

Abstract
Sodium-dependent transporters are inhibited indirectly by the Na-K-ATPase inhibitor ouabain. Here we report stimulation of sodium-hydrogen exchange (NHE) in ouabain-treated cells. BCECF was used to measure cytoplasmic pH in cultured rat optic nerve astrocytes. Ammonium chloride was applied to acid load the cells. On removal of ammonium chloride, cytoplasmic pH fell abruptly, then gradually recovered toward baseline. Ouabain (1 μM) did not change cell sodium content, but the rate of pH recovery increased by 68%. Ouabain speeded pH recovery both in the presence and absence of bicarbonate. In bicarbonate-free medium, dimethylamiloride, an NHE inhibitor, eliminated the effect of 1 μM ouabain on pH recovery. Western blot analysis showed an NHE1 immunoreactive band but not NHE2, NHE3, or NHE4. Immunoprecipitation studies showed phosphorylation of NHE1 in cells treated with 1 μM ouabain. Ouabain evoked an increase of cAMP, and the effect of 1 μM ouabain on pH recovery was abolished by H-89, a protein kinase A inhibitor. 8-Bromoadenosine-cAMP increased the pH recovery rate, and this recovery was not further increased by ouabain. Although 1 μM ouabain did not alter cytoplasmic calcium concentration, it stimulated calcium entry after store depletion, a response abolished by 2-APB. Ouabain-induced stimulation of pH recovery was suppressed by inhibitors of capacitative calcium entry, SKF-96365, and 2-APB, as well as the cytoplasmic calcium chelator BAPTA. The cAMP increase in ouabain-treated cells was abolished by BAPTA and 2-APB. Taken together, the results are consistent with increased capacitative calcium entry and subsequent cAMP-PKA-dependent stimulation of NHE1 in ouabain-treated cells.
Keywords: astrocytes; adenosine 3′,5′-cyclic monophosphate; capacitative calcium entry; pH recovery
important functions of astrocytes include buffering of potassium ions released from electrically active neurons (3), uptake of extracellular glutamate (45), fixation of exogenous ammonium by glutamine synthetase (17, 39), and regulation of extracellular pH (29). Neural activity as well as anoxia causes changes of extracellular pH in the optic nerve (18, 44). Glial acid-base transporters are thought to oppose pH shifts and preserve homeostasis. Mechanisms available to astrocytes for extracellular pH and intracellular pH regulation are one and the same. They include sodium-hydrogen exchange (NHE), sodium-dependent bicarbonate/chloride exchanger, sodium-independent bicarbonate/chloride exchanger, vacuolar proton ATPase (36), and Na+-HCO3− cotransporter (48). One NHE isoform, NHE1, is found in most cell types and consequently is thought to play a key role in cytoplasmic pH regulation. Clearly, the ability of NHE to shift protons outward depends on the difference in the concentration of sodium that exists across the plasma membrane. The sodium ion concentration inside the cell is approximately one-tenth the extracellular concentration, and sodium entering via the NHE provides the driving force needed to shift protons outward. It follows that the rate of NHE-mediated proton export is slowed when cytoplasmic sodium concentration rises. However, various hormones modulate NHE function without necessarily causing a change in cytoplasmic sodium concentration (31, 42).
Ouabain-like cardiac glycosides and ouabain itself bind to Na-K-ATPase in a very selective manner and inhibit Na-K-ATPase activity (1, 4, 7). At sufficient concentrations, cardiac glycosides suppress Na-K-ATPase-mediated sodium export, causing cytoplasmic sodium concentration to rise. As a result, transporters that depend on the sodium gradient, including NHEs, become inhibited indirectly. NHE inhibition would be expected to lead to a decrease in cytoplasmic pH but, curiously, an endogenous ouabain-like compound isolated from the urine of healthy volunteers has been shown to increase cytoplasmic pH in cultured vascular smooth muscle (37). Cell responses to ouabain can be more far reaching than alteration of cytoplasmic sodium and potassium concentration. In cells such as thymocytes, ouabain causes cytoplasmic calcium to rise (20). This is interesting because interactions between calcium balance and cytoplasmic pH have been reported in several cell types (9, 13, 41), and, indeed, in mast cells the ouabain-induced increase of cytoplasmic calcium concentration is paralleled by an increase of cytoplasmic pH (33).
Here we examined the effect of ouabain on rat optic nerve astrocytes. The results suggest ouabain treatment stimulates NHE, but the effect appeared not to be directly dependent on changes in cell-wide cytoplasmic sodium concentration. Instead, the findings were consistent with protein kinase A-mediated activation of NHE1 as a result of ouabain-induced stimulation of capacitative calcium entry and an increase of cAMP.
MATERIALS AND METHODS
Reagents.
DMEM/F12, BCECF-AM, fura-2 AM, BAPTA-AM, and pluronic F-127 were purchased from Invitrogen (Carlsbad, CA). KB-R7943 was obtained from Tocris Biosciences (Ellisville, MO). All other chemicals were purchased from Sigma (St. Louis, MO).
Polyclonal rabbit anti-glial fibrillary acidic protein was obtained from DakoCytomation (Carpinteria, CA). Mouse anti-NHE1 antibody was obtained from BD Transduction Laboratories (Lexington, KY). Rabbit anti-rat NHE2, NHE3, and NHE4 antibodies were obtained from Alpha Diagnostic International (San Antonio, TX). p44/42 MAP kinase rabbit monoclonal antibody, phospho-p44/42 MAPK (Thr202/Tyr204) mouse monoclonal antibody and phospho-(Ser) 14-3-3 binding motif mouse monoclonal antibody were obtained from Cell Signaling Technology (Danvers, MA). Rabbit anti-Na-K-ATPase α2 antibody, rat brain, and kidney microsomal preparations were obtained from Upstate Laboratories (Lake Placid, NY). Rat skeletal muscle lysate was obtained from Zyagen Laboratories (San Diego, CA). Alexa Fluor 680 goat anti-mouse secondary antibody was purchased from Invitrogen. Goat anti-rabbit IR dye 800 conjugated secondary antibody was obtained from Rockland (Gilbertsville, PA). The cAMP [125I] radioimmunoassay kit was obtained from Perkin-Elmer Life and Analytical Sciences (Shelton, CT).
Solutions.
Cytoplasmic pH, cAMP, sodium, and calcium measurements were carried out on cells bathed in bicarbonate-containing Krebs′ solution (in mM) NaCl, 119; KCl, 4.7; KH2PO4, 1.2; NaHCO3, 25; dextrose, 5.5; MgCl2, 1; CaCl2, 2.5 with a pH adjusted to 7.4 and an osmolarity of ∼300 mosM. As specified, cells were exposed to a modified Krebs solution containing 20 mM ammonium chloride added in substitution for an equimolar amount of NaCl. The composition of bicarbonate-free HEPES buffer was (in mM) 115 NaCl, 5 KCl, 2.5 CaCl2, 1.2 MgSO4, 2 NaH2PO4, 32.2 HEPES, 10 glucose, with a pH adjusted to 7.4 and an osmolarity of ∼300 mosM. Test agents were dissolved either in dimethylsulfoxide (0.06% final concentration) or in water according to solubility and were added to the Krebs solution to obtain intended final concentration. Control cells received only the vehicle. Nigericin was dissolved in methanol.
Cell culture.
Optic nerve astrocytes were isolated and cultured according to a modification of a previous method (24). Eyes from 1- to 5-day-old rat pups were obtained from Hilltop Laboratories (Scottsdale, PA) and washed in DMEM/F12 containing penicillin (100 U/ml) and streptomycin (0.1 mg/ml). The optic nerve was dissected, plated onto 35 mm culture dishes, and maintained at 37°C in a humidified atmosphere of 95% air-5% CO2 in DMEM/F12 medium containing fetal bovine serum (10%), penicillin (100 U/ml), streptomycin (0.1 mg/ml), and epidermal growth factor (5 ng/ml). The medium was changed every alternate day. After 7–8 days, when enough cells had spread out onto the petri dish, the remnant of the optic nerve was removed, and the cells were immunocytochemically stained for expression of glial fibrillary acidic protein (GFAP) to confirm astrocyte identity. Cells that did not stain positively for GFAP were discarded. Cells were then trypsinized and passaged. Confluence usually obtained in 2 wk. Third and fourth passage cells were used in the present study.
Measurement of cytoplasmic pH and calcium concentration.
Cytoplasmic pH was recorded in cells loaded with BCECF by measuring the fluorescence intensity at alternating excitation wavelengths of 460 nm and 488 nm using a method described previously (27). Cells grown on 35-mm culture dishes were loaded for 3 min with 3 μM BCECF-AM in Krebs solution. After several washes, the BCECF-loaded cells were superfused for 5 min with control Krebs solution equilibrated with 95% air-5% CO2 at 37°C. The superfusate was then switched to Krebs solution containing 20 mM ammonium chloride. After 5 min, the superfusate was switched back to control Krebs solution. The rate of pH recovery during the first 2.5 min was calculated. The rate was roughly linear. At the end of each experiment the relationship between pH and the 488/460 fluorescence ratio was established by exposing the cells to a series of potassium-rich pH buffers containing 10 μM nigericin added to equilibrate cytoplasmic pH with the superfusate.
Using a strategy similar to that used for pH measurements, cytoplasmic calcium concentration was measured in cells loaded with fura-2 AM. The ratio of fluorescence intensity was determined at 340 nm versus 380 nm using the published method (24).
Western blot analysis.
Western blot analysis was performed following the method described previously (51). Briefly, the cells were homogenized in RIPA buffer containing 50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 10 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 10 mM sodium fluoride, 10% glycerol, 1% Triton X-100, 1% sodium deoxycholate, Complete Mini Protease Inhibitor Cocktail tablets (Roche Diagnostics, Indianapolis, IN) (3 tabs/20 ml), and 1 μM phenylmethyl-sulfonylfluoride. The solubilized material was added to Laemmli buffer and separated by electrophoresis on a 7.5% SDS-polyacrylamide minigel. Proteins were then transferred by electrophoresis to nitrocellulose membrane and then blocked for 1 h with Odyssey blocking buffer (LI-COR, Lincoln, NE). After blocking, the nitrocellulose membranes were incubated overnight at 4°C with anti-NHE1 (1:1,000), anti-NHE2 (1:1,000), anti-NHE3 (1:1,000), anti-NHE4 (1:1,000), or rabbit anti-Na-K-ATPase α2 (1:5,000) antibodies dissolved in the blocking buffer. After five washes in TTBS [30 mM Tris, 150 mM NaCl, and 0.5% (vol/vol) Tween 20 at pH 7.4], the nitrocellulose membranes were then incubated for 1 h either with secondary antibody conjugated with IR Dye 680 goat anti-mouse secondary antibody (1:20,000) (Alexa-Fluor, Invitrogen) for NHE1 or IR Dye 800 goat anti-rabbit secondary antibody for NHE2, -3, and -4 (Rockland). Protein bands were visualized by infrared laser scanning (Odyssey, LI-COR). For ERK1/2 and phospho-ERK1/2 analysis, samples were resolved on a 10% SDS-polyacrylamide minigel and probed with p44/42 MAP kinase rabbit monoclonal and phospho-p44/42 MAPK (Thr202/Tyr204) mouse monoclonal antibodies.
The phosphorylation of NHE1 was determined using a published method (50). Cells were lysed in ice-cold immunoprecipitation lysis buffer (pH 7.5) containing (in mM) 20 Tris·HCl, 150 NaCl, 1 EDTA, 1 EGTA, 2.5 sodium pyrophosphate, 1 β-glycerophosphate, 1 Na3VO4, 100 NaF, 1% Triton X-100, 0.1% SDS, and protease inhibitors. The samples were centrifuged at 14,000 g for 60 min at 4°C. Supernatant (250 μg protein) was incubated overnight at 4°C with mouse monoclonal antibody against phospho-Ser 14-3-3 protein binding motif (1:20). Immunocomplexes were mixed with protein G (Sigma) for 2 h at 4°C and washed with the lysis buffer without SDS. The immunocomplexes were dissociated from beads with the Laemmli buffer and heated for 5 min at 70°C. Protein sample was then immunoblotted for NHE1 as described above. It was not feasible to run a loading control because there is no reference protein that we can say with assurance remains unchanged. An equal amount of protein from control and treated cells was used. The assumption was made that the immunoprecipitation and Western blot efficiency was similar in the treated and control samples. Protein concentration was measured using a BCA protein assay kit (Pierce, Rockford, IL) based on a method described by Smith and colleagues (49) using bovine serum albumin as the standard.
Measurement of cell sodium.
Sodium was measured by atomic absorption spectrophotometry following a published method (26). Briefly, cells grown on culture dishes were washed with ice-cold isotonic magnesium chloride solution (100 mM MgCl2, pH adjusted to 7.4 with Tris base). The magnesium chloride solution was then removed, and the cells were digested in 30% nitric acid. The acid digest was diluted with deionized water, and the sodium content of the diluted cell lysate was measured by using an atomic absorption spectrophotometer (Analyst 100; Perkin-Elmer, Norwalk, CT) at wavelength of 566.5 nm.
Measurement of cAMP.
The effect of ouabain on cAMP concentration was measured in cells incubated in normal Krebs solution (pH 7.4) for 12.5 min. In a different experiment, ouabain-treated cells were subjected to same perturbation as in the case of pH measurement, i.e., 5-min exposure to modified Krebs solution containing 20 mM ammonium chloride, followed by a 2.5-min recovery in control Krebs solution. Preparation of sample from cultured cells and measurement of cAMP in the sample was carried out as described previously (47) using a cAMP [125I] radioimmunoassay kit (Perkin-Elmer Life and Analytical Sciences). Briefly, the Krebs solution was removed, and 700 μl chilled trichloroacetic acid (TCA; 6%) was added to each culture dish. The cells were scraped from the culture dish, and the cell/TCA mixture was frozen at −20°C. The samples were thawed rapidly at 37°C and sonicated for 2 min in a Misonix 3000 sonicator (power setting 2). The freeze, thaw, sonication cycle was repeated four times, and the mixture was centrifuged at 5,000 g for 20 min at room temperature. The supernatant was transferred to a 2 ml Eppendorf tube, the pellet was washed with 200 μl chilled TCA, and the mixture was centrifuged again. The supernatant was removed and added to the first supernatant. Pooled supernatant was used for cAMP assay, and the pellets were used for protein assay.
Data analysis.
One-way analysis of variance was used to compare multiple groups of data, and Student's t-test was used to compare paired data. A P value of <0.05 was considered as significant.
RESULTS
Effect of ouabain and 5-(N,N-dimethyl)amiloride hydrochloride on pH recovery.
Exposure of the cells to bicarbonate buffer containing 20 mM ammonium chloride caused a sharp increase in pH from the baseline value of 7.24 ± 0.01 (n = 37) to 8.25 ± 0.03. Removal of the ammonium chloride-containing test solution caused a sharp decrease in cytoplasmic pH to 5.88 ± 0.01, followed by a period of gradual recovery toward baseline (Fig. 1A). In control cells, the recovery was roughly linear during the initial 2.5 min. The recovery rate was 2.81 ± 0.15 × 10−3 pH U/s.
Fig. 1.
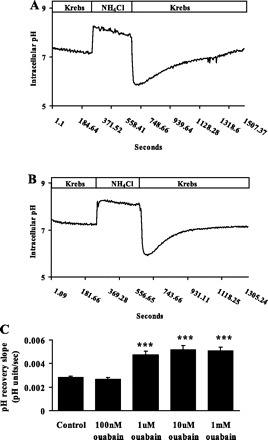
Cytoplasmic pH response of rat optic nerve astrocytes exposed to 20 mM ammonium chloride for 5 min. On removal of the ammonium chloride, pH fell sharply and then recovered toward the baseline. Typical recordings of pH recovery are shown for control cells (A) and in the presence of ouabain (1 μM; B). C: combined data on the rate of pH recovery measured in the presence of different ouabain concentrations maintained throughout the experiment. Values are means ± SE of results from 6–37 independent experiments. ***P < 0.001, significant difference from control.
In the presence of 1 μM ouabain, the rate of pH recovery during the initial 2.5 min was significantly increased to 4.73 ± 0.3 × 10−3 pH (n = 10) U/s. (Fig. 1, B and C). Baseline pH remained unchanged (Table 1). 1 μm, 10 μm, and 1 mM ouabain caused similar stimulation of pH recovery (Fig. 1C), although the two higher concentrations caused a small increase in baseline pH (Table 1). Dimethylamiloride (DMA), a recognized NHE inhibitor, significantly reduced the effect of 1 μM ouabain on pH recovery rate (Fig. 2).
Table 1.
Baseline cytoplasmic pH
| Experimental Condition | Baseline pH |
|---|---|
| Control | 7.24±0.01 (n = 37) |
| 100 nM ouabain | 7.22±0.01 (n = 6) |
| 1 μM ouabain | 7.23±0.02 (n = 10) |
| 10 μM ouabain | 7.32±0.01 (n = 6)† |
| 1 mM ouabain | 7.31±0.02 (n = 9)* |
| 1 mM 8-Br-cAMP | 7.21±0.02 (n = 10) |
| 100 μM DMA | 7.22±0.02 (n = 10) |
| 10 μM H-89 | 7.21±0.01 (n = 10) |
| 50 μM SKF-96365 | 7.22±0.01 (n = 4) |
| 50 μM 2-APB | 7.23±0.01 (n = 4) |
| 50 μM BAPTA-AM | 7.21±0.01 (n = 5) |
| 10 μM KB-R7943 | 7.20±0.01 (n = 6) |
| 50 μM PD-98059 | 7.21±0.01 (n = 6) |
Values are means ± SE of data from 4 to 37 independent experiments; in each experiment, data from 15–30 individual cells were averaged and considered as n = 1. Baseline pH was measured in bicarbonate-containing buffer. 8-Br-cAMP, 8-bromoadenosine-cAMP; DMA, 5-(N,N-dimethyl)amiloride hydrochloride.
P < 0.01,
P < 0.001, significant difference from control.
Fig. 2.
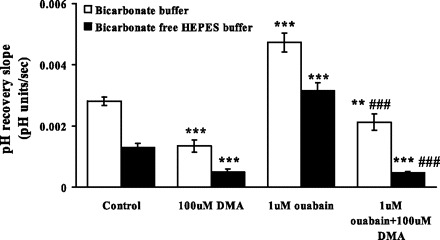
The effect of dimethylamiloride (DMA; 100 μM) and ouabain (1 μM) on the rate of cytoplasmic pH recovery after ammonium chloride removal in the presence and in the absence of bicarbonate. DMA was introduced after establishment of a steady baseline pH but before the addition of ammonium chloride. Ouabain was added 5 min after addition of DMA. DMA and ouabain were present throughout the rest of the experiment. Values are means ± SE of results from 3–37 independent experiments. **P < 0.05, ***P < 0.001, significant difference from respective control. ###P < 0.001, significant difference from 1 μM ouabain treatment.
To dissect the contribution of NHE and bicarbonate transporters to the ouabain response, studies were carried out in bicarbonate-free HEPES buffer. In the absence of bicarbonate, 1 μM ouabain still caused a marked increase in the pH recovery rate (Fig. 2). Baseline pH was not changed in bicarbonate-free buffer. Because the ouabain effect on pH recovery persisted in bicarbonate-free buffer, studies were conducted using DMA to examine the contribution of NHE. DMA (100 μM) did not change the baseline pH, but the pH recovery rate was reduced to 0.5 ± 0.1 × 10−3 pH U/s (n = 3). DMA eliminated the ability of 1 μM ouabain to increase the pH recovery rate (Fig. 2).
Expression of NHE1 and Na-K-ATPase α2.
Cultured astrocytes were subjected to Western blot analysis for NHE1, NHE2, NHE3, and NHE4. NHE1 was detected as an immunoreactive band at ∼110 kDa (Fig. 3A). Immunoreactive bands were not detected for NHE2, -3, or -4 (data not shown). In addition to the housekeeping Na-K-ATPase α1 isoform, glial cells express Na-K-ATPase α2. A dense Na-K-ATPase α2 immunoreactive band was detected in cultured rat optic nerve astrocytes (Fig. 3B).
Fig. 3.
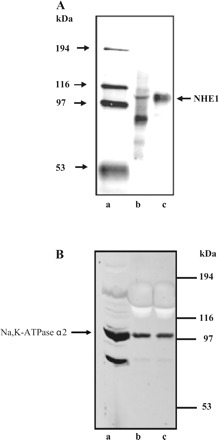
Detection of Na/H exchanger (NHE) isoform 1 (NHE1) (A) and Na-K-ATPase α2 (B) by Western blot analysis. A: lane a, molecular mass markers; lane b, rat kidney microsomes (10 μg); lane c, astrocyte lysate (15 μg). B: lane a, rat brain microsomes; lane b, rat skeletal muscle microsomes; lane c, astrocyte lysate (22 μg/lane).
Effect of ouabain on cell sodium.
Studies were conducted to examine the sodium content of cells subjected to an experimental protocol like that used in pH studies: a 5-min period of exposure to 20 mM ammonium chloride followed by a 2.5-min recovery period in control Krebs solution. The sodium content detected in control cells was similar to the sodium content detected in cells exposed to 1 μM ouabain throughout the course of the experiment. This agrees with an earlier report that 1 μM ouabain caused no detectable increase of sodium concentration in astrocytes loaded with the sodium-sensitive dye SBFI (24). At concentrations of 1 mM and 10 mM, ouabain increased cell sodium content significantly (Fig. 4).
Fig. 4.
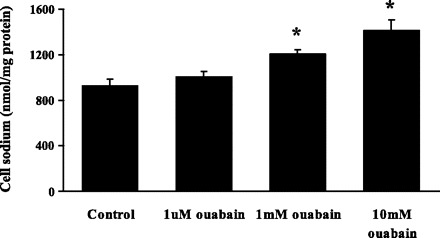
The effect of ouabain on the sodium content of cultured rat optic nerve astrocytes subjected to a 5-min period of exposure to 20 mM ammonium chloride followed by a 2.5-min recovery period in Krebs solution. Treated cells received ouabain (1 μM, 1 mM, or 10 mM) throughout the experiment. Values are means ± SE of results from 4 independent experiments. *P < 0.01, significant difference from control.
Effect of 8-bromoadenosine-cAMP and H-89.
8-Bromoadenosine-cAMP (8-Br-cAMP) is a cell-permeable analog of cAMP and a recognized activator of protein kinase A. When cells were exposed to 8-Br-cAMP (1 mM) throughout the experiment, the pH recovery rate following a 5-min exposure to ammonium chloride increased significantly. The rate of pH recovery observed in the presence of 8-Br-cAMP was similar to the rate of recovery observed in cells exposed to 1 μM ouabain. When 1 mM 8-Br-cAMP was added together with 1 μM ouabain, the rate of pH recovery was not significantly different from the rate of recovery measured in cells exposed to 1 μM ouabain or 1 mM 8-Br-cAMP alone (Fig. 5).
Fig. 5.
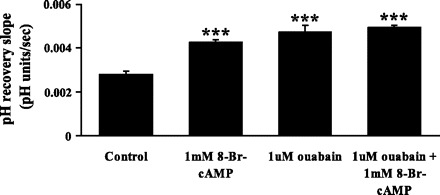
The effect of 8-bromoadenosine-cAMP (8-Br-cAMP; 1 mM) and ouabain (1 μM) on the rate of cytoplasmic pH recovery after ammonium chloride removal. Ouabain was present throughout the experiment. 8-Br-cAMP was introduced after establishment of a steady baseline pH but before the addition of ammonium chloride. Values are means ± SE of results from 10–37 independent experiments. ***P < 0.001, significant difference from control.
Some cells were exposed to 10 μM H-89, an inhibitor of cAMP-dependent protein kinase, protein kinase A. When added together with 1 μM ouabain, 10 μM H-89 abolished the ouabain-induced increase in the rate of pH recovery (Fig. 6). In cells exposed to H-89 alone, baseline pH was unchanged (Table 1), but the rate of pH recovery was lower than in control cells.
Fig. 6.
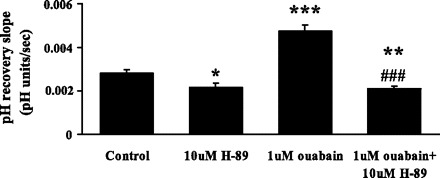
The effect of protein kinase A inhibitor H-89 (10 μM) and ouabain (1 μM) on the rate of cytoplasmic pH recovery after ammonium chloride removal. H-89 was introduced after establishment of a steady baseline pH but before the addition of ammonium chloride. Ouabain was added 5 min after H-89. H-89 and ouabain were present throughout the rest of the experiment. Values are means ± SE of results from 10–37 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, significant difference from control. ###P < 0.001, significant difference from 1 μM ouabain.
Effect of ouabain on intracellular cAMP.
Cyclic AMP was measured in control cells and in cells subjected to an episode of acidification like that used in the pH studies, i.e., a 5-min exposure to 20 mM ammonium chloride followed by a 2.5-min recovery period, representing the midpoint of the pH recovery phase. Ammonium chloride exposure did not alter cAMP. In contrast, ouabain (1 μM) increased cAMP by ∼60% (Fig. 7A). The magnitude of the increase was similar in cells exposed to 1 mM ouabain. Forskolin (10 μM), a direct adenylate cyclase stimulator, increased cAMP to a similar extent as ouabain (Fig. 7B).
Fig. 7.
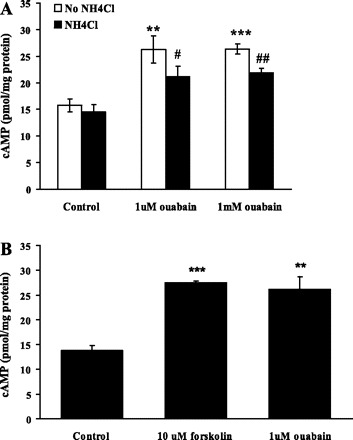
A: the effect of ouabain on cAMP in control cells and in cells subjected to 20 mM ammonium chloride exposure for 5 min followed by a 2.5-min recovery period in Krebs solution. Treated cells received ouabain (1 μM or 1 mM) throughout the experiment. B: the effect of forskolin on cAMP. Cells were treated with forskolin (10 μM) or ouabain (1 μM) without exposure to ammonium chloride. Values are means ± SE of results from 4 independent experiments. **P < 0.01, ***P < 0.001, significant difference from control where experiments were conducted without ammonium chloride treatment. #P < 0.05, ##P < 0.01, significant difference from control where the cells were treated with ammonium chloride.
Effect of BAPTA, SKF-96365, and 2-APB on pH recovery.
The calcium chelator BAPTA abolished the ouabain-induced stimulation of pH recovery and reduced the rate to a level significantly lower than that observed in control cells. Added alone BAPTA reduced the rate of pH recovery by ∼50% (Fig. 8A). When the cells were exposed to inhibitors of capacitative calcium entry, SKF-96365 or 2-APB (50 μM each), the rate of pH recovery in ouabain-treated cells was markedly reduced. The rate was not different from the rate observed in control cells (Fig. 8, B and C).
Fig. 8.
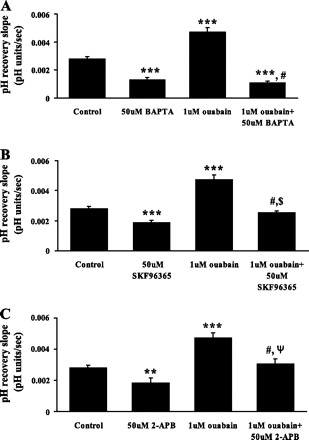
The effect of the calcium chelator BAPTA-AM (A) and the capacitative calcium entry inhibitors SKF-96365 (B) or 2-APB (C) on the rate of cytoplasmic pH recovery after ammonium chloride removal in control and ouabain-treated cells. Cells were preincubated 15 min with 50 μM BAPTA-AM, SKF-96365, or 2-APB, and the pH recovery was measured in the continued presence of the respective drug. Values are means ± SE of results from 5–37 independent experiments. ***P < 0.001, **P < 0.05, significant difference from control. #P < 0.001, significant difference from ouabain treatment. $P < 0.01, significant difference from SKF-96365 (50 μM). ΨP < 0.05, significant difference from 2-APB.
Effect of ouabain on calcium.
In cells loaded with fura-2, cytoplasmic calcium concentration was measured continuously while the cells were exposed to 20 mM ammonium chloride for 5 min and then allowed to recover on removal of ammonium chloride. Cytoplasmic calcium concentration and pH increased at the start of the ammonium chloride exposure, cytoplasmic calcium was then declined toward the baseline even though pH remained elevated. Throughout the course of the experiment, no significant difference in cytoplasmic calcium concentration was observed between control cells and cells exposed to 1 μM ouabain (Fig. 9A). Although 1 μM ouabain did not cause a detectable change of calcium concentration, it stimulated calcium entry when applied after store depletion. After the establishment of a stable baseline in calcium-free buffer, cells were exposed to 100 μM ATP. This caused a transient increase of cytoplasmic calcium concentration, a response we interpret as purinergic receptor-mediated emptying of calcium stores. Subsequently, 2.5 mM calcium was added to the bathing medium to allow capacitative calcium entry (Fig. 9B). Some cells were exposed to 1 μM ouabain before addition of calcium. Ouabain caused a significant increase in capacitative calcium entry (Fig. 9C). Capacitative calcium entry in ouabain-treated cells was abolished by 2-APB (Fig. 9, D and E).
Fig. 9.
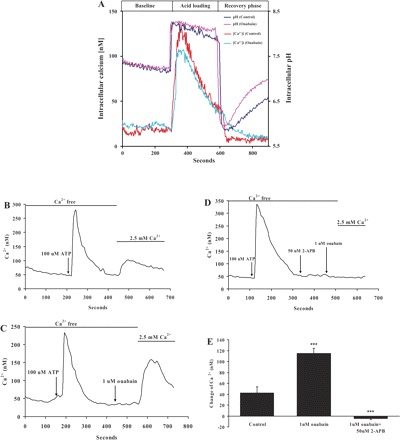
The effect of ouabain on calcium. A: in cells loaded with fura-2, cytoplasmic calcium concentration was measured continuously while the cells were exposed to 20 mM ammonium chloride for 5 min and then allowed to recover. Cytoplasmic calcium concentration and pH increased at the start of the ammonium chloride exposure, then calcium declined toward the baseline even though pH remained elevated. Throughout the course of the experiment, no significant difference was observed between cytoplasmic calcium concentration in control and ouabain-treated cells. B–D: capacitative calcium entry. B: a stable baseline was obtained in a calcium-free buffer, and the cells were then exposed to 100 μM ATP to empty the stores. Subsequently, 2.5 mM calcium was added to the bathing medium to initiate capacitative calcium entry. C: capacitative calcium entry in cells exposed to 1 μM ouabain. D: ouabain-stimulated capacitative calcium entry was abolished by 2-APB (50 μM). E: means ± SE of results from 4 independent experiments. ***P < 0.001, significant difference from control.
To exclude the possibility that ouabain response to pH is related to calcium entry by reverse mode sodium-calcium exchange, cells were exposed to KB-R7943. Ouabain-stimulated pH recovery was unaffected in the presence of 10 μM KB-R7943 (Fig. 10).
Fig. 10.
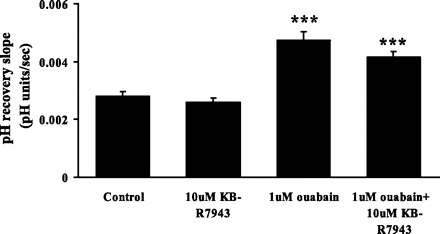
The effect of KB-R7943 on the rate of cytoplasmic pH recovery after ammonium chloride removal in control and ouabain-treated cells. KB-R7943 (10 μM) was added to the cells 5 min before addition of ouabain (1 μM), and both drugs were present throughout the rest of the experiment. KB-R7943 (10 μM) alone did not have any effect on baseline pH or on the pH recovery rate. Values are means ± SE of results from 4 independent experiments. ***P < 0.001, significant difference from control.
Effect of BAPTA and 2-APB on intracellular cAMP.
BAPTA and 2-APB both abolish the ouabain-induced stimulation of pH recovery rate. Because the pH recovery response is associated with an increase of cAMP, studies were conducted to measure cAMP in cells exposed to BAPTA or 2-APB. Both BAPTA and 2-APB abolished the increase of cAMP elicited by 1 μM ouabain (Fig. 11). Added alone, BAPTA and 2-APB each reduced the resting level of cAMP.
Fig. 11.
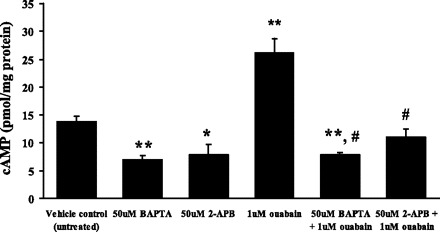
cAMP measured in cells exposed to ouabain in the presence or absence of either BAPTA or 2-APB. The measurements were made in cells subjected to 20 mM ammonium chloride exposure for 5 min followed by a 2.5-min recovery period in Krebs solution. BAPTA (50 μM) or 2-APB (50 μM) was added to the cells 15 min before addition of ouabain. Values are means ± SE of results from 4 independent experiments. *P < 0.05, **P < 0.01, significant difference with respect to the vehicle control group. #P < 0.001, significant difference with respect to the 1 μM ouabain group.
NHE1 phosphorylation.
Studies were conducted to examine NHE1 phosphorylation in cells subjected to an experimental protocol like that used in pH studies: a 5-min period of exposure to 20 mM ammonium chloride followed by a 2.5-min recovery period in control Krebs solution. Following this protocol, the cells were lysed, and the lysate was subjected to immunoprecipitation using a monoclonal antibody against phosphoserine. The precipitate was then used for Western blot analysis with an antibody directed against NHE1. Material isolated from cells exposed to 1 μM ouabain showed an increase in NHE1 band intensity (Fig. 12). The increased abundance of NHE1 in the immunoprecipitate is consistent with increased serine phosphorylation of NHE1. A similar increase in NHE1 band density was observed in cells exposed to ouabain without ammonium chloride.
Fig. 12.
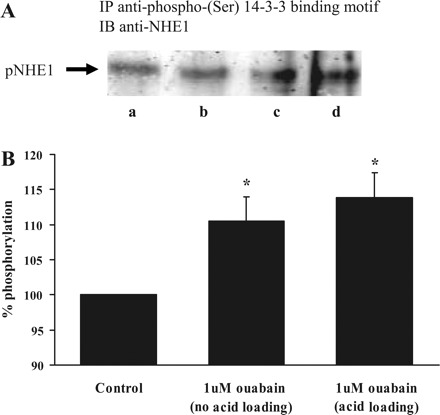
Western blot analysis of phosphorylated (p)NHE1. Cells were subjected to a 5-min period of exposure to 20 mM ammonium chloride followed by 2.5-min recovery in Krebs solution. A different group of cells was not exposed to ammonium chloride. In each case, some cells were exposed to 1 μM ouabain. Following treatment, the cells were lysed, and the lysate was subjected to immunoprecipitation (IP) using a monoclonal antibody directed against phosphoserine. The precipitate was then subjected to Western blot (IB) analysis for NHE1. A: results from the following groups: lane a, vehicle control without acid loading; lane b, vehicle control with acid loading; lane c, 1 μM ouabain without acid loading; lane d, 1 μM ouabain with acid loading. B: quantified data on band density to indicate difference between the control (expressed as 100%) and ouabain-treated groups (n = 3 independent experiments). *P < 0.05, significant difference from control.
Effect of ERK inhibitor PD-98059.
Concentrations of ouabain in the nanomolar range activate MAPK/ERK signaling in some cells (32). Studies were conducted to examine the possible involvement of MAPK/ERK in the astrocyte response to 1 μM ouabain. The ERK inhibitor PD-98059 failed to alter ouabain-induced stimulation of pH recovery (Fig. 13A). Western blot analysis revealed significant ERK phosphorylation in control cells but ouabain-treated cells showed no difference in ERK phosphorylation (Fig. 13, B and C).
Fig. 13.
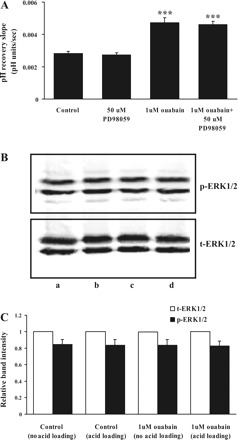
A: the effect of PD-98059 on the rate of cytoplasmic pH recovery after ammonium chloride removal in control and ouabain-treated cells. PD-98059 (50 μM) was added to the cells 5 min before addition of ouabain (1 μM), and both drugs were present throughout the rest of the experiment. Values are means ± SE of results from 4 independent experiments. ***P < 0.001, significant difference from control. B: Western blot analysis of ERK1/2 phosphorylation in cells exposed to 1 μM ouabain. Top: phospho-ERK1/2. Bottom: total (t)ERK1/2. The results show the following groups: lane a, control without acid loading; lane b, control with acid loading; lane c, 1 μM ouabain without acid loading; lane d, 1 μM ouabain with acid loading (20 μg protein/lane). C: quantified data on band density (means ± SE).
DISCUSSION
Ouabain increases the rate of cytoplasmic pH recovery after an acid load. The ouabain effect was observed both in the presence and absence of bicarbonate and was suppressed by the NHE inhibitor DMA. The findings are consistent with stimulation of NHE1 following an increase in cAMP and activation of protein kinase A. It is noteworthy that the rate of cytoplasmic pH recovery was changed by ouabain at a concentration understood to stimulate capacitative calcium entry by a mechanism linked to the inhibition of the Na-K-ATPase α2 isoform (24).
The response to 1 μM ouabain was not associated with a detectable cell-wide increase in calcium concentration. Despite this, the calcium chelator BAPTA abolished the ouabain-induced increase in pH recovery rate. The ability of both SKF-96365 and 2-APB to abolish the effect of ouabain on pH recovery points to the requirement for capacitative calcium entry. Consistent with the earlier proposal that 1 μM ouabain stimulates capacitative calcium entry, ouabain-treated cells displayed a larger increase of cytoplasmic calcium than control cells when external calcium was made available following store depletion. 2-APB entirely prevented this calcium response. The mechanism responsible for the increase in capacitative calcium entry is not fully understood. It has been argued that Na-K-ATPase α2 inhibition could alter local ion concentrations sufficiently to permit calcium entry via reverse-mode sodium-calcium exchange (7). On this basis, KB-R7943 should prevent 1 μM ouabain-induced stimulation of pH recovery. However, our data show that KB-R7943 has no effect on ouabain-induced stimulation of pH recovery.
Ouabain caused an increase in the rate of pH recovery both in bicarbonate-containing and bicarbonate-free solutions. In bicarbonate-free solution, DMA abolished the effect of ouabain on the pH recovery. It is likely that the principal cellular alkalinizing mechanism is the NHE. Taken together, the findings are consistent with the idea that ouabain-induced stimulation of pH recovery is due to activation of NHE1, the only isoform detected in these cells. The experiments in bicarbonate-free solution argue against the possibility that ouabain changes the rate of pH recovery by changing the function of a bicarbonate-dependent acid extruder or bicarbonate-dependent acid loader even though it has been reported that chloride bicarbonate exchange can be modulated via PKA (10). This is noteworthy since astrocytes are known to possess a functional NHE and also sodium- and bicarbonate-coupled acid extrusion mechanism (15).
Immunoprecipitation experiments indicated 1 μM ouabain caused significant phosphorylation of the NHE1 polypeptide. The findings reinforce the argument that NHE1 is activated. There was no evidence for a change in ERK activation in ouabain-treated cells, and the ERK1/2 inhibitor PD-98059 failed to suppress ouabain-induced stimulation of pH recovery. The results make it unlikely that ERK and its downstream effector p90RSK contributed to the observed NHE1 activation in ouabain-treated cells, although this mechanism of NHE activation occurs in acidotic rat ventricular myocytes (25).
cAMP-dependent stimulation of NHE1 in astrocytes has not been widely reported. Support for the notion that the mechanism involves an increase in cAMP comes from the finding that 8-Br-cAMP and ouabain each stimulated pH recovery to a similar degree but no further increase was observed when they were added together. Moreover, H-89, an inhibitor of cAMP-dependent protein kinase (PKA), abolished the effect of 1 μM ouabain on pH recovery. The findings are consistent with a previous report that the activity of rat NHE1 was stimulated by cAMP (31, 42). Not all NHE isoforms respond similarly. The activity of rat NHE3 is inhibited by cAMP (31, 52, 54). NHE1 is the isoform present in most cells (22) and is the predominant NHE isoform in rat astrocytes (6).
Neither DMA, H-89, nor 8-Br-cAMP altered baseline cytoplasmic pH. However, reduction of the rate of cytoplasmic pH recovery by DMA points to a significant level of NHE-mediated proton export. A reduction in the rate of pH recovery also was found in cells exposed to H-89 alone. This suggests that cAMP causes a basal degree of NHE stimulation. The fact that H-89 abolished the effect of ouabain on pH recovery constituted evidence that NHE1 plays an important role in regulating pH under conditions of intracellular acidosis. Such a role for NHE1 in acidosis has been proposed under pathologic conditions such as severe ischemia (5). It is interesting to note that in the ischemic heart, the concentration of endogenous ouabain increases several fold (19).
An increase in cAMP caused by ouabain is not a unique response of astrocytes. Such a cAMP rise has been reported in different tissues, including dog renal cortex (53), goldfish intestinal mucosa (2), mouse pancreatic islets (21), murine epithelioid and fibroblastic cell lines (34), rat brain (38), and rat renal collecting tubule cells in culture (28). In each of these reported studies, the concentration of ouabain was likely enough to inhibit Na-K-ATPase to a degree where cytoplasmic sodium increased. In such an instance, a rise in cAMP may be linked to the change in cytoplasmic sodium or to cell volume changes that may accompany the abnormal cytoplasmic ion composition. In the present study, however, an increase of cAMP was observed in astrocytes exposed to 1 μM ouabain, a concentration that did not change total cell sodium content determined by atomic absorption spectrophotometry and does not detectably alter cell-wide cytoplasmic sodium concentration in SBFI-loaded cells (24). The ability of both SKF-96365 and 2-APB to abolish the effect of ouabain on cAMP rise points to the involvement of capacitative calcium entry, which is stimulated by 1 μM ouabain. Capacitative calcium influx has been reported to be coupled with stimulation of a calcium-sensitive isoform of adenylate cyclase (type III) in bovine granulosa cells (12). Although changes of intracellular/extracellular pH can affect adenylate cyclase activity (43), cAMP was not altered by ammonium chloride-induced acidification. The identity of calcium-sensitive adenylate cyclase isoforms in optic nerve astrocytes remains unknown.
In cultured vascular smooth muscle cells, an endogenous ouabain-like factor increases pH (37). This is consistent with our finding that a low concentration of ouabain stimulates proton export and raises the possibility that endogenous ouabain may indirectly influence pH regulation. Endogenous ouabain-like factors are found in a wide range of mammalian tissues and plasma, and there is growing appreciation for the notion that they can act as hormones (11, 46). A role for endogenous ouabain has been proposed in blood pressure regulation and sodium excretion (8, 46). In this respect, it is worth mentioning that ouabain-like factor has been detected in ocular tissues (35). In apparent contrast to our findings, there is a report that 100 μM ouabain failed to change the rate of pH recovery after ammonium chloride exposure in mouse astrocytes bathed in bicarbonate-containing buffer (16). Based on studies with low sodium solutions and amiloride, the authors suggested that in the presence of bicarbonate, NHE contributes minimally to pH recovery in mouse astrocytes. This differs from the present study on rat optic nerve astrocytes, where the major contribution of NHE is evidenced by ∼50% inhibition of pH recovery with DMA.
Ouabain is a specific inhibitor of Na-K-ATPase, and there is no evidence to suggest a direct interaction of ouabain and NHE. On the other hand, NHE-mediated proton export uses the sodium gradient established by Na-K-ATPase, so ouabain would be expected to increase sodium in the cell and thus diminish NHE activity. Astrocytes express the Na-K-ATPase α2 isoform as well as the ubiquitous α1 isoform. The rat α1 isoform of Na-K-ATPase, which is responsible for regulating bulk cytosolic sodium, has a very low sensitivity to ouabain; the IC50 is 1,000-fold higher than α2 isoform (30, 40). Consequently, 1 μM ouabain is sufficient to inhibit the α2 isoform but not α1. Perhaps because α1 remains active, 1 μM ouabain did not cause a detectable change in cell sodium. The rise in cell sodium caused by concentrations of ouabain in the millimolar range was quite modest. This modest increase is not surprising given low plasma membrane sodium permeability and the fact that rat Na-K-ATPase α1 activity is not completely inhibited by millimolar ouabain concentrations (40). pH recovery after ammonium chloride removal was also stimulated in cells exposed to high (10 μM–1 mM) concentrations of ouabain even though cell sodium was increased. We suggest that the cAMP- and PKA-mediated stimulation of NHE activity was sufficient to overcome the inhibitory influence exerted by the modest increase in cell sodium. The fact that 1 μM and 1 mM ouabain have a similar stimulatory effect on pH recovery and on cellular cAMP content but a dissimilar effect on cell sodium suggests linkage of the ouabain-induced pH stimulation to cAMP.
Neural activity and factors such as anoxia have a tendency to cause changes of extracellular pH in the optic nerve (18, 44). Astrocytes use NHE for regulation of both extracellular pH and cytoplasmic pH (29). On this basis, altered NHE activity could impact the efficiency of pH homeostasis. Although ouabain-like factors are secreted from adrenal cortex (11, 23) and ischemia causes local release (19), the likelihood of endogenous cardiac glycosides attaining a micromolar concentration is remote. However, it is possible that NHE1 activity could be altered by other circumstances that alter Na-K-ATPase α2 activity, capacitative calcium entry, or cAMP. Both in vivo and in vitro studies of astrocytes have shown that an acidic cytoplasmic pH is cytotoxic (14).
In summary, we show that ouabain phosphorylates and stimulates NHE1 in cultured optic nerve astrocytes. The results are consistent with protein kinase A-dependent stimulation of NHE1 that occurs as the consequence of cAMP elevation linked to an increase of capacitative calcium entry in ouabain-treated cells.
GRANTS
This research was supported by National Institutes of Health Grant EY-014069.
Acknowledgments
The cooperation of Dr. Shigeo Tamiya, Dr. Mansim Okafor, and Betty M. Nunn in the Department of Ophthalmology and Visual Sciences at the University of Louisville, Louisville, Kentucky, is gratefully acknowledged.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Bagrov AY, Fedorova OV, Austin-Lane JL, Dmitrieva RI, Anderson DE. Endogenous marinobufagenin-like immunoreactive factor and Na+, K+ ATPase inhibition during voluntary hypoventilation. Hypertension 26: 781–788, 1995 [DOI] [PubMed] [Google Scholar]
- 2.Bakker R, Groot JA. cAMP-mediated effects of ouabain and theophylline on paracellular ion selectivity. Am J Physiol Gastrointest Liver Physiol 246: G213–G217, 1984 [DOI] [PubMed] [Google Scholar]
- 3.Barres BA, Chun LLY, Corey DP. Ion channels in vertebrate glia. Annu Rev Neurosci 13: 441–474, 1990 [DOI] [PubMed] [Google Scholar]
- 4.Baudouin-Legros M, Brouillard F, Tondelier D, Hinzpeter A, Edelman A. Effect of ouabain on CFTR gene expression in human Calu-3 cells. Am J Physiol Cell Physiol 284: C620–C626, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Beauloye C, Bertrand L, Krause U, Marsin AS, Dresselaers T, Vanstapel F, Vanoverschelde JL, Hue L. No-flow ischemia inhibits insulin signaling in heart by decreasing intracellular pH. Circ Res 88: 513–519, 2001 [DOI] [PubMed] [Google Scholar]
- 6.Benos DJ, McPherson S, Hahn BH, Chaikin MA, Benveniste EN. Cytokines and HIV envelope glycoprotein gp120 stimulate Na+/H+ exchange in astrocytes. J Biol Chem 269: 13811–13816, 1994 [PubMed] [Google Scholar]
- 7.Blaustein MP, Juhaszova M, Golovina VA. The cellular mechanism of action of cardiotonic steroids: a new hypothesis. Clin Exp Hypertens 20: 691–703, 1998 [DOI] [PubMed] [Google Scholar]
- 8.Blaustein MP, Zhang J, Chen L, Hamilton BP. How does salt retention raise blood pressure? Am J Physiol Regul Integr Comp Physiol 290: R514–R523, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Bondarenko A, Svichar N, Chesler M. Role of Na+-H+ and Na+-Ca2+ exchange in hypoxia-related acute astrocyte death. Glia 49: 143–152, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Brett CL, Kelly T, Sheldon C, Church J. Regulation of Cl−-HCO3− exchangers by cAMP-dependent protein kinase in adult rat hippocampal CA1 neurons. J Physiol 545: 837–853, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buckalew VM. Endogenous digitalis-like factors. An historical overview. Front Biosci 10: 2325–2334, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Burnay MM, Vallotton MB, Capponi AM, Rossier MF. Angiotensin II potentiates adrenocorticotrophic hormone-induced cAMP formation in bovine adrenal glomerulosa cells through a capacitative calcium influx. Biochem J 330: 21–27, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carafoli E. Intracellular calcium homeostasis. Annu Rev Biochem 56: 395–433, 1987 [DOI] [PubMed] [Google Scholar]
- 14.Chesler M. Failure and function of intracellular pH regulation in acute hypoxic-ischemic injury of astrocytes. Glia 50: 398–406, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Chesler M. Regulation and modulation of pH in the brain. Physiol Rev 83: 1183–1221, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Chow SY, Yen-Chow YC, White HS, Woodbury DM. pH regulation after acid load in primary cultures of mouse astrocytes. Brain Res Dev Brain Res 60: 69–78, 1991 [DOI] [PubMed] [Google Scholar]
- 17.Cooper AJ, Plum F. Biochemistry and physiology of brain ammonia. Physiol Rev 67: 440–519, 1987 [DOI] [PubMed] [Google Scholar]
- 18.Davis PK, Carlini WG, Ransom BR, Black JA, Waxman SG. Carbonic anhydrase activity develops postnatally in the rat optic nerve. Brain Res 428: 291–298, 1987 [DOI] [PubMed] [Google Scholar]
- 19.D'Urso G, Frascarelli S, Balzan S, Zucchi R, Montali U. Production of ouabain-like factor in normal and ischemic rat heart. J Cardiovasc Pharmacol 43: 657–662, 2004 [DOI] [PubMed] [Google Scholar]
- 20.Echevarria-Lima J, de Araujo EG, de Meis L, Rumjanek VM. Ca2+ mobilization induced by ouabain in thymocytes involves intracellular and extracellular Ca2+ pools. Hypertension 41: 1386–1392, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Gagerman E, Hellman B, Taljedal IB. Effects of ouabain on insulin release, adenosine 3′,5′-monophosphate and guanine 3′,5′-monophosphate in pancreatic islets. Endocrinology 104: 1000–1002, 1979 [DOI] [PubMed] [Google Scholar]
- 22.Grinstein S, Rotin D, Mason MJ. Na+/H+ exchange and growth factor-induced cytosolic pH changes. Role in cellular proliferation. Biochim Biophys Acta 988: 73–97, 1989 [DOI] [PubMed] [Google Scholar]
- 23.Hamlyn JM, Lu ZR, Manunta P, Ludens JH, Kimura K, Shah JR, Laredo J, Hamilton JP, Hamilton MJ, Hamilton BP. Observations on the nature, biosynthesis, secretion and significance of endogenous ouabain. Clin Exp Hypertens 20: 523–533, 1998 [DOI] [PubMed] [Google Scholar]
- 24.Hartford AK, Messer ML, Moseley AE, Lingrel JB, Delamere NA. Na,K-ATPase alpha 2 inhibition alters calcium responses in optic nerve astrocytes. Glia 45: 229–237, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Haworth RS, McCann C, Snabaitis AK, Roberts NA, Avkiran M. Stimulation of the plasma membrane Na+/H+ exchanger NHE1 by sustained intracellular acidosis: evidence for a novel mechanism mediated by the ERK pathway. J Biol Chem 278: 31676–31684, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Hou Y, Delamere NA. Influence of ANG II on cytoplasmic sodium in cultured rabbit nonpigmented ciliary epithelium. Am J Physiol Cell Physiol 283: C552–C559, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Hou Y, Wu Q, Delamere NA. H+ATPase-mediated cytoplasmic pH-responses associated with elevation of cytoplasmic calcium in cultured rabbit nonpigmented ciliary epithelium. J Membr Biol 182: 81–90, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Ishikawa S, Saito T. Effect of ouabain on cellular free calcium and cellular cyclic AMP production in response to arginine vasopressin in rat renal papillary collecting tubule cells in culture. J Endocrinol 121: 467–477, 1989 [DOI] [PubMed] [Google Scholar]
- 29.Jendelova P, Sykova E. Role of glia in K+ and pH homeostasis in the neonatal rat spinal cord. Glia 4: 56–63, 1991 [DOI] [PubMed] [Google Scholar]
- 30.Juhaszova M, Blaustein MP. Na+ pump low and high ouabain affinity alpha subunit isoforms are differently distributed in cells. Proc Natl Acad Sci USA 94: 1800–1805, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kandasamy RA, Yu FH, Harris R, Boucher A, Hanrahan JW, Orlowski J. Plasma membrane Na+/H+ exchanger isoforms (NHE-1, -2, and -3) are differentially responsive to second messenger agonists of the protein kinase A and C pathways. J Biol Chem 270: 29209–29216, 1995 [DOI] [PubMed] [Google Scholar]
- 32.Kotova O, Al-Khalili L, Talia S, Hooke C, Fedorova OV, Bagrov AY, Chibalin AV. Cardiotonic steroids stimulate glycogen synthesis in human skeletal muscle cells via a Src- and ERK1/2-dependent mechanism. J Biol Chem 281: 20085–20094, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Lago J, Alfonso A, Vieytes MR, Botana LM. Ouabain-induced enhancement of rat mast cells response. Modulation by protein phosphorylation and intracellular pH. Cell Signal 13: 515–524, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Lelievre L, Paraf A, Charlemagne D, Sheppard JR. Plasma membrane studies on drug sensitive and resistant cell lines. I. Cross resistance and membrane enzyme coordination (ouabain/cAMP/Na+/K+ ATPase/adenylate cyclase). Exp Cell Res 104: 191–197, 1977 [DOI] [PubMed] [Google Scholar]
- 35.Lichtstein D, Gati I, Samuelov S, Berson D, Rozenman Y, Landau L, Deutsch J. Identification of digitalis-like compounds in human cataractous lenses. Eur J Biochem 216: 261–268, 1993 [DOI] [PubMed] [Google Scholar]
- 36.Mellergard P, Ouyang YB, Siesjo BK. Intracellular pH regulation in cultured rat astrocytes in CO2/HCO3−-containing media. Exp Brain Res 95: 371–380, 1993 [DOI] [PubMed] [Google Scholar]
- 37.Meyer-Lehnert H, Wanning C, Michel H, Backer A, Kramer HJ. Cellular mechanisms of action of a ouabain-like factor in vascular smooth-muscle cells. J Cardiovasc Pharmacol 22, Suppl 2: S16–S19, 1993 [DOI] [PubMed] [Google Scholar]
- 38.Mork A, Geisler A. A comparative study on the effects of tetracyclines and lithium on the cyclic AMP second messenger system in rat brain. Progr Neuropsychopharmacol Biol Psychiatry 19: 157–169, 1995 [DOI] [PubMed] [Google Scholar]
- 39.Norenberg MD, Martinez-Hernandez A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res 161: 303–310, 1979 [DOI] [PubMed] [Google Scholar]
- 40.Obrien WJ, Lingrel JB, Wallick ET. Ouabain Binding kinetics of the rat alpha two and alpha three isoforms of the sodium-potassium adenosine triphosphate. Arch Biochem Biophys 310: 32–39, 1994 [DOI] [PubMed] [Google Scholar]
- 41.Orchard CH, Kentish JC. Effects of changes of pH on the contractile function of cardiac muscle. Am J Physiol Cell Physiol 258: C967–C981, 1990 [DOI] [PubMed] [Google Scholar]
- 42.Pedersen SF, King SA, Rigor RR, Zhuang Z, Warren JM, Cala PM. Molecular cloning of NHE1 from winter flounder RBCs: activation by osmotic shrinkage, cAMP, and calyculin A. Am J Physiol Cell Physiol 284: C1561–C1576, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Poggioli J, Lazar G, Houillier P, Gardin JP, Paillard M. Acute variations in extracellular pH modulate transduction pathways of PTH in rat proximal tubule. Am J Physiol Cell Physiol 263: C941–C947, 1992 [DOI] [PubMed] [Google Scholar]
- 44.Ransom BR, Philbin DM Jr. Anoxia-induced extracellular ionic changes in CNS white matter: the role of glial cells. Can J Physiol Pharmacol 70, Suppl: S181–S189, 1992 [DOI] [PubMed] [Google Scholar]
- 45.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675–686, 1996 [DOI] [PubMed] [Google Scholar]
- 46.Schoner W. Endogenous cardiac glycosides, a new class of steroid hormones. Eur J Biochem 269: 2440–2448, 2002 [DOI] [PubMed] [Google Scholar]
- 47.Shahidullah M, Wilson WS, Millar C. Effects of timolol, terbutaline and forskolin on IOP, aqueous humour formation and ciliary cyclic AMP levels in the bovine eye. Curr Eye Res 14: 519–528, 1995 [DOI] [PubMed] [Google Scholar]
- 48.Shrode LD, Putnam RW. Intracellular pH regulation in primary rat astrocytes and C6 glioma cells. Glia 12: 196–210, 1994 [DOI] [PubMed] [Google Scholar]
- 49.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem 150: 76–85, 1985 [DOI] [PubMed] [Google Scholar]
- 50.Snabaitis AK, D'Mello R, Dashnyam S, Avkiran M. A novel role for protein phosphatase 2A in receptor-mediated regulation of the cardiac sarcolemmal Na+/H+ exchanger NHE1. J Biol Chem 281: 20252–20262, 2006 [DOI] [PubMed] [Google Scholar]
- 51.Tamiya S, Delamere NA. The influence of sodium-calcium exchange inhibitors on rabbit lens ion balance and transparency. Exp Eye Res 83: 1089–1095, 2006 [DOI] [PubMed] [Google Scholar]
- 52.Thwaites DT, Kennedy DJ, Raldua D, Anderson CMH, Mendoza ME, Bladen CL, Simmons NL. H/dipeptide absorption across the human intestinal epithelium is controlled indirectly via a functional Na/H exchanger. Gastroenterology 122: 1322–1333, 2002 [DOI] [PubMed] [Google Scholar]
- 53.Westenfelder C, Sack EM, Stuart E, Earnest WR, Baranowski RL, and Kurtzman NA. Effect of ouabain on basal and PTH-stimulated cAMP production in dog renal cortex (Abstract). Kidney Int 16: 844, 1979 [Google Scholar]
- 54.Yun CH, Oh S, Zizak M, Steplock D, Tsao S, Tse CM, Weinman EJ, Donowitz M. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci USA 94: 3010–3015, 1997. [Erratum. Proc Natl Acad Sci USA 94: 10006, 1997.]. [DOI] [PMC free article] [PubMed] [Google Scholar]