

Abstract
Investigation of the structure/function relationships of the sodium-glucose transporter (SGLT1) is crucial to understanding the cotransporter mechanism. In the present study, we used cysteine-scanning mutagenesis and chemical modification by methanethiosulfonate (MTS) derivatives to test whether predicted transmembrane IV participates in sugar binding. Five charged and polar residues (K139, Q142, T156, K157, and D161) and two glucose/galactose malabsorption missense mutations (I147 and S159) were replaced with cysteine. Mutants I147C, T156C, and K157C exhibited sufficient expression to be studied in detail using the two-electrode voltage-clamp method in Xenopus laevis oocytes and COS-7 cells. I147C was similar in function to wild-type and was not studied further. Mutation of lysine-157 to cysteine (K157C) causes loss of phloridzin and α-methyl-d-glucopyranoside (αMG) binding. These functions are restored by chemical modification with positively charged (2-aminoethyl) methanethiosulfonate hydrobromide (MTSEA). Mutation of threonine-156 to cysteine (T156C) reduces the affinity of αMG and phloridzin for T156C by ∼5-fold and ∼20-fold, respectively. In addition, phloridzin protects cysteine-156 in T156C from alkylation by MTSEA. Therefore, the presence of a positive charge or a polar residue at 157 and 156, respectively, affects sugar binding and sugar-induced Na+ currents.
Keywords: cysteine scanning mutagenesis, chemical modification by methanethiosulfonate reagents
the high-affinity sodium-glucose cotransporter (SGLT1) belongs to the homologous family of Na+/solute symporters, SLC5 (22). It is a secondary active transporter that uses the sodium electrochemical gradient to transport sugar substrates uphill against a concentration gradient (2, 9, 22). SGLT1 is expressed most abundantly at the mucosal surface of the small intestine and serves as the principal uptake pathway for glucose derived from dietary sources. Dysfunctional mutations in SGLT1 cause intestinal glucose/galactose malabsorption (22). For many years, SGLT1 has served as a model system for studying the molecular basis of ion-coupled cotransporters. In pursuit of this objective, structure/function studies have formed an important experimental strategy to identify key residues participating in cotransporter function.
Application of the substituted cysteine accessibility method (SCAM) has shown that the Na+ interaction domain is located in the NH2-terminal half of SGLT1 and involves residues 163, 166, 170, and 173 in the putative external loop joining transmembranes (TM) IV-V (7, 8, 10, 11). The polar residues at position 176 hydrogen bond to the hydroxyl group on the β-phenyl ring of phloridzin (18). There is also evidence that D454 in the putative external loop joining TM X-XI is involved in the coupling of Na+ and sugar in the transport process (4). The sugar binding domain, on the other hand, has been localized to the COOH-terminal half of SGLT1 (16). Q457, located in the putative external loop joining TM X-XI, appears to be particularly important because sugar transport is abolished by reaction of methanethiosulfonate (MTS) reagents and maleimides with Q457C (13). However, under these conditions, the transporter still binds Na+ and sugar (13). These results suggest that although residue 457 is at or near the sugar-translocation site, other unidentified residue(s) must be involved in the interaction with sugar ligand. Interestingly, there is evidence that A166 is important for the interaction between the Na+ and sugar pathways and that helices TM IV-V are close to TM X-XI (14). Therefore, the aim of the present investigation was to explore whether residues within predicted TM IV participate in sugar interaction.
In the 34 different missense mutations that were identified for glucose/galactose malabsorption (GGM) (22), four mutations are located in TM IV and the putative loop joining TM IV-V (R135W, L147R, S159P, and A166T). In the region of TM IV, 19 of 24 residues (138-161) are conserved in all members of SGLT1 and SGLT2. Previous studies have shown that several residues important for substrate and cation recognition are located in the putative external loop joining TM IV-V in the NH2-terminal half of the transporter (7, 8, 10, 11, 14). Therefore, it is possible that TM IV may be important for SGLT1 function.
The present study describes the results from detailed investigation of two single cysteine mutants, K157C and T156C, which are conserved across species in SGLT1 and SGLT2. Our results suggest that manipulation of positive charge and polarity at positions 157 and 156, respectively, significantly affects sugar binding and sugar-induced Na+ currents.
MATERIALS AND METHODS
Molecular Biology
The cysteine mutations were generated via the megaprimer protocol of polymerase chain reaction mutagenesis as described previously and confirmed by sequencing (21).
Oocyte Preparation and Injection
Xenopus laevis were prepared as described previously (10). The oocytes were injected with 60 ng cDNA [empty vector pMT4, wild-type (WT) rSGLT1 or mutant rSGLT1]. The injected oocytes for the electrophysiology were stored at 16–18°C for 4 days or more before study. The present study was reviewed and approved by the Committee on Animal Research at the University of Toronto, Toronto, and was conducted in accordance with the committee's guidelines.
Electrophysiology
Voltage clamping and recordings were performed using a GeneClamp 500 amplifier, Digidata 1200B interface, and pClamp 9.0 data acquisition software (Axon Instruments, Union City, CA) as described previously (10). The oocytes were constantly superfused with a voltage-clamping solution consisting of 100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 10 mM HEPES-Tris base (pH 7.4) and held at a holding potential, Vh, of −50 mV, then was subjected to a series of voltage test pulses, Vt. The current responses were recorded with a sampling interval of 20 μs for pre-steady-state and steady-state experiments. The sampling frequency was 50 KHz. Results were filtered via a 1-kHz, 5-point Gaussian filter. Additional curve fitting was performed in ORIGIN 7.0 with the Levenberg-Marquardt algorithm; n is the number of observations.
Transient Current Measurements
The rSGLT1 pre-steady-state currents were determined as described previously (10). The pre-steady-state currents for each Vt were integrated over the entire course of the trace to calculate the total charge transferred by the cotransporter. The charge, Q, was plotted as a function of the test pulses, and these Q (Vt) curves were fitted to the two-state Boltzmann relation
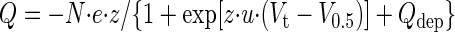 |
(1) |
where Q is the total charge transferred, Qdep is the charge due to depolarizing pulses, e is the elementary charge, z is the apparent valence of the movable charge, V0.5 is the potential at which half of the total charge transfer is complete, and N is the number of cotransporters expressed at the surface. The term u = F/RT; F is Faraday's constant, R is the gas constant, and T is absolute temperature.
Steady-state parameters were determined with the difference in the steady-state currents obtained before and after exposure to the substrate as described previously (10). Steady-state currents were acquired with test pulses of 300-ms duration. The final 100 ms of a test pulse were selected and the average current value of this range was acquired. The average current values were plotted versus [substrate] and the following equation was fit to the curve
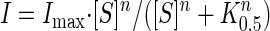 |
(2) |
where S is the substrate of investigation (Na+, αMG), Imax is the maximal current induced at saturating substrate concentration ([S]), n is the Hill coefficient, and K0.5 is the Michaelis constant, which is the [S] at which the I = Imax/2, which serves as an approximation of substrate affinity. The calculation of substrate affinity values used the Imax values of −150 mV test pulses.
Protocols for Chemical Modification
Cysteine-specific reagents (1 mM) (2-aminoethyl) methanethiosulfonate hydrobromide (MTSEA), anionic sodium(2-sulfonatoethyl)methanethiosulfonate (MTSES), or [(2-trimethylammonium)ethyl] methanethiosulfonate bromide (MTSET) (Toronto Research Chemicals, Toronto, ON, Canada) were dissolved in a voltage-clamping solution consisting of 100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 10 mM HEPES-Tris base (pH 7.4) immediately before use. The oocytes expressing mutant were labeled with the bath solution including cysteine-specific reagents for 10 min, with membrane clamped at −50 mV.
Cell Transfection and Western Blot Detection
NH2-terminal myc-tagged WT and mutants used for COS-7 transfections were prepared as described previously (21). Nontransfected cells and COS-7 cells transfected with vector alone served as controls. Proteins samples were resolved on 10% SDS-PAGE and transferred to nitrocellulose. The myc-epitope was detected with mouse monoclonal 9E10 (anti-c-myc, 1:1,000) antibody (Berkeley Antibody), followed by peroxidase-conjugated anti-mouse IgG (1:200,000) (Sigma). Immunoblots were developed by chemiluminescence, and area analysis was performed using the public domain NIH Image program (developed at National Institutes of Health). Western blot analysis for β-actin was performed to check equal loading.
Labeling of Surface-Expressed T156C in COS-7 Cells With Biotin-MTSEA
MTS compounds (Toronto Research Chemicals) were prepared fresh in (DMSO) and diluted to 1 mM concentrations in PBS (pH 7.4) immediately before use. Various protocols were used as described in results. Typically, cells in each well (1 × 105 cells/well, 12-well plate) were first preincubated for varying times at room temperature with either 500 μl PBS (control), 1.35 mM phloridzin, 1 mM MTSEA-alone, or in different combinations. The cells in each well were then washed (over 10–20 s) and immediately exposed to 500 μl 1 mM biotin-MTSEA for varying times up to 10 min at room temperature. Cells in each well were then washed three times with 2 ml cold PBS and individual wells were scraped into 0.5 ml lysis buffer, (50 mM Tris·HCl, 150 mM NaCl, 1% Triton, 1% SDS, 1 mM EDTA, and protease inhibitor cocktail). Samples were rocked at 4°C for 30 min, and the insoluble protein was removed by centrifugation at 14,000 rpm for 15 min. Biotin-labeled proteins were isolated from the cell lysates with immobilized streptavidin-agarose (Sigma) (10% total volume, ∼50 μl) by incubating overnight at 4°C with gentle agitation. The beads were washed, and the biotinylated protein was eluted from the beads by the addition of 50 μl SDS-PAGE sample buffer (4% SDS) at 100°C for 3 min.
Statistical Comparisons of Means
Data are presented with means ± SE. A one-way analysis of variance followed by Tukey's honestly significant difference post hoc test was applied to the entire data set using SPSS software (SPSS, Chicago, IL) to determine whether significant differences existed between mean values. Statistical significance was accepted at an α-level of P < 0.05.
RESULTS
In the present study, we used SCAM and chemical modification by MTS derivatives to study the role of residues of K139-D161 that lie within predicted TM IV (Fig. 1). Five charged and polar residues (K139, Q142, T156, K157, and D161) and two GGM missense mutations (I147 and S159) (22) were replaced with cysteine. Several of these (I147C, T156C, and K157C) exhibited sufficient expression to be studied in detail using the two-electrode voltage-clamp method in Xenopus laevis oocytes. I147C was similar in function to WT and was not studied further (unpublished observations).
Fig. 1.

The secondary topology model of the high-affinity Na+-glucose cotransporter SGLT1 (19). The functional importance of residues (T156, K157, F163, A166, Q170, L173, D176, D454, and Q457) and domains is indicated.
Characterization of K157C
Pre-steady-state behavior of K157C compared with WT.
Figure 2 shows representative pre-steady-state currents from oocytes expressing WT or mutant K157C in the presence of 100 mM Na+. Saturating concentrations of the competitive inhibitor phloridzin (200 μM) (Fig. 2, A and B) or sugar substrate, αMG (10 mM) (Fig. 2, A and C), eliminate WT transient currents, indicating that binding of phloridzin and αMG are intact. Transfection with empty vector (pMT4) does not give rise to pre-steady-state currents (Fig. 2D). The specific phloridzin-sensitive transient currents for WT are shown in Fig. 2E. In contrast to WT, the transient current of mutant K157C remains essentially unaffected by exposure to either phloridzin (Fig. 2, H and I) or αMG (Fig. 2, H and J). The phloridzin-sensitive transient currents for the mutant K157C are shown in Fig. 2F and are significantly lower than WT (Fig. 2E). The phloridzin binding of mutant K157C could be studied by the effect of the external phloridzin on the pre-steady-state charge movement. The pre-steady-state charge movement of mutant K157C, which shows the loss of phloridzin binding for mutant K157C, was obtained by integrating the phloridzin-sensitive pre-steady-state currents (Fig. 2G).
Fig. 2.

Representative pre-steady-state transient currents generated with a voltage clamp over a range of −150 mV to +70 mV in oocytes expressing wild-type (WT) and mutant K157C, in the presence and absence of 0.2 mM phloridzin (PZ) or 10 mM α-methyl-d-glucopyranoside (αMG). In all cases, the Na+ concentration was 100 mM, except for K, in which currents were measured at 0 mM Na+. A: WT transient currents. B: WT transient currents after exposure to PZ. C: WT transient currents after exposure to αMG. D: transient currents from an oocyte injected with empty pMT4 plasmid. E: WT-specific PZ-sensitive transient currents (IWT). F: K157C-specific PZ-sensitive transient currents (IK157C). G: charge transfer (Q) characteristics of WT rSGLT1 (n = 3) compared with K157C rSGLT1 (n = 5). H: K157C transient currents. I: K157C transient currents after exposure to PZ. J: K157C transient currents after exposure to αMG. K: K157C transient currents in 0 mM Na+.
Site-directed alkylation of cysteine-157 (K157C) rescued activity of SGLT1.
Since mutation of positively charged lysine-157 to neutral cysteine abolishes both phloridzin and αMG binding, we developed a rescue strategy designed to recover phloridzin and αMG binding by restoring a positive charge at position 157. Accordingly, we exposed oocytes expressing K157C to the positively charged MTS derivative, MTSEA, which mimics the lysine side chain at position 157 (Fig. 3). Previous work has established that exposure of oocytes expressing WT to various MTS derivatives does not alter SGLT1 function (9).
Fig. 3.

Rescue strategy for activity of SGLT1 by site-directed alkylation of cysteine-157 (K157C).
Figure 4 shows that following exposure to MTSEA (Fig. 4A), phloridzin (Fig. 4B), and αMG (Fig. 4C), inhibition of mutant K157C transient currents are restored, demonstrating that exposure of K157C to MTSEA rescues phloridzin and αMG binding. The net phloridzin-sensitive K157C transient currents (obtained by subtracting the currents in Fig. 4A from Fig. 4B) are shown in Fig. 4E. We attempted to study phloridzin and αMG binding in mutant K157R. We found that there was insufficient expression of mutant K157R in oocytes to permit further investigation (confirmed by Western blot analyses of COS-7 cells; unpublished observations).
Fig. 4.

Representative pre-steady-state transient currents generated with a voltage clamp over a range of −150 mV to +70 mV in oocytes expressing mutant K157C after reaction with (2-aminoethyl) methanethiosulfonate hydrobromide (MTSEA), in the presence and absence of 0.2 mM PZ or 10 mM αMG. In all cases, the Na+ concentration was 100 mM, except for D, in which currents were measured at 0 mM Na+. A: K157C-MTSEA transient currents. B: K157C-MTSEA transient currents after exposure to PZ. C: K157C-MTSEA transient currents after exposure to αMG. D: K157C-MTSEA transient currents in 0 mM Na+. E: K157C-MTSEA-specific PZ-sensitive transient currents (IK157C-MTSEA).
The pre-steady-state currents of mutant K157C-MTSEA for each Vt (Fig. 4E) were integrated over the entire course of the trace to calculate the total charge transferred by the cotransporter. The charge, Q, was then plotted as a function of the test pulses, and these Q (Vt) curves were fitted to the two-state Boltzmann relation. The V0.5 value of K157C-MTSEA is −9.2 ± 1.4 mV (n = 5), whereas the V0.5 value of WT is −1.5 ± 5.1 mV (n = 5). The z-values of the two are also comparable (0.88 for K157C-MTSEA vs. 1.01 for WT). The pre-steady-state Boltzmann parameters in 100 mM Na+ for both WT and rescued K157C-MTSEA are very similar, suggesting equivalent voltage sensitivity.
To determine the extent of the rescue achieved by restoring a positive charge at position 157, we estimated mutant K157C expression relative to WT. SGLT1 pre-steady-state currents yields three time constants τ (τfast, τmedium, and τslow) (9, 12), and the oocyte capacitive membrane current, which is not relevant to transporter activity, is <1 ms (2, 9). Loo et al. (12) estimated WT hSGLT1 expression by adding the charge transferred associated with the medium and slow decays (Qmed and Qslow). We used the same approach to calculate the charge transferred in nC from the medium and slow decays for K157C and rWT (K157C 3.8 ± 0.7 nC, n = 8 vs. WT 12.8 ± 0.4 nC, n = 3). From this ratio, the estimated expression of mutant K157C is ∼30% of WT. Thus we estimate the rescue of K157C sugar binding activity following chemical modification by MTSEA to be >85% (Fig. 5).
Fig. 5.

Results demonstrating the effects on K157C charge transfer in oocytes treated with various 1 mM sulfhydryl-specific reagents (MTS derivatives) in 100 mM Na+ (n ≥ 3 for each compound tested). The error bars represent SE. A one-way analysis of variance (ANOVA) was conducted to determine whether significant differences existed in the maximal charge transfer (Qmax) of K157C (capacitive currents removed), empty pMT4 + PZ, K157C + PZ, K157C + αMG, and K157C reacted with various MTS reagents + PZ or αMG. Following ANOVA, Tukey's honestly significant difference (HSD) post hoc test was applied to the entire data set shown in lines 1–14. MTSES, anionic sodium(2-sulfonatoethyl)methanethiosulfonate; MTSET, [(2-trimethylammonium)ethyl] methanethiosulfonate bromide. Statistical significance was accepted at an α-level of P < 0.05. *Significant statistical pairwise comparisons between lines 3 and 5 and between lines 4 and 6.
Figure 5 also shows the results following chemical modification of cysteine-157 in K157C by two membrane-impermeant MTS derivatives (positively charged MTSET and negatively charged MTSES). In both cases, rescue of phloridzin- and αMG-dependent charge transfer is much less than that achieved following treatment with MTSEA (Fig. 5; lines 7 and 11 versus line 5; lines 9 and 13 versus line 6), but pretreatment with MTSET or MTSES blocks the ability of MTSEA to restore K157C phloridzin and αMG binding (Fig. 5; lines 8 and 12 versus line 5; lines 10 and 14 versus line 6). Since the MTS reagents did not affect WT rSGLT1 (10), the changes observed in Fig. 5 must arise from alkylation of cysteine-157 after exposure to MTSEA. Therefore, the inability of MTSET or MTSES to rescue K157C sugar binding is not due to inaccessibility to cysteine-157 because MTSEA rescued sugar binding. Since MTSET and MTSES are both membrane-impermeant and since chemical modification of cysteine-157 in K157C by either derivative blocks reactivity to MTSEA, this indicates that cysteine-157 is located exofacially.
Figure 6, A and B, shows that the charge transfer following reaction of K157C with MTSEA (i.e., of K157C-MTSEA) depends on the sodium concentration in the bath solution. αMG-induced Na+ currents are also Na+ dependent (Fig. 6C). Na+ dependence of K157C-MTSEA thus mirrors Na+ dependence of WT rSGLT1 (8) and provides further evidence that MTSEA exposure restores sugar-induced Na+ currents and phloridzin binding.
Fig. 6.

Comparison of the effects of 100 mM Na+ (▪), 40 mM Na+ (•), and 10 mM Na+ (▴) on mutant K157C reacted with 1 mM MTSEA in the same oocyte. A: effects on PZ-dependent charge transfer. B: effects on αMG-dependent charge transfer. C: effects on αMG-induced Na+ currents in the presence of varying concentrations of Na+.
To determine whether reactivity of K157C with MTSEA depends on sodium, oocytes were exposed to MTSEA in either 100 mM Na+ or 0 mM Na+ (choline buffer). As shown in Fig. 7, A and B, reactivity of 1 mM MTSEA with cysteine-157 in K157C is not influenced by the presence of Na+. K157C-MTSEA, labeled in the absence of Na+, restores both phloridzin and αMG binding. Furthermore, when the same oocyte is pretreated with MTSEA in the absence of Na+ and is then treated with MTSEA in the presence of 100 mM Na+, there is no additive effect on rescue of phloridzin and αMG binding.
Fig. 7.

Typical results demonstrating the effects of pretreatment with 1 mM MTSEA on mutant K157C charge transfer in the absence of Na+ (choline replacement) or in the presence of 100 mM Na+. A: effects on charge transfer. B: effects on αMG binding. C: effects on αMG-induced Na+ currents.
Steady-state αMG-induced Na+ currents of K157C and K157C-MTSEA.
Figure 8A shows the maximal current induced by 10 mM αMG for K157C and for K157C reacted with various MTS reagents. Neither K157C-MTSET nor K157C-MTSES rescues sugar binding and sugar-induced Na+ currents (Fig. 8A, lines 6 and 8 versus line 4). This is in contrast to the effect of chemical modification by MTSEA. As shown in Fig. 6C, reaction with MTSEA (K157C-MTSEA) restores Na+-dependent sugar-induced Na+ currents, and the Na+ leak is very small (unpublished observations). However, when the same oocyte that was pretreated with MTSET or MTSES is then reacted with MTSEA in the presence of Na+, sugar binding and sugar-induced Na+ currents were not restored (Fig. 8A, lines 7 and 9 versus line 5). Although MTSET or MTSES does not affect sugar binding and sugar-induced Na+ currents, pretreatment with these agents is able to block reactivity of MTSEA, thus indicating that MTSET and MTSES are able to alkylate cysteine-157 in K157C.
Fig. 8.

A: MTSEA rescue of K157C activity. Figure displays maximal αMG-induced Na+ currents (Imax) measured at −150 mV in oocytes expressing WT whose expression is ∼3 times higher than that of K157C, or empty pMT4 plasmid, or mutant K157C before or after reaction with MTS compounds. Error bars represent SE. The number of independent experiments is given in parentheses. ANOVA was conducted to determine whether significant differences existed in the Imax levels of WT, empty pMT4, empty pMT4-MTSEA, K157C, and K157C reacted with various MTS reagents. Following ANOVA, Tukey's HSD post hoc test was applied to the entire data set shown in lines 1-9. Statistical significance was accepted at an α-level of P < 0.05. *Difference compared with Imax of K157C before and following rescue with MTSEA at −150 mV (P < 0.05). B: αMG apparent substrate concentration at 0.5 Imax (K0.5) of WT (10), T156C, and K157C-MTSEA for voltage dependence (n ≥ 3). Error bars represent SE.
To determine the affinity of rescued K157C-MTSEA for sugar substrate αMG, we measured αMG-induced steady-state currents at various sugar concentrations over a range of holding potentials, and the resulting curves were fitted to the Michaelis-Menten relation. As shown in Fig. 8B, the K0.5 of restored mutant K157C-MTSEA activity exhibited voltage dependence from −150 mV to −50 mV, similar to WT rSGLT1 (K0.5 is 0.15 ± 0.02 mM at −150 mV) (10). The αMG apparent K0.5 of K157C-MTSEA was 2.65 ± 0.93 mM (V = −150 mV) and 5.81 ± 2.46 mM (V = −50 mV) (significantly different, P ≅ 0.05). K157C-MTSEA Imax was 28.99 ± 5.99 nA (V = −150 mV) and 6.48 ± 1.68 nA (V = −50 mV), (significantly different, P < 0.05). Therefore the αMG affinity of K157C-MTSEA is decreased about 18-fold compared with WT.
In summary, mutation of the positively charged lysine residue at position 157 abolishes sugar and phloridzin binding. Replacement of the positive charge by MTSEA, however, restores activity.
Characterization of T156C
Given the substantial effects on sugar binding and sugar-induced Na+ currents that arise following mutation of lysine-157 to neutral cysteine, we investigated whether mutation of the neighboring residue, threonine-156, would affect SGLT1-sugar interaction.
Steady-state kinetics of T156C.
A plot of the apparent αMG affinity as functions of V is shown in Fig. 8B. The affinity of T156C for αMG (at V = −50 mV, K0.5 = 0.95 ± 0.06 mM) was decreased about 5-fold compared with WT. When Na+ dependence of T156C sugar-induced Na+ currents was analyzed by fitting the Hill equation, K0.5 ranged from 28.9 ± 1.4 mM at −30 mV to 22.9 ± 3.7 mM at −150 mV. The corresponding values for n, the Hill coefficient, ranged from 1.0 to 2.0. The Na+ affinities of T156C were decreased about 6-fold at −150 mV and same at −30 mV compared with WT (11, 17).
Apparent affinity of T156C for phloridzin.
Binding of phloridzin to WT SGLT1 inhibited αMG-induced currents by competing for the sugar-binding site, reduced the leak current by locking the transporter in a nontransporting state, and eliminated the charge movements that give rise to the transient currents. By measuring the amount of transient current eliminated as a function of phloridzin concentration, an estimate of the apparent affinity of the transporter for phloridzin could be made.
We have determined that the phloridzin binding affinity of T156C was decreased ∼20-fold compared with WT (Krofchick D and Silverman M, unpublished observations). These values for phloridzin binding are consistent with the apparent reduced affinity for αMG demonstrated in Fig. 8B. Therefore, a threonine-to-cysteine mutation at position 156 causes a significant reduction in binding affinity for both sugar and phloridzin.
Chemical modification of T156C by MTS reagents.
The T156C I-V curves in the absence or presence of 2 mM MTSEA are shown in Fig. 9A. After reacting mutant T156C with MTSEA, the Na+ leak current is increased ∼10-fold, and the αMG-induced Na+ current is almost abolished (Fig. 9A). These results are similar to those reported for mutant Q457C (13). This effect was observed in the absence of Na+ (choline+ replacing Na+) as well as in its presence (unpublished observations). The fluorophore TMR6M, which blocks sugar-induced Na+ currents in Q457C (11), also abolishes αMG-induced Na+ currents in T156C (Fig. 9B), but the Na+ leak currents of T156C are very small (unpublished observations). A similar exposure (10 min) to 2 mM MTSEA was previously shown to have no effect on the function of WT SGLT1 (10). When phloridzin was infused at the same time as MTSEA, phloridzin conferred partial protection against MTSEA modification of T156C. Phloridzin protection was only observed when inhibitor was present together with MTSEA in the infusing solution. It has not been possible to definitively test whether saturating concentrations of αMG could provide similar protection since prolonged exposure to high concentrations of αMG leads to paradoxical inactivation of the transporter.
Fig. 9.

A: steady-state currents induced by 20 mM αMG before and after MTSEA exposure and also after MTSEA exposure in the presence of 2 mM PZ in an oocyte expressing mutant T156C. B: steady-state currents induced by 20 mM αMG before and after TMR6M exposure in an oocyte expressing mutant T156C.
We then studied the apparent competition between phloridzin and MTSEA with respect to MTSEA reactivity with cysteine-156 (21). COS-7 cells expressing WT SGLT1 and T156C were each reacted with biotin-MTSEA. Fig. 10A demonstrates that biotin-MTSEA specifically labels cells expressing T156C, but not WT SGLT1. These results demonstrate that MTSEA has reacted with cysteine-156 in T156C. We then developed an assay system based on competition of MTSEA and biotin-MTSEA for cysteine-156. Fig. 10B shows that MTSEA prevents biotin-MTSEA from reacting with T156C, but it takes ∼6–7 min for maximal MTSEA effects to be observed. Phloridzin was able to protect the mutant T156C from reacting with MTSEA because after removal of phloridzin/MTSEA, then cells could still react with biotin-MTSEA.
Fig. 10.

A: specificity of biotin-MTSEA reaction to the cysteine at 156. Top: COS-7 cells were transfected with empty plasmid (pMT4), NH2-terminal myc-tagged WT, or NH2-terminal myc-tagged mutant T156C. Forty-eight hours after transfection, cells were reacted with biotin-MTSEA for 10 min at room temperature. The reaction was stopped by washing with PBS. Lysates were collected and reacted with streptavidin-agarose, and bound proteins were subjected to immunoblot analysis with anti-myc antibody. Bottom: before reacting with streptavidin, aliquots of each lysate were also immunoblotted with anti-myc antibody to verify expression and are shown directly below the corresponding sample. B: PZ protection of MTSEA accessibility/reactivity to cysteine-156 in mutant T156C. Top: COS-7 cells expressing myc-tagged mutant T156C were preexposed to either MTSEA alone or MTSEA in the presence of 1.35 mM PZ for the indicated times. Cells were then washed and treated for 10 min with biotin-MTSEA. The reaction was stopped by washing with PBS, and cells were allowed to recover for 10 min at 37°C. Cell lysates were collected and reacted with streptavidin-agarose, and bound proteins were subjected to immunoblot analysis with anti-myc antibody. Bottom: aliquots of each cell lysate were also immunoblotted with anti-myc antibody before reacting with streptavidin to verify equal expression of the myc-tagged mutants and are shown directly below the corresponding sample.
In summary, mutation of the polar amino acid to a neutral residue (T156C) reduces the affinity for both αMG and phloridzin. Conversely, phloridzin protects cysteine-156 from alkylation by MTSEA.
DISCUSSION
We identify two residues (threonine-156 and lysine-157) in predicted TM IV that are important for sugar binding. Wright and coworkers (16) have previously proposed the sugar domain to be established by TM X-XIII in the COOH-terminal half of SGLT1. Their laboratory also demonstrated that reacting Q457C with MTSEA inhibited sugar transport, but the mutant could still bind sugar (11). These results suggest that other residues may be important for binding of sugar. Recently, Puntheeranurak et al. (19) expressed rSGLT1 in COS-7 and G6D3 cells and found that mutants C255 (in the putative external loops joining TM VI-VII) and C608 (the putative external loops joining TM XIII-XIV) formed a disulfide bridge. These results suggest that the putative external loops joining TM VIII-IX are involved in sugar binding. Gagnon et al. (5), using the Xenopus laevis oocytes expression system, showed that mutants C255 and C511 form a disulfide bridge, suggesting the putative loop TM VI-VII is close to TM XI-XIII. Despite the discrepancy between these two sets of observations (which may be due to the different experimental systems used), both studies suggest that the putative loop TM VI-VII also participates as part of the extracellular binding pocket for sugar.
Lysine-157 is conserved in all members of both SGLT1 and SGLT2. The finding that mutation of lysine-157 to alanine in hSGLT1 apparently results in “impaired transport” (20) supports the notion that lysine-157 is an important residue in sugar binding. In the present study, we also found that lysine-157 is important for sugar binding. Mutation of positively charged lysine to cysteine abolishes sugar and phloridzin binding. Restoration of the positive charge, however, by chemical modification of cysteine-157 with MTSEA rescues sugar binding and sugar-induced Na+ currents. These results suggest that sugar-induced Na+ currents may indirectly represent sugar transport or could represent sensing of glucose by SGLT1 as shown for human SGLT3 by Diez-Sampedro et al. (3). In contrast to the functional rescue by MTSEA, reaction of K157C with either anionic MTSES or bulky cationic MTSET did not lead to rescue of sugar binding even though both were able to label cysteine-157 (Fig. 5). We suggest the rescue of sugar binding by chemical modification of K157C with MTSEA is a result of MTSEA's ability to mimic the guanidino group of lysine (Fig. 3).
The properties of mutant T156C also suggest that TM IV participates in sugar binding. We observe an approximate 5-fold decrease in αMG affinity and at least a 20-fold reduction in phloridzin affinity. The mutant shows a modest reduction in Na+ affinity primarily at hyperpolarizing potentials, and reduced voltage dependency (unpublished observations). Interestingly, phloridzin protects mutant T156C from alkylation by MTSEA. Finally, αMG-induced Na+ currents for the mutant T156C are blocked after exposure to TMR6M. These results are similar to those observed for Q457C (13).
Alkylation of mutants T156C and K157C with MTSEA is independent of Na+, whereas alkylation of Q457C with MTSEA depends on the presence of Na+. Similarly, MTSEA also reacts with A166C in the absence or the presence of Na+ (11). In this regard, alkylation of mutants T156C, K157C, and A166C is similar to that of mutant G527 (TM XIII) of hSGLT1 (6) and the sodium/dicarboxylate cotransporter (15), all of which are also independent of Na+. However, phloridzin binding and sugar-induced Na+ currents of mutants T156C, K157C-MTSEA, and Q457C are all dependent on sodium. Binding of Na+ appears to produce a conformational change in the TM XI region of mutant Q457 compared with TM IV and/or the putative external loop between TM IV and V region of mutants T156, K157, and A166. A large conformational change in the region of TM XI (Q457) may be required to allow access to extracellular ligand in the late stages of the transport cycle.
Finally, there are two alternative interpretations of the collective data presented in the present study. One possibility is that both lysine-157 and threonine-156 directly participate in binding of sugar substrate. This is plausible in view of the evidence derived from the crystallographic structure of lactose permease, that positively charged residues can participate in hydrogen bonding with sugar ligands (1). Another possibility is the mutation of the charge and polarity at positions 157 and 156 causes a conformational change in SGLT1, which alters sugar interaction with its binding pocket in the protein. Whichever interpretation is correct, there seems to be reciprocity in that phloridzin protects against alkylation of MTSEA at position 156 due to either the glycoside moiety of phloridzin competing directly for binding to position 156 or phloridzin occupancy of the sugar binding site at some other location which induces a conformational change that prevents MTSEA accessibility to position 156. Taken together, these results suggest that TM IV participates in sugar interaction with SGLT1.
GRANTS
This work was supported by Canadian Institutes of Health Research Grant MOP-15267.
Acknowledgments
We thank Dr. P. Backx and Dr. R. Tsushima for helpful discussion.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science 301: 610–615, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Chen XZ, Coady MJ, Lapointe JY. Fast voltage clamp discloses a new component of presteady-state currents from the Na+-glucose cotransporter. Biophys J 71: 2544–2552, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diez-Sampedro A, Hirayama BA, Osswald C, Gorboulev V, Baumgarten K, Volk C, Wright EM, Koepsell H. A glucose sensor hiding in a family of transporters. Proc Natl Acad Sci USA 100: 11753–11758, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diez-Sampedro A, Loo DD, Wright EM, Zampighi GA, Hirayama BA. Coupled sodium/glucose cotransport by SGLT1 requires a negative charge at position 454. Biochemistry 43: 13175–13184, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Gagnon DG, Bissonnette P, Lapointe JY. Identification of a disulfide bridge linking the fourth and the seventh extracellular loops of the Na+/glucose cotransporter. J Gen Physiol 127: 145–158, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirayama BA, Loo DD, Diez-Sampedro A, Leung DW, Meinild AK, Lai-Bing M, Turk E, Wright EM. Sodium-dependent reorganization of the sugar-binding site of SGLT1. Biochemistry 46: 13391–13406, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Huntley SA, Krofchick D, Silverman M. A glutamine to glutamate mutation at position 170 (Q170E) in the rabbit Na+/glucose cotransporter, rSGLT1, enhances binding affinity for Na+. Biochemistry 45: 4653–4663, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Huntley SA, Krofchick D, Silverman M. Position 170 of rabbit Na+/glucose cotransporter (rSGLT1) lies in the Na+ pathway: modulation of polarity/charge at this site regulates charge transfer and carrier turnover. Biophys J 87: 295–310, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krofchick D, Silverman M. Investigating the conformational states of the rabbit Na+/glucose cotransporter. Biophys J 84: 3690–3702, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo B, Silverman M. Cysteine scanning mutagenesis of the segment between putative transmembrane helices IV and V of the high affinity Na+/Glucose cotransporter SGLT1. Evidence that this region participates in the Na+ and voltage dependence of the transporter. J Biol Chem 273: 29341–29351, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Lo B, Silverman M. Replacement of Ala-166 with cysteine in the high affinity rabbit sodium/glucose transporter alters transport kinetics and allows methanethiosulfonate ethylamine to inhibit transporter function. J Biol Chem 273: 903–909, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Loo DD, Hirayama BA, Cha A, Bezanilla F, Wright EM. Perturbation analysis of the voltage-sensitive conformational changes of the Na+/glucose cotransporter. J Gen Physiol 125: 13–36, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loo DD, Hirayama BA, Gallardo EM, Lam JT, Turk E, Wright EM. Conformational changes couple Na+ and glucose transport. Proc Natl Acad Sci USA 95: 7789–7794, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meinild AK, Loo DD, Hirayama BA, Gallardo E, Wright EM. Evidence for the involvement of Ala 166 in coupling Na+ to sugar transport through the human Na+/glucose cotransporter. Biochemistry 40: 11897–11904, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Pajor AM, Randolph KM. Conformationally sensitive residues in extracellular loop 5 of the Na+/dicarboxylate co-transporter. J Biol Chem 280: 18728–18735, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panayotova-Heiermann M, Eskandari S, Turk E, Zampighi GA, Wright EM. Five transmembrane helices form the sugar pathway through the Na+/glucose cotransporter. J Biol Chem 272: 20324–20327, 1997. [DOI] [PubMed] [Google Scholar]
- 17.Panayotova-Heiermann M, Loo DD, Lam JT, Wright EM. Neutralization of conservative charged transmembrane residues in the Na+/glucose cotransporter SGLT1. Biochemistry 37: 10522–10528, 1998. [DOI] [PubMed] [Google Scholar]
- 18.Panayotova-Heiermann M, Loo DD, Lostao MP, Wright EM. Sodium/d-glucose cotransporter charge movements involve polar residues. J Biol Chem 269: 21016–21020, 1994. [PubMed] [Google Scholar]
- 19.Puntheeranurak T, Kasch M, Xia X, Hinterdorfer P, Kinne RK. Three surface subdomains form the vestibule of the Na+/glucose cotransporter SGLT1. J Biol Chem 282: 25222–25230, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Turk E, Kerner CJ, Lostao MP, Wright EM. Membrane topology of the human Na+/glucose cotransporter SGLT1. J Biol Chem 271: 1925–1934, 1996. [DOI] [PubMed] [Google Scholar]
- 21.Vayro S, Lo B, Silverman M. Functional studies of the rabbit intestinal Na+/glucose carrier (SGLT1) expressed in COS-7 cells: evaluation of the mutant A166C indicates this region is important for Na+-activation of the carrier. Biochem J 332: 119–125, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright EM, Hirayama BA, Loo DF. Active sugar transport in health and disease. J Intern Med 261: 32–43, 2007. [DOI] [PubMed] [Google Scholar]