the results of Azpiazu and Gautam (34), who showed that a common population of G proteins is shared by two different GPCRs.
Limits of sensitivity
One advantage of using FRAP as opposed to RET to study protein-protein interactions is that the former technique can provide evidence for the absence of a stable interaction. The inability of a protein to restrict the mobility of a potential binding partner is meaningful, whereas the absence of a RET signal between two proteins does not rule out the possibility that the two are associated. Nevertheless, it is important to carefully consider the limits of sensitivity of our approach. Several workers (35,36,37,38) have developed methods to extract kinetic information from FRAP curves that are altered due to binding to an immobile partner. It can be shown that a full reaction-diffusion system reduces to an effective diffusion approximation when binding and unbinding rates are fast compared with diffusion (38). In this case, molecules will appear to diffuse with a slowed effective diffusion coefficient, Deff, which is given by:
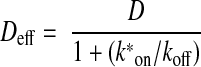 |
where D is the coefficient of diffusion (without binding), koff is the dissociation rate constant, and k*on is the pseudo-first-order association rate constant, which is defined as the product of the concentration of immobile binding sites and the association rate constant kon. The ratio k*on/koff is then a unitless pseudo-equilibrium constant and is equal to the ratio of bound to free molecules. Therefore, for a given ratio of bound/free molecules (in our case G proteins), it is possible to predict the effect of binding on FRAP. Figure 6 shows FRAP curves (taken from Fig. 3) superimposed on a least-squares fit to an empirical equation that is based on a 1-dimensional approximation of 2-dimensional diffusion (10). Two simulated FRAP curves are also shown where k*on/koff=1 (fraction bound=0.5) and k*on/koff=0.25 (fraction bound=0.2). As is evident from these curves, a bound fraction of 0.5 would be easily distinguished from the unbound case, whereas a bound fraction of 0.2 would be close to the limits of detection of this method. Virtually identical results were obtained when a more complicated 2-dimensional equation was used to simulate FRAP curves (data not shown) (39). From this exercise, we conclude that under the conditions of our study no more than 20% of the labeled G proteins present at the plasma membrane were bound to C-α2ARs at a given point in time. This estimate is an upper limit, because it is based on the assumption that binding and unbinding are rapid compared with diffusion and the characteristic fluorescence recovery time. If instead binding and unbinding were slow compared with diffusion (i.e., if R-G complexes were stable for tens of seconds), then the slowing of fluorescence recovery would be even greater than that shown in Fig. 6 and the detection limit would be lowered.
Limitations
An important distinction between our experiments and the physiological situation is that in most cells endogenous G proteins are present in large excess over any single type of GPCR (28). Thus, relatively weak R-G precoupling (such as might have escaped detection in our experiments) could still allow a substantial fraction of α2ARs to be associated with G proteins provided the latter are sufficiently abundant. It is possible that transient R-G precoupling that eluded detection in our experiments can account for basal RET between α2ARs and G proteins detected in other studies (4, 6). In any case, we emphasize that our results do not rule out the existence of physiologically relevant precoupled R-G complexes but place an upper limit on their abundance and stability. It is also important to consider the likelihood that other cells may possess components or compartments not found in HEK 293 or CHO-K1 cells that scaffold or restrict the mobility of GPCRs and G proteins (28). Although it is likely that we would have detected a stable direct interaction between C-α2ARs and G proteins, we can not rule out the possibility that indirect interactions or co-compartmentalization occur and are functionally significant in other cells. At present, our conclusions are also limited to interactions between α2ARs and their G proteins. We focused on α2ARs in the present study because several previous studies (4,5,6) obtained evidence for R-G precoupling using this receptor. It is certainly possible that other GPCRs (and other G proteins) can form more stable R-G complexes than α2ARs. The FRAP method used here should be useful for testing differences between GPCRs with respect to G protein association.
Acknowledgments
We thank Drs. Huanmian Chen and Steve Ikeda (National Institute on Alcohol Abuse and Alcoholism, Rockville, MD, USA) and Dr. Lily Jan (University of California, San Francisco, CA, USA) for gifts of plasmid DNA.We thank Omer Dushek and Daniel Coombs (University of British Columbia, Vancouver, BC, Canada) for providing simulated FRAP curves generated using the full reaction-diffusion equation. This work was supported by grants from the National Science Foundation (MCB 0620024 to N.A.L.) and the U. S. National Institutes of Health (GM078319 to N.A.L.).
References
- Tolkovsky A M, Levitzki A. Mode of coupling between the beta-adrenergic receptor and adenylate cyclase in turkey erythrocytes. Biochemistry. 1978;17:3795. doi: 10.1021/bi00611a020. [DOI] [PubMed] [Google Scholar]
- Kwok-Keung Fung B, Stryer L. Photolyzed rhodopsin catalyzes the exchange of GTP for bound GDP in retinal rod outer segments. Proc Natl Acad Sci U S A. 1980;77:2500–2504. doi: 10.1073/pnas.77.5.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh E N, Jr, Lamb T D. Amplification and kinetics of the activation steps in phototransduction. Biochim Biophys Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- Gales C, Van Durm J J, Schaak S, Pontier S, Percherancier Y, Audet M, Paris H, Bouvier M. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat Struct Mol Biol. 2006;13:778–786. doi: 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- Neubig R R, Gantzos R D, Thomsen W J. Mechanism of agonist and antagonist binding to alpha 2 adrenergic receptors: evidence for a precoupled receptor-guanine nucleotide protein complex. Biochemistry. 1988;27:2374–2384. doi: 10.1021/bi00407a019. [DOI] [PubMed] [Google Scholar]
- Nobles M, Benians A, Tinker A. Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc Natl Acad Sci U S A. 2005;102:18706–18711. doi: 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip F, Sengupta P, Scarlata S. Signaling through a G Protein-coupled receptor and its corresponding G protein follows a stoichiometrically limited model. J Biol Chem. 2007;282:19203–19216. doi: 10.1074/jbc.M701558200. [DOI] [PubMed] [Google Scholar]
- Digby G J, Lober R M, Sethi P R, Lambert N A. Some G protein heterotrimers physically dissociate in living cells. Proc Natl Acad Sci U S A. 2006;103:17789–17794. doi: 10.1073/pnas.0607116103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes T R, Tang L, Mervine S M, Sabo J L, Yost E A, Devreotes P N, Berlot C H. Visualization of G protein betagamma dimers using bimolecular fluorescence complementation demonstrates roles for both beta and gamma in subcellular targeting. J Biol Chem. 2004;279:30279–30286. doi: 10.1074/jbc.M401432200. [DOI] [PubMed] [Google Scholar]
- Ellenberg J, Siggia E D, Moreira J E, Smith C L, Presley J F, Worman H J, Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein P, Frank M, Hoffmann C, Lohse M J, Bunemann M. Dynamics of receptor/G protein coupling in living cells. EMBO J. 2005;24:4106–4114. doi: 10.1038/sj.emboj.7600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Fernandez J M, Abogadie F C, Milligan G, Delmas P, Brown D A. Multiple pertussis toxin-sensitive G-proteins can couple receptors to GIRK channels in rat sympathetic neurons when expressed heterologously, but only native G(i)-proteins do so in situ. Eur J Neurosci. 2001;14:283–292. doi: 10.1046/j.0953-816x.2001.01642.x. [DOI] [PubMed] [Google Scholar]
- Gales C, Rebois R V, Hogue M, Trieu P, Breit A, Hebert T E, Bouvier M. Real-time monitoring of receptor and G-protein interactions in living cells. Nat Methods. 2005;2:177–184. doi: 10.1038/nmeth743. [DOI] [PubMed] [Google Scholar]
- Akgoz M, Kalyanaraman V, Gautam N. Receptor-mediated reversible translocation of the G protein betagamma complex from the plasma membrane to the Golgi complex. J Biol Chem. 2004;279:51541–51544. doi: 10.1074/jbc.M410639200. [DOI] [PubMed] [Google Scholar]
- Akgoz M, Kalyanaraman V, Gautam N. G protein betagamma complex translocation from plasma membrane to Golgi complex is influenced by receptor gamma subunit interaction. Cell Signal. 2006;18:1758–1768. doi: 10.1016/j.cellsig.2006.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lean A, Stadel J M, Lefkowitz R J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- Bernstein L S, Ramineni S, Hague C, Cladman W, Chidiac P, Levey A I, Hepler J R. RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J Biol Chem. 2004;279:21248–21256. doi: 10.1074/jbc.M312407200. [DOI] [PubMed] [Google Scholar]
- Hague C, Bernstein L S, Ramineni S, Chen Z, Minneman K P, Hepler J R. Selective inhibition of alpha1A-adrenergic receptor signaling by RGS2 association with the receptor third intracellular loop. J Biol Chem. 2005;280:27289–27295. doi: 10.1074/jbc.M502365200. [DOI] [PubMed] [Google Scholar]
- Wang X, Zeng W, Soyombo A A, Tang W, Ross E M, Barnes A P, Milgram S L, Penninger J M, Allen P B, Greengard P, Muallem S. Spinophilin regulates Ca2+ signalling by binding the N-terminal domain of RGS2 and the third intracellular loop of G-protein-coupled receptors. Nat Cell Biol. 2005;7:405–411. doi: 10.1038/ncb1237. [DOI] [PubMed] [Google Scholar]
- Benians A, Nobles M, Hosny S, Tinker A. Regulators of G-protein signaling form a quaternary complex with the agonist, receptor, and G-protein. A novel explanation for the acceleration of signaling activation kinetics. J Biol Chem. 2005;280:13383–13394. doi: 10.1074/jbc.M410163200. [DOI] [PubMed] [Google Scholar]
- Jeong S W, Ikeda S R. Differential regulation of G protein-gated inwardly rectifying K(+) channel kinetics by distinct domains of RGS8. J Physiol. 2001;535:335–347. doi: 10.1111/j.1469-7793.2001.00335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Pacheco M A, Doupnik C A. Gating properties of GIRK channels activated by Galpha(o)- and Galpha(i)-coupled muscarinic m2 receptors in Xenopus oocytes: the role of receptor precoupling in RGS modulation. J Physiol. 2002;545:355–373. doi: 10.1113/jphysiol.2002.032151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupnik C A, Davidson N, Lester H A, Kofuji P. RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc Natl Acad Sci U S A. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh O, Kubo Y, Miyatani Y, Asano T, Nakata H. RGS8 accelerates G-protein-mediated modulation of K+ currents. Nature. 1997;390:525–529. doi: 10.1038/37385. [DOI] [PubMed] [Google Scholar]
- Gilman A G. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Neubig R R. Membrane organization in G-protein mechanisms. FASEB J. 1994;8:939–946. doi: 10.1096/fasebj.8.12.8088459. [DOI] [PubMed] [Google Scholar]
- Graeser D, Neubig R R. Compartmentation of receptors and guanine nucleotide-binding proteins in NG108–15 cells: lack of cross-talk in agonist binding among the alpha 2-adrenergic, muscarinic, and opiate receptors. Mol Pharmacol. 1993;43:434–443. [PubMed] [Google Scholar]
- Ostrom R S, Post S R, Insel P A. Stoichiometry and compartmentation in G protein-coupled receptor signaling: implications for therapeutic interventions involving G(s) J Pharmacol Exp Ther. 2000;294:407–412. [PubMed] [Google Scholar]
- Rebois R V, Hebert T E. Protein complexes involved in heptahelical receptor-mediated signal transduction. Recept Channels. 2003;9:169–194. [PubMed] [Google Scholar]
- Ayoub M A, Maurel D, Binet V, Fink M, Prezeau L, Ansanay H, Pin J P. Real-time analysis of agonist-induced activation of protease-activated receptor 1/Galphai1 protein complex measured by bioluminescence resonance energy transfer in living cells. Mol Pharmacol. 2007;71:1329–1340. doi: 10.1124/mol.106.030304. [DOI] [PubMed] [Google Scholar]
- 31.Vogel, S. S., Thaler, C., and Koushik, S. V. (2006) Fanciful FRET. Sci. STKE331, re2 [DOI] [PubMed] [Google Scholar]
- Zacharias D A, Violin J D, Newton A C, Tsien R Y. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- Limbird L E, Gill D M, Lefkowitz R J. Agonist-promoted coupling of the beta-adrenergic receptor with the guanine nucleotide regulatory protein of the adenylate cyclase system. Proc Natl Acad Sci U S A. 1980;77:775–779. doi: 10.1073/pnas.77.2.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azpiazu I, Gautam N. A fluorescence resonance energy transfer-based sensor indicates that receptor access to a G protein is unrestricted in a living mammalian cell. J Biol Chem. 2004;279:27709–27718. doi: 10.1074/jbc.M403712200. [DOI] [PubMed] [Google Scholar]
- Carrero G, McDonald D, Crawford E, de Vries G, Hendzel M J. Using FRAP and mathematical modeling to determine the in vivo kinetics of nuclear proteins. Methods. 2003;29:14–28. doi: 10.1016/s1046-2023(02)00288-8. [DOI] [PubMed] [Google Scholar]
- Phair R D, Gorski S A, Misteli T. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol. 2004;375:393–414. doi: 10.1016/s0076-6879(03)75025-3. [DOI] [PubMed] [Google Scholar]
- Sprague B L, McNally J G. FRAP analysis of binding: proper and fitting. Trends Cell Biol. 2005;15:84–91. doi: 10.1016/j.tcb.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Sprague B L, Pego R L, Stavreva D A, McNally J G. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys J. 2004;86:3473–3495. doi: 10.1529/biophysj.103.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dushek O, Coombs D. Improving parameter estimation for cell surface FRAP data. J Biochem Biophys Methods. 2007;70:1224–1231. doi: 10.1016/j.jbbm.2007.07.002. [DOI] [PubMed] [Google Scholar]