

Abstract
Germ-line mutations in the human BRCA2 gene confer susceptibility to breast cancer. Efforts to elucidate its function have revealed a putative transcriptional activation domain and in vitro interaction with the DNA repair protein RAD51. Other studies have indicated that RAD51 physically associates with the p53 tumor suppressor protein. Here we show that the BRCA2 gene product is a 460-kDa nuclear phosphoprotein, which forms in vivo complexes with both p53 and RAD51. Moreover, exogenous BRCA2 expression in cancer cells inhibits p53’s transcriptional activity, and RAD51 coexpression enhances BRCA2’s inhibitory effects. These findings demonstrate that BRCA2 physically and functionally interacts with two key components of cell cycle control and DNA repair pathways. Thus, BRCA2 likely participates with p53 and RAD51 in maintaining genome integrity.
Germ-line mutations in the human BRCA1 and BRCA2 breast cancer suppressor genes confer susceptibility to breast and ovarian cancers (1–4). Mutations in BRCA1 and BRCA2 are believed to be responsible for most hereditary breast cancers (3–10), which account for 5–10% of all breast cancer cases (11). About 52% of the families with four or more breast cancer cases have inherited mutations in BRCA1, and 32% possess BRCA2 mutations (12). In contrast, somatic mutations in BRCA1 and BRCA2 are rare in sporadic cases of breast cancer (5, 13–15). Both BRCA1 and BRCA2 are up-regulated in proliferating and differentiating cells, expressed in a cell cycle-dependent manner peaking at the G1/S boundary (19–21), widely expressed during development (22–24), and essential for early embryonic development (25–29). Recent evidence indicates that BRCA1 is involved in transcriptional regulation (30, 31).
The BRCA2 gene is composed of 27 exons and encodes a predicted 384-kDa protein possessing no obvious homology to other sequences in publicly available databases (4). There are eight repetitive units in the BRCA2 protein sequence (BRC motifs; refs. 16 and 17) and a potential nuclear localization signal within its C terminus (18). BRCA2 also possesses a putative transcriptional activation domain in exon 3, suggesting a role of BRCA2 in the regulation of gene expression (32). Evidence for a possible function of BRCA2 in DNA repair has been suggested by recent findings that Brca2 mutant mouse embryos are defective in DNA repair (27, 33, 34). Moreover, yeast two-hybrid and glutathione S-transferase (GST) pull-down analysis revealed that BRCA2 interacts in vitro with RAD51, a protein involved in DNA double-strand break repair and homologous recombination (27, 35, 36). Binding sites for RAD51 have been mapped to each of the eight BRC motifs in human BRCA2 (36) and to a C-terminal region of mouse Brca2 (27, 35).
Structural and functional characterization of the endogenous BRCA2 protein have been hampered by the large size of the protein and the lack of suitable immunological reagents for its detection. In the present studies, we generated polyclonal and monoclonal antibodies to characterize endogenous BRCA2 and identify proteins that form complexes with this protein. We demonstrate that BRCA2 is a nuclear phosphoprotein that associates in vivo with a significant portion of the endogenous pool of RAD51. Because an immediate cellular reponse to DNA damage is p53-mediated cell cycle arrest (37), and RAD51 has been reported to physically associate with p53 (38, 39), we also investigated whether a physical and functional relationship could be detected between BRCA2 and p53.
MATERIALS AND METHODS
Generation of BRCA2 Antibodies.
Polyclonal antibodies were raised in rabbits, and mAbs were generated in mice. The peptides were conjugated to keyhole limpet hemacyanin. The three GST-fusion regions were generated by reverse transcription–PCR amplification using the following primers: 5′-GGTCAAGTTCTTTAGCTACAGGATCCACCC-3′ and 5′-CTCCATCTGGGCTCCATGTCGACCTGAAAG-3′ for GSTB1, 5′-CCTGCAACTTGTTACAGAATTCAGTCCCC-3′ and 5′-AGGGTGAAGAGCTAGTCTCGAGTTCCTCAA-3′ for GSTB2, and 5′-GTCAGTGAATCCACTAGAATTCCTCCCACC-3′ and 5′-TGGTCTTGAACTCCTGGCCTCGAGCACT-3′ for GSTB3. These regions then were subcloned into the GST-fusion protein expression vector pGEX-5X-2 or pGEX-4T-2 (Pharmacia) and verified by sequencing. All antibodies were affinity-purified.
Plasmid Constructs.
pGFPB2 containing human full-length BRCA2 cDNA fused in-frame with green fluorescent protein (GFP) was constructed as follows. A fragment at the N terminus of BRCA2 was generated from pUCBRCA2 by PCR amplification using a high-fidelity PCR system (Boehringer Mennheim) and the following primers: 5′-GGCCAGATCTATGCCTATTGGATCCAAAGAGAGG-3′ and 5′-GGCCGTCGACTGCTTGTTTATCACCTGTGTCTCC-3′. This fragment was subcloned into the pEGFPC1 vector (CLONTECH) at BglII and SalI sites to generate pGFPB2N. A Sse8387I–SalI BRCA2 fragment released from pUCBRCA2 then was ligated with pGFPB2N digested with BglII and Sse8387I to generate pGFPB2. Sequencing confirmed the identity of the fragment generated by PCR. A human full-length RAD51 cDNA with BamHI and SalI flanking was amplified from a human testis cDNA library (CLONTECH) by using Pfu DNA polymerase (Stratagene) and the following primers: 5′-GGCCGGATCCATGGCAATGCAGATGCAGCTTG-3′ and 5′-GGCCGTCGACTCTTTGGCATCTCCCACTCCAT-3′. Cloning of this cDNA fragment into pcDNA3HA generated an N-terminal hemagglutinin (HA)-tagged human Rad51 construct, pHA-RAD51. A BglII–BamHI fragment containing two copies of the consensus p53 binding sequence and the adenovirus major late TATA box from p50–2 (a gift from A. J. Levine, Princeton Univ., Princeton, NJ) were cloned into the pGL2 vector (Promega) to generate a luciferase reporter plasmid PGluc. GAL4luc and GAL4-VP16 were a gift from R. Davis (Univ. of Massachusetts, Worcester).
Fluorescence Microscopy.
pGFPB2 was transiently transfected into 293T cells in 6-well plates containing coverslips using Lipofectamine (GIBCO). Twenty-four hours after transfection, cells were incubated with Hoechst stain (bisBenzimide Trihydrochloride Hoechst No. 33258; 5 μg/ml; Sigma) for 15 min to label nuclei. Coverslips were transferred to a Sykes-Moore perfusion chamber (Belco) mounted on the stage of a Nikon Eclipse E600 microscope and maintained at 37°C. Images were collected by using a Nikon Plan Apo ×100 oil immersion objective lens (numerical aperture = 1.4), a 1,000 × 800 pixel back-illuminated cooled charge-coupled device camera (Princeton Instruments, Trenton, NJ), and Metamorph software (Universal Imaging, Media, PA).
Luciferase Assays.
Plasmid DNA was transiently transfected into cells by using Lipofectamine PLUS (GIBCO). Approximately 2 × 106 cells were cotransfected with 1 μg of reporter plasmid and 1 μg of effector plasmid except for pGFPB2 (5 μg). Cells were harvested 48 hr after transfection, and luciferase activity was measured by using a luciferase assay kit (Promega). The transfection efficiency was normalized by measuring β-galactosidase activities after the cotransfection of MFG-β-galactosidase plasmid.
Immunoprecipitation and Immunoblotting.
Cell lysates were prepared in lysis buffer (50 mM Tris⋅HCl, pH 8.0/120 mM NaCl/0.5% Nonidet P-40/10 μg/ml aprotinin/10 μg/ml leupeptin/1 mM phenylmethylsulfonyl fluoride/1 mM sodium orthovanadate). In some cases, cells were labeled with [35S]methionine (0.1 mCi/ml) or [32P]phosphoric acid (0.25 mCi/ml; NEN) for 3 hr. The total protein content of the lysates was determined by bicinchoninic acid assay (Pierce). Phosphatase treatment of immunoprecipitates was performed as described (40). Cell lysates or immunoprecipitates were electrophoresed on 4%, 6%, or 10% SDS-polyacrylamide gels and transferred onto a poly(vinylidene difluoride) membrane (Millipore). ECL or ECL Plus (Amersham) was used for signal detection. Anti-GFP antibody was obtained from CLONTECH, antibodies against p53 (PAb421), DNA-dependent protein kinase, or RAD51 from Calbiochem, antibodies against the HA epitope or p53 (DO-1) from Santa Cruz Biotechnology, and anti-retinoblastoma protein (RB) antibody from PharMingen.
RESULTS
Characterization of the BRCA2 Gene Product.
To facilitate the study of BRCA2 structure and function, we generated a series of polyclonal and monoclonal antibodies by using both peptides and GST fusion proteins corresponding to various regions of the molecule (Fig. 1A). On Western blots of 293T cell lysates, affinity-purified polyclonal antibodies against both the N-terminal (anti-N19) and C-terminal peptides (anti-C15) recognized a band at a molecular mass much greater than that of the 220-kDa myosin standard (Fig. 1B). This band also was detected in MCF7 cells (see Fig. 3C, lane 3) and comigrated with 460-kDa DNA-dependent protein kinase (Fig. 1C, lane 9). However, it was not detected by anti-C15 in CAPAN-1 cells (Fig. 1C, lane 4), a human pancreatic tumor cell line that has lost one BRCA2 allele and contains the 6174delT mutation in the remaining allele (41).
Figure 1.

Characterization of BRCA2 protein. (A) Diagram showing the relative positions of peptides (N19 and C15) and GST-fusion proteins (GSTB1, GSTB2, and GSTB3) used to generate antibodies against BRCA2. Among antibodies generated against GST-fusion proteins, we selected a mAb generated against GSTB2 for use in this study. (B) Lysates (50 μg of total protein) of 293T cells transfected with pGFPB2 (lanes 1, 3, 5, 7, and 9) or control vector pEGFPC1 (lanes 2, 4, 6, 8, and 10) were subjected to 4% SDS/PAGE, and immunoblotted with the indicated antibodies with (lanes 3, 4, 7, and 8) or without (lanes 1, 2, 5, 6, 9, and 10) homologous peptide competition. The GFP tag at the N terminus of BRCA2 may interfere with recognition of BRCA2 by anti-N19, which is specific for the N terminus of BRCA2, based on the relatively stronger signal for GFP-BRCA2 using anti-C15. (C) Cell lysates (50 μg of total protein; lanes 1–4), mAb B2 immunoprecipitates (from 1 mg of total protein; lanes 5–8), and DNA-dependent protein kinase immunoprecipitates (1 mg of total protein; lane 9) were resolved on the same 4% SDS/PAGE gel, and immunoblotted with anti-C15 or anti-DNA-protein kinase. Cells included 293T transfected with pEGFPC1 (lanes 1 and 5) or pGFPB2 (lanes 2 and 6), MCF7 (lanes 3, 7, and 9), and CAPAN-1 (lanes 4 and 8). (D) mAb B2 immunprecipitates from 2 mg of total protein of CAPAN-1 (lane 1) and MCF7 (lane 2) were resolved by 5% SDS/PAGE and immunoblotted with anti-N19. (E) MCF7 (lane 1) and CAPAN-1 (lane 2) cell lysates were labeled with [32P]phosphoric acid, immunoprecipitated with mAb B2, resolved on a 4% SDS/PAGE, and exposed to Kodak X-Omat AR film. (F) BRCA2 or RB immunoprecipitates of MCF7 cells were untreated (lanes 1 and 3) or treated (lanes 2 and 4) with lambda-phosphatase, separated by 4% (BRCA2) or 8% (RB) SDS/PAGE, and immunoblotted with anti-C15 or anti-RB.
Figure 3.
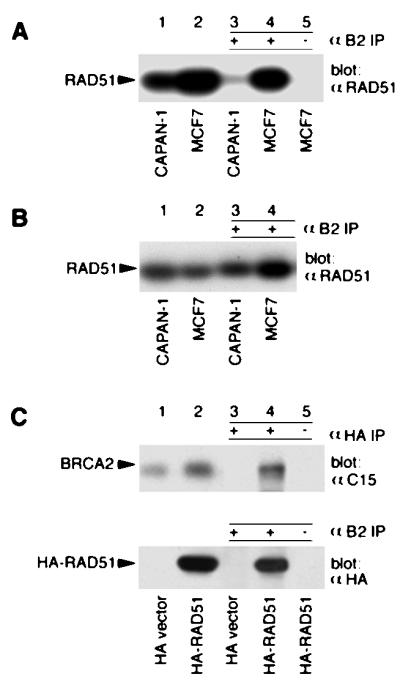
In vivo interactions of BRCA2 with RAD51. (A) Cell lysates (50 μg of total protein; lanes 1 and 2) and mAb B2 (lanes 3 and 4) or anti-IgG (lane 5) immunoprecipitates (from 2.5 mg of total protein) were resolved by 10% SDS/PAGE, and immunoblotted with anti-RAD51. (B) Cell lysates (50 μg of total protein for CAPAN-1, and 25 μg of total protein for MCF7; lanes 1 and 2) and mAb B2 immunoprecipitates (from 4 mg of total protein for CAPAN-1, and 2 mg of total protein for MCF7; lanes 3 and 4) were resolved by 10% SDS/PAGE and immunoblotted with anti-RAD51. (C) 293T cells transfected with pHA-RAD51 or the control HA vector were lysed and immunoprecipitated with anti-HA, mAb B2, or anti-IgG. Lysates (lanes 1 and 2) and immunoprecipitates (lanes 3–5) were separated by 4% SDS/PAGE and immunoblotted with anti-C15, or 10% SDS/PAGE and immunoblotted with anti-HA. Anti-IgG immunoprecipitates from HA-RAD51 transfected cells were used as a specificity control (lane 5).
To confirm that this band reflected the authentic BRCA2 protein, we transfected 293T cells with the plasmid pGFPB2 containing full-length BRCA2 cDNA fused at its N terminus to GFP or with the control vector pEGFPC1. Both anti-N19 and anti-C15 specifically recognized a band in pGFPB2-transfected, but not in vector-transfected, 293T cells at the predicted size of the GFP-BRCA2 fusion protein, approximately 27 kDa higher in mass than the native BRCA2 protein (Fig. 1B, lanes 1 and 5). A protein of the same size also was recognized by a polyclonal antibody against GFP (Fig. 1B, lane 9), confirming its identity as GFP-BRCA2. Moreover, all of these bands were specifically competed by the homologous peptides (Fig. 1B). Further confirmation that the proteins we had identified represent the authentic BRCA2 protein was provided by immunoprecipitation/Western blot experiments. MCF7, CAPAN-1, or transfected 293T cell lysates were immunoprecipitated with a mAb (mAb B2) raised against the GSTB2 region of BRCA2 (Fig. 1A), and then immunoblotted with anti-C15. The expected 460-kDa BRCA2 protein was detected in MCF7 and 293T cells but not in CAPAN-1 cells, and the 487-kDa GFP-BRCA2 fusion protein was detected only in pGFPB2-transfected 293T cells (Fig. 1C). All of these findings firmly established the identity of the 460-kDa protein as the product of the BRCA2 gene.
When BRCA2 was immunoprecipitated with mAb B2 from CAPAN-1 or MCF cell lysates and immunoblotted with anti-N19, a truncated BRCA2 protein was specifically detected in CAPAN-1 cells (Fig. 1D, lane 1). This truncated species migrated above the 220-kDa myosin standard and had an estimated molecular mass of 230 kDa, nearly matching the predicted size of 224 kDa. These results indicate that the mutated BRCA2 gene in CAPAN-1 cells encodes a stable truncated protein.
It should be noted that both anti-C15 and mAb B2 recognized a BRCA2 doublet in MCF7 and 293T cells (Fig. 1 B and C), suggesting the possibility of posttranslational processing, such as phosphorylation. Evidence that BRCA2 was phosphorylated derived from [32P]-labeling experiments, in which a [32P]-labeled band at 460 kDa was detected in autoradiographs of mAb B2 immunoprecipitates of MCF7, but not CAPAN-1 cells (Fig. 1E). However, when mAb B2 immunoprecipitates from MCF7 cells were treated with lambda-phosphatase, the BRCA2 doublet was not altered. Under the same conditions, the RB doublet, which is known to result from phosphorylation (42), was reduced to a single band (Fig. 1F). Thus, phosphorylation was unlikely to account for the BRCA2 doublet. It also should be noted that anti-N19 recognized only the upper band of the BRCA2 doublet, and that GFP-BRCA2 migrated as a single band as well (Fig. 1 B and C). Thus, the doublet likely resulted from BRCA2 amino-terminal processing or as an alternative product of the BRCA2 gene.
Exogenously Expressed BRCA2 Localizes to the Nucleus.
BRCA2 reportedly possesses a potential nuclear localization signal in its C terminus (18), and recently Bertwistle et al. (43) reported that BRCA2 was enriched in the nuclear fraction derived from MCF7 cells. Although our antibodies recognized GFP-BRCA2 overexpressed in transfected cells, it was not possible to reliably detect endogenous BRCA2 by standard immunofluorescent methods (unpublished data). However, the availability of the GFP-tagged BRCA2 construct made it possible to localize the transfected GFP-BRCA2 protein in live 293T cells by fluorescence microscopy. Although GFP alone was distributed throughout the cell (data not shown), GFP-BRCA2 primarily was localized to the Hoechst-labeled nuclei (Fig. 2). These results suggest a nuclear localization of the BRCA2 protein.
Figure 2.
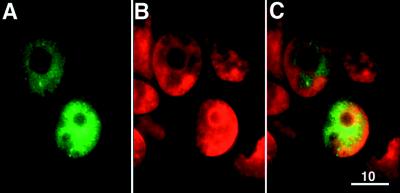
Subcellular localization of exogenously expressed BRCA2. Representative images of live 293T cells transfected with pGFPB2. (A) Image showing the green signal of GFP-BRCA2 in transfected 293T cells. (B) Image showing the nuclei of cells in the same field labeled with Hoechst. Hoechst labeling is indicated as red color instead of its authentic blue color. (C) Overlay of the images in A and B. (Bar = 10 μm.)
In vivo Interaction of BRCA2 with RAD51.
BRCA2 has been reported to interact with RAD51 in vitro (27, 35, 36). To investigate whether these proteins form a complex in vivo, BRCA2 was immunoprecipitated with mAb B2 from MCF7 or CAPAN-1 cell lysates and immunoblotted with anti-RAD51. As shown in Fig. 3A, endogenous RAD51 was readily detected in a complex with endogenous BRCA2 in MCF7 cells (Fig. 3A, lane 4), whereas there was a faint signal for RAD51 coimmunoprecipitated with the truncated BRCA2 protein in CAPAN-1 cells (Fig. 3A, lane 3). As a specificity control, anti-IgG failed to immunoprecipitate complexes containing RAD51 from MCF7 cells (Fig. 3A, lane 5). The signal intensities for RAD51 in 50 μg of cell lysates (Fig. 3A, lane 2) and mAb B2 immunoprecipitates from 2.5 mg of cell lysates (Fig. 3A, lane 4) were comparable. Thus, approximately 2% of RAD51 was present in mAb B2 immunoprecipitates. When adjusted for the approximately 10% efficiency of BRCA2 immunoprecipitation by mAb B2 (Fig. 1C), we estimate that at least 10–20% of RAD51 was associated with BRCA2 in MCF7 cells.
The levels of both the mutant BRCA2 protein and RAD51 were substantially lower in CAPAN-1 cells (Fig. 1D, lane 1; Fig. 3A, lane 1) than those of the corresponding proteins in MCF7 cells (Fig. 1D, lane 2; Fig. 3A, lane 2). Thus, to compare the ability of the truncated BRCA2 protein to interact RAD51 with that of the wild-type BRCA2 protein, we increased the amount of CAPAN-1 cell lysate used for coimmunoprecipitation to give similar levels of the proteins of interest to those in MCF7 cells (Fig. 3B, lanes 1 and 2). Under these conditions, the amount of RAD51 coimmunoprecipitated with the truncated BRCA2 in CAPAN-1 cells (Fig. 3B, lane 3) was comparable to that observed with wild-type BRCA2 in MCF7 cells (Fig. 3B, lane 4). These findings are consistent with the in vitro mapping of RAD51 binding sites to each of the eight BRC motifs in human BRCA2 (36) retained by the mutant BRCA2 product in CAPAN-1 cells.
We also transfected 293T cells with pHA-RAD51, an N-terminal HA epitope-tagged RAD51 construct. HA-RAD51 then was immunoprecipitated with anti-HA and immunoblotted with anti-C15. As shown in Fig. 3C, BRCA2 was detected in HA-RAD51 immunoprecipitates from pHA-RAD51 but not the control vector-transfected 293T cells. The presence of HA-RAD51 in BRCA2 immunoprecipitates was demonstrated in pHA-RAD51-transfected 293T cells as well (Fig. 3C, Lower). As specificity controls, none of the proteins were detected in immunoprecipitates with anti-IgG under the same conditions (Fig. 3C, lane 5). All of these results establish that complexes containing BRCA2 and RAD51 occur in vivo.
BRCA2 Forms a Complex with p53 in vivo.
BRCA2’s nuclear localization and its putative transcriptional activation domain (32) suggest that this protein may associate with a transcriptional complex. Because p53-mediated cell cycle arrest is induced after DNA damage to allow DNA repair (37), we sought to investigate whether BRCA2 and p53 might be physically present in the same complex. To do so, endogenous p53 was immunoprecipitated from MCF7 cell lysates by mAb DO-1 and immunoblotted with anti-C15. As shown in Fig. 4A, endogenous BRCA2 was readily detectable in p53-containing immunoprecipitates of MCF7 cells. In contrast, neither CAPAN-1 nor a p53 negative bladder cancer cell line, EJ, showed evidence of such immunocomplexes (Fig. 4A, lanes 4 and 6). The IgG heavy chain has a gel mobility similar to p53, making it difficult to reliably detect p53 by immunoblotting after immunoprecipitation. To perform reciprocal experiments to determine whether endogenous p53 was present in BRCA2 immunoprecipitates, we labeled cells with [35S]methionine. BRCA2-containing complexes first were immunoprecipitated with anti-C15, eluted, and then immunoprecipitated with DO-1. As shown in Fig. 4B, a [35S]-labeled 53-kDa band was observed in autoradiographs of BRCA2/p53 double immunoprecipitates of MCF7, but not CAPAN-1 or EJ cells (Fig. 4B, lanes 4 and 6). These findings establish that BRCA2 and p53 exist in vivo in the same complexes.
Figure 4.
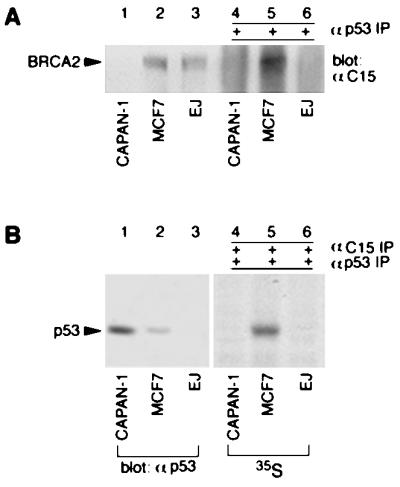
BRCA2 forms a complex with p53 in vivo. (A) Cell lysates (40 μg of total protein; lanes 1–3) and p53 immunoprecipitates (4 mg of total protein; lanes 4–6) were resolved by 6% SDS/PAGE and immunoblotted with anti-C15. (B) Cell lysates (lanes 1–3) were separated by 10% SDS/PAGE and immunoblotted with anti-p53 PAb421. Anti-C15 immunoprecipitates from [35S]-methionine-labeled cells were eluted and reprecipitated with anti-p53 DO-1. The double immunoprecipitates were resolved by 10% SDS/PAGE and subjected to fluorography (lanes 4–6).
BRCA2 Inhibits p53 Transcriptional Activity.
To investigate possible functional interactions between BRCA2 and p53, we tested whether BRCA2 might act as a regulator for p53’s transcriptional activity. To do so, MCF7 cells were transfected with pGFPB2 and PGluc, a luciferase reporter plasmid containing two copies of the p53-responsive element. As shown in Fig. 5, BRCA2 caused a decrease in the basal activation of p53-responsive elements by endogenous p53, when compared with transfection with the control vector pEGFPC1. BRCA2 also caused marked inhibition of exogenous p53 stimulation of PGluc. Cotransfection with BRCA2 had a similar inhibitory effect on exogenous p53-stimulated transcriptional activity in the osteosarcoma cell line, SAOS2, and the small cell lung carcinoma cell line, H1299 (data not shown). We found no difference in the level of p53 protein level expressed in cells in the presence or absence of cotransfection with BRCA2 (data not shown), excluding the possibility that inhibition by BRCA2 was the result of changes in p53 protein level. As a specificity control, GAL4luc, a luciferase reporter containing four copies of GAL4 responsive elements, was not inhibited by cotransfection of BRCA2 with GAL4-VP16, a vector expressing the GAL4 DNA binding domain fused to the transactivation domain of VP16 (data not shown). Thus, BRCA2 was not a general transcriptional inhibitor but appeared to specifically inhibit p53 transcriptional activity.
Figure 5.

RAD51 enhances BRCA2 inhibition of p53’s transcriptional activity. MCF7 cells were cotransfected with the indicated plasmids and PGluc. Forty-eight hours after transfection, luciferase activity was measured as described in Materials and Methods.
RAD51 Enhances BRCA2 Inhibition of p53 Transcriptional Activity.
If the inhibitory effect of BRCA2 on p53’s transcriptional activity reflects an intersection of cell cycle checkpoint and DNA repair pathways, we reasoned that RAD51 might affect p53’s transcriptional activity as well. As shown in Fig. 5, RAD51 had no effect on the activation of p53-responsive elements by endogenous p53 in MCF7 cells or in response to exogenous p53. However, cotransfection of RAD51 with BRCA2 caused nearly complete inhibition of PGluc activation by either endogenous or exogenous p53. These results indicate that RAD51 enhances BRCA2 inhibition of p53’s transcriptional activity.
DISCUSSION
Abnormalities caused by targeted disruption of the Brca2 gene include increased sensitivity to DNA damage induced by ionizing irradiation, UV light, and other genotoxic agents (27, 33, 34). The accumulation of double-strand DNA breaks and chromosomal abnormalities combined with the lack of obvious checkpoint or apoptotic response abnormalities in Brca2 mutant cells have implied a role of BRCA2 in DNA repair (33, 34). Recent findings that BRCA2 and RAD51 interact in vitro have suggested further that BRCA2 may be involved in RAD51-mediated repair pathways (27, 35, 36). In this study, we identified the BRCA2 gene product as a 460-kDa nuclear phosphoprotein that forms a complex with RAD51 in vivo. While this manuscript was in preparation, Chen et al. (44) reported detection of BRCA2 as a nuclear protein, consistent with our findings. They also reported detection of immunocomplexes containing BRCA2 and RAD51 (44). Our findings established that a major fraction of endogenous RAD51 is associated with the endogenous BRCA2 protein, implying that this interaction is functionally significant.
We demonstrated further that CAPAN-1 tumor cells, which contain a BRCA2 mutation producing a truncated product, express a single BRCA2 immunoreactive species of around 230 kDa, consistent with the predicted size of the mutant. Of note, the mutant protein, though expressed at low level, interacted with RAD51 as efficiently as the wild-type BRCA2. This mutant would be predicted to retain all eight repetitive BRC motifs that have been mapped in vitro to be RAD51 binding sites in human BRCA2 (36). These results point to a possible mechanism by which truncated BRCA2 mutants found commonly in tumors (Breast Cancer Information Core) actually may sequester RAD51 in a nonfunctional complex.
The p53 tumor suppressor gene is known to mediate cell cycle arrest after DNA damage (37), and there are reports that p53 can be detected in complexes with RAD51 (38, 39). We established here that p53 exists in in vivo complexes containing BRCA2. Moreover, a functional interaction between BRCA2 and p53 was demonstrated by evidence that BRCA2 inhibits p53 transcriptional activity. These results are consistent with the possibility that BRCA2 may act to limit the length or severity of p53-mediated cell cycle arrest after DNA damage. Other studies have shown that Brca2 mutant mouse embryos exhibit a growth arrest phenotype (27–29, 33, 34), and that this phenotype is less severe in Brca2/p53 double mutant mouse embryos (28). In addition, Brca2 mutant embryo cells exhibit increased p21WAF1/CIP1 levels associated with a defect in cell proliferation (29, 33, 34). All of these findings are consistent with a role of BRCA2 in down-regulating p53 transcriptional activity.
BRCA2, RAD51, and BRCA1 have similar patterns of expression (45), and mouse mutants of each of these genes exhibit embryonic lethality (25–29, 46, 47) that can be partially rescued in a p53 null background (28, 47, 48). These findings imply that all of these molecules have related functions. BRCA1 and BRCA2 have been reported to have transcriptional activity potential (30–32). Although RAD51 has been shown to be a component of the RNA polymerase II holoenzyme (49), it is not itself known to have transcriptional activity (45). We did not detect any effect of RAD51 on p53 transcriptional activity. However, we did observe cooperative down-regulation of p53’s activity by RAD51 and BRCA2, providing evidence that RAD51 is coupled to transcription pathways through its interactions with BRCA2 and p53. Thus, BRCA2 appears to serve as a regulator linking both cell cycle control and DNA repair pathways.
Acknowledgments
We are indebted to Dr. S. V. Tavtigian, Dr. A. K. C. Wong, and Dr. Paul Bartel for the full-length BRCA2 cDNA, and Dr. Alan D. Marmorstein for advice and assistance on fluorescence microscopy. We also thank Ms. Mutsuko Ouchi for technical assistance and the members of the Aaronson Laboratory for helpful discussion. This work was supported by National Institutes of Health Specialized Program of Research Excellence in breast cancer 1P50CA68425 (S.A.A.) and an Incentive Award from Mount Sinai School of Medicine (T.O.). L.Y.M. is a recipient of Postdoctoral Fellowship DAMD17-97-1-7317 from the Department of the Army.
ABBREVIATIONS
- GST
glutathione S-transferase
- GFP
green fluorescent protein
- HA
hemagglutinin
- RB
retinoblastoma protein
References
- 1.Miki Y, Swensen J, Shattuck-Eidens D, Futreal P A, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett L M, Ding W, et al. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 2.Wooster R, Neuhausen S L, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D, et al. Science. 1994;265:2088–2090. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- 3.Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G, et al. Nature (London) 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 4.Tavtigian S V, Simard J, Rommens J, Couch F, Shattuckeidens D, Neuhausen S, Merajver S, Thorlacius S, Offit K, Stoppalyonnet D, et al. Nat Genet. 1996;12:333–337. doi: 10.1038/ng0396-333. [DOI] [PubMed] [Google Scholar]
- 5.Futreal P A, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett L M, Haugen-Strano A, Swensen J, Miki Y, et al. Science. 1994;266:120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- 6.Friedman L S, Ostermeyer E A, Szabo C I, Dowd P, Lynch E D, Rowell S E, King M-C. Nat Genet. 1994;8:399–404. doi: 10.1038/ng1294-399. [DOI] [PubMed] [Google Scholar]
- 7.Castilla L H, Couch F J, Erdos M R, Hoskins K F, Calzone K, Garber J E, Boyd J, Lubin M B, Deshano M L, Brody L C, et al. Nat Genet. 1994;8:387–391. doi: 10.1038/ng1294-387. [DOI] [PubMed] [Google Scholar]
- 8.Simard J, Tonin P, Durocher F, Morgan K, Rommens J, Gingras S, Samson C, Leblanc J-F, Belanger C, Dion F, et al. Nat Genet. 1994;8:392–398. doi: 10.1038/ng1294-392. [DOI] [PubMed] [Google Scholar]
- 9.Phelan C M, Lancaster J M, Tonin P, Gumbs C, Cochran C, Carter R, Ghadirian P, Perret C, Moslehi R, Dion F, et al. Nat Genet. 1996;13:120–122. doi: 10.1038/ng0596-120. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi H, Chiu H-C, Bandera C A, Behbakht K, Liu P C, Couch F J, Weber B L, LiVolsi V A, Furusato M, Rebane B A, et al. Cancer Res. 1996;56:2738–2741. [PubMed] [Google Scholar]
- 11.Claus E B, Risch N, Thompson W D. Am J Hum Genet. 1991;48:232–242. [PMC free article] [PubMed] [Google Scholar]
- 12.Ford D, Easton D F, Stratton M, Narod S, Goldgar D, Devilee P, Bishop D T, Weber B, Lenoir G, Chang-Claude J, et al. Am J Hum Genet. 1998;62:676–689. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lancaster J M, Wooster R, Mangion J, Phelan C M, Cochran C, Gumbs C, Seal S, Barfoot R, Collins N, Bignell G, et al. Nat Genet. 1996;13:238–240. doi: 10.1038/ng0696-238. [DOI] [PubMed] [Google Scholar]
- 14.Miki Y, Katagiri T, Kasumi F, Yoshimoto T, Nakamura Y. Nat Genet. 1996;13:245–247. doi: 10.1038/ng0696-245. [DOI] [PubMed] [Google Scholar]
- 15.Teng D H-F, Bogden R, Mitchell J, Baumgard M, Bell R, Berry S, Davis T, Ha P C, Kehrer R, Jammulapati S, et al. Nat Genet. 1996;13:241–244. doi: 10.1038/ng0696-241. [DOI] [PubMed] [Google Scholar]
- 16.Bork P, Blomberg N, Nilges M. Nat Genet. 1996;13:22–23. doi: 10.1038/ng0596-22. [DOI] [PubMed] [Google Scholar]
- 17.Bignell G, Micklem G, Stratton M R, Ashworth A, Wooster R. Hum Mol Genet. 1997;6:53–58. doi: 10.1093/hmg/6.1.53. [DOI] [PubMed] [Google Scholar]
- 18.McAllister K A, Haugen-Strano A, Hagevik S, Brownlee H A, Collins N K, Futreal P A, Bennett L M, Wiseman R W. Cancer Res. 1997;57:3121–3125. [PubMed] [Google Scholar]
- 19.Rajan J V, Wang M, Marquis S T, Chodosh L A. Proc Natl Acad Sci USA. 1996;93:13078–13083. doi: 10.1073/pnas.93.23.13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaughn J P, Cirisano F D, Huper G, Berchuck A, Futreal P A, Marks J R, Igelhart D. Cancer Res. 1996;56:4590–4594. [PubMed] [Google Scholar]
- 21.Gudas J M, Li T, Nguyen H, Jensen D, Rauscher III F J, Cowan K H. Cell Growth Differ. 1996;7:717–723. [PubMed] [Google Scholar]
- 22.Lane T F, Deng C, Elson A, Lyu M S, Kozak C A, Leder P. Genes Dev. 1995;9:2712–2722. doi: 10.1101/gad.9.21.2712. [DOI] [PubMed] [Google Scholar]
- 23.Marquis S T, Rajan J V, Wynshaw-Boris A, Xu J, Yin G-Y, Abel K J, Weber B L, Chodosh L A. Nat Genet. 1995;11:1–26. doi: 10.1038/ng0995-17. [DOI] [PubMed] [Google Scholar]
- 24.Rajan J V, Marquis S T, Gardner H P, Chodosh L A. Dev Biol. 1997;184:385–401. doi: 10.1006/dbio.1997.8526. [DOI] [PubMed] [Google Scholar]
- 25.Gowen L C, Johnson B L, Latour A M, Sulik K K, Koller B H. Nat Genet. 1996;12:191–194. doi: 10.1038/ng0296-191. [DOI] [PubMed] [Google Scholar]
- 26.Hakem R, Pompa J L D L, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, et al. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- 27.Sharan S K, Morimatsu M, Albrecht U, Lim D-S, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A. Nature (London) 1997;386:804–810. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- 28.Ludwig T, Chapman D L, Papaioannou V E, Efstratiadis A. Genes Dev. 1997;11:1226–1241. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki A, de la Pompa J L, Hakem R, Elia A, Yoshida R, Mo R, Nishina H, Chuang T, Wakeham A, Itie A, et al. Genes Dev. 1997;11:1242–1252. doi: 10.1101/gad.11.10.1242. [DOI] [PubMed] [Google Scholar]
- 30.Chapman M S, Verma I M. Nature (London) 1996;382:678–679. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]
- 31.Monteiro A N, August A, Hanafusa H. Proc Natl Acad Sci USA. 1996;93:13595–13599. doi: 10.1073/pnas.93.24.13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milner J, Ponder B, Hughes-Davies L, Seltmann M, Kouzarides T. Nature (London) 1997;386:772–773. doi: 10.1038/386772a0. [DOI] [PubMed] [Google Scholar]
- 33.Connor F, Bertwistle D, Mee P J, Ross G M, Swift S, Grigorieva E, Tybulewicz V L J, Ashworth A. Nat Genet. 1997;17:423–430. doi: 10.1038/ng1297-423. [DOI] [PubMed] [Google Scholar]
- 34.Patel K J, Yu V P C C, Lee H, Corcoran A, Thistlethwaite F C, Evans M J, Colledge W H, Friedman L S, Ponder B A J, Venkitaraman A R. Mol Cell. 1998;1:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- 35.Mizuta R, LaSalle J M, Cheng H-L, Shinohara A, Ogawa H, Copeland N, Jenkins N A, LaLande M, Alt F W. Proc Natl Acad Sci USA. 1997;94:6927–6932. doi: 10.1073/pnas.94.13.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong A K C, Pero R, Ormonde P A, Tavtigian S V, Bartel P L. J Biol Chem. 1997;272:31941–31944. doi: 10.1074/jbc.272.51.31941. [DOI] [PubMed] [Google Scholar]
- 37.Levine A J. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 38.Stürzbecher H-W, Donzelmann B, Henning W, Knippschild U, Buchhop S. EMBO J. 1996;15:1992–2002. [PMC free article] [PubMed] [Google Scholar]
- 39.Buchhop S, Gibson M K, Wang X W, Wagner P, Stürzbecher H-W, Donzelmann B, Henning W, Knippschild U, Harris C C. Nucleic Acids Res. 1997;19:3868–3874. doi: 10.1093/nar/25.19.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scully R, Chen J, Ochs R L, Keegan K, Hoekstra M, Feunteun J, Livingston D M. Cell. 1997;90:1–20. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 41.Goggins M, Schutte M, Lu J, Moskaluk C A, Weinstein C L, Petersen G M, Yeo C J, Jackson C E, Lynch H T, Hruban R H, Kern S E. Cancer Res. 1996;56:5360–5364. [PubMed] [Google Scholar]
- 42.Sherr C J. Trends Cell Biol. 1994;4:15–18. doi: 10.1016/0962-8924(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 43.Bertwistle D, Swift S, Marston N J, Jackson L E, Crossland S, Crompton M R, Marshall C J, Ashworth A. Cancer Res. 1997;57:5485–5488. [PubMed] [Google Scholar]
- 44.Chen P-L, Chen C-F, Chen Y, Xiao J, Sharp Z D, Lee W-H. Proc Natl Acad Sci USA. 1998;95:5287–5292. doi: 10.1073/pnas.95.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Tombline G, Weber B L. Cell. 1998;92:433–436. doi: 10.1016/s0092-8674(00)80936-8. [DOI] [PubMed] [Google Scholar]
- 46.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Proc Natl Acad Sci USA. 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lim D-S, Hasty P. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hakem R, Pompa J L D L, Elia A, Potter J, Mak T W. Nat Genet. 1997;16:298–302. doi: 10.1038/ng0797-298. [DOI] [PubMed] [Google Scholar]
- 49.Maldonado E, Shieklattar R, Sheldon M, Cho H, Drapkin R, Rickert P, Lees E, Anderson C W, Linn S, Reinberg D. Nature (London) 1996;381:86–89. doi: 10.1038/381086a0. [DOI] [PubMed] [Google Scholar]