

Abstract
The presence of endotoxin from Gram-negative bacteria signals the innate immune system to up-regulate bacterial clearance and/or killing mechanisms. Paradoxically, such responses also contribute to septic shock, a clinical problem occurring with high frequency in Gram-negative septicemia. CD14 is a receptor for endotoxin (lipopolysaccharide, LPS) and is thought to have an essential role in innate immune responses to infection and thereby in the development of septic shock. Using a novel rabbit model of endotoxic shock produced by multiple exposures to endotoxin, we show that anti-rabbit CD14 mAb, which blocks LPS-CD14 binding, protects against organ injury and death even when the antibody is administered after initial exposures to LPS. In contrast, anti-rabbit tumor necrosis factor mAb treatment fails to protect when administered after LPS injections. These results support the concept that anti-CD14 treatment provides a new therapeutic window for the prevention of pathophysiologic changes that result from cumulative exposures to LPS during septic shock in man.
Gram-negative (GN) bacterial infections are associated with the development of septic shock, a serious clinical problem estimated to result in >100,000 deaths annually in the United States (1–3). Endotoxin (lipopolysaccharide, LPS), a glycolipid component of the outer membrane of all GN organisms, is generally acknowledged to play a central role in the development of septic shock (4–6). This syndrome is characterized by hypotension, coagulopathy, organ failure, and death (1, 4, 7, 8). The prevailing view is that the underlying mechanisms responsible for these complex pathophysiologic changes involve cellular injury caused by an array of proinflammatory mediators released from cells of the innate immune system (9–12). The innate immune system has evolved mechanisms to recognize trace amounts of LPS via a cell surface receptor that initiates cell activation after binding LPS. Numerous studies provide support for the concept that two proteins, LPS-binding protein (LBP) and CD14 are essential for LPS-induced cellular activation (13). Mice with targeted deletions of the gene encoding either CD14 or LBP are resistant to the lethal effects of LPS supporting the contention that LBP and CD14 are essential elements of a nonredundant LPS recognition system (14–16).
Effective approaches for the treatment of septic shock have lagged behind the substantive advances made in understanding host mechanisms involved in the response to infection (17). It is likely that the complexity of host responses in sepsis makes the relationship between the timing of delivery and the efficacy of potential therapeutics for septic shock a critical issue. For example, such considerations may serve to limit the effectiveness of treatments involving neutralization of single mediators such as tumor necrosis factor (TNF) or interleukin-1 that might only appear at certain stages in the sequence of events leading to septic shock (17). Here, we have investigated the effects of mAbs to either rabbit (rab) CD14 or rabbit TNF in a model of endotoxin shock in rabbits in which three sequential exposures to LPS administered during a 24-hr period results in progressive injury marked by death between 24 and 48 hr. The characteristics of this model contrast significantly with acute lethal bolus injection models (18, 19). It is likely that the model used here more closely reflects the development of septic shock in patients occurring over time as a consequence of repeated exposures to LPS.
Herein, we show that a mAb to rabbit CD14 that blocks LPS binding and subsequent cell activation offers remarkable protection even when administered after several exposures to LPS. In contrast, a mAb to rabbit TNF that neutralizes the biological activity of this mediator fails to demonstrate the same degree of protection. These data suggest that therapy by using anti-CD14 monoclonal treatment in patients with GN septicemia and shock may be useful even after the patient has been exposed LPS and in settings where other therapies have failed to be efficacious.
MATERIALS AND METHODS
Experimental Model.
A three-injection endotoxemic model was used as described by Mathison et al. (20). In brief, all of the rabbits (n = 108), except for the control group (n = 15), received i.v. injections of Re595 LPS (5 μg/kg) via the marginal ear vein at times 0, 5, and 24 hr. The rough form of LPS designated Re595 was purified from Salmonella minnesota bacteria (20). Stock samples of Re595 at 5 mg/ml were kept frozen at −20°C. Just before an i.v. injection, an aliquot was thawed, sonicated, and diluted with 0.9% NaCl to a concentration of 10 μg/ml. New Zealand White rabbits were obtained from Western Oregon Rabbit Company, Philomath, OR, and allowed 10 days for equilibration before use. Three days before the start of experiments, a 3.5 French polyurethane cannula (MRE-040, Braintree Scientific) with an accessible subcutaneous port (Jelco Intermittent injection cap, Critikon, Tampa, FL) was placed into the aortic arch via the left carotid artery by using aseptic technique and halothane anesthesia. Animals had free access to food and water and were unrestrained throughout the length of each study. Blood samples (1.3 ml total) were obtained, and mean arterial pressures (MAPs) were measured by using a model P10EZ transducer (Viggo-Spectramed, Oxnard, CA) at various time points throughout each 48-hr experiment. Blood was collected in pyrogen-free glass tubes and also in 1-ml syringes that had been rinsed with heparin (250 units/ml in saline). Serum was harvested within 6 hr of blood collection and stored at −20°C until cytokine assays were performed. After the MAP was measured and the blood sample collected, 1.3 ml of heparinized (10 units/ml) saline was flushed through the injection port and cannula. Surviving animals at 48 hr were killed, and organ tissue from all animals was harvested for histological examination.
Animals treated with 5 mg/kg of anti-rabCD14 included (i) prophylactic treatment (i.v. doses at times −1, +4, and +23 hr; n = 13) and (ii) delayed treatment groups (i.v. doses at times +4 and +23 hr; n = 10), +8 and +23 hr (n = 6), +12 and +23 hr (n = 5), or +23 hr (n = 5). For the anti-rabTNF studies, groups receiving 5 mg/kg of anti-rabTNF included (i) prophylactic treatment (i.v. doses at times −1, +4, and +23 hr; n = 11) and (ii) delayed treatment (i.v. doses at times +4 and +23 hr; n = 12). Control animals that did not receive any LPS included a sham surgery group (n = 4), an anti-CD14 alone group (5 mg/kg i.v. doses at times −1, +4, and +23 hr; n = 6), and an anti-rabTNF alone group (5 mg/kg i.v. doses at times −1, +4, and +23 hr; n = 5). Untreated animals included those receiving LPS alone (n = 22) or an isotype-matched mAb (i.v. doses of 5 mg/kg at times −1, +4, and +23 hr; n = 4) or at times +4 and +23 hr (n = 5). Experiments were performed by using groups of 12 rabbits with treatment determined in advance and randomly assigned to the animals. Each group included animals receiving LPS alone, animals treated with antibody and LPS, and control animals receiving antibody alone. During the 1-yr course of these studies, there was no discernable change in the nature of injury induced by multiple LPS injections alone or the protection from LPS-induced injury afforded by anti-CD14 antibody treatment.
Cytokine Assays.
IL-8 levels in the blood were determined by using a sandwich ELISA with goat anti-rabbit IL-8 IgG (21). TNF activity was measured by using a cytolytic assay with Actinomycin D-treated L929 cells (20). Functional activity of the mAbs was tested in an ex vivo rabbit whole blood assay, and the in vitro production of TNF was measured by using WEHI 164 clone 13 cell cytotoxicity assay (22, 23).
mAb Production.
mAb to rabbit CD14 was generated by somatic cell fusion between the mouse myeloma cell line (P3-x63A, 8.653) and spleen cells from BALB/c mice immunized with transfected Chinese hamster ovary cells expressing cell-surface rabbit CD14 (GenBank accession no. M90488) (24). The anti-rabCD14-specific hybridomas were identified by ELISA against purified rabbit CD14 and subcloned by limiting dilution. A clone designated 1116 1a6 (IgG2a) was isolated that blocked TNF production in an ex vivo rabbit whole blood assay (22). Anti-rabTNF was produced as described above by using mice immunized with recombinant rabbit TNF. Recombinant rabbit TNF-α was expressed and purified as described (25). IgG fractions were purified from conditioned medium by using immobilized Protein G (GIBCO/BRL) and stored at −20°C until the day of use. The anti-rabTNF was shown to block TNF bioactivity (22, 23) by using either natural or recombinant sources of TNF. All tissue culture and glassware used was pyrogen-free, and reagents/antibodies were endotoxin-free as determined by a chromogenic LAL (BioWhittaker).
Flow Cytometry.
Analysis of fluorescein isothiocyanate (FITC)-Re595 LPS binding to elicited rabbit peritoneal exudate macrophages and Chinese hamster ovary (CHO)-rabCD14 cells was performed as described (20) and analyzed by using the FACScan and CellQuest software (Beckton Dickinson).
Hematology.
Heparinized whole blood was diluted 1:20 in 3% acetic acid and leukocyte counts were determined manually by using a hemocytometer. Blood smears were stained by using Hema 3 reagents (Curtin Matheson Scientific, Houston), and differential counts were performed.
Histology.
Fresh tissue samples were harvested from the lung, liver, kidney, spleen, and adrenal gland of each animal. Tissue was fixed in 10% formalin and sectioned from paraffin blocks by using standard histological technique. Slides were stained with hematoxylin-eosin or periodic acid/Schiff reagent.
Statistical Analysis.
Data are expressed as mean ± standard error (SE). Organ injury scores and mortality data were evaluated by using the two-tailed Fisher Exact test. The Student t test was used for evaluation of the MAP and white blood cell (WBC) count data. Cytokine assays, hematology studies, and evaluation of histopathology preparations were blind, and specimen identity was revealed only on completion of analysis.
RESULTS AND DISCUSSION
Anti-Rabbit CD14 mAb.
We screened a panel of murine hybridomas secreting anti-rabCD14 mAbs first for binding to recombinant rabbit CD14. Positive clones were further screened in two functional assays that reflect LPS binding to CD14 and LPS-induced monocyte activation in an ex vivo whole blood assay. Using these assays, a single mAb was selected that inhibited FITC-LPS binding to rabbit CD14 expressed by stably transfected CHO-K1 cells or elicited rabbit peritoneal exudate macrophages. This same clone secreted a mAb that when added to rabbit whole blood blocked LPS-induced TNF release in this ex vivo whole blood assay (Figs. 1 A and B). Thus, we identified a hybridoma that had properties, which would prevent LPS binding to CD14 as well as subsequent cellular activation events triggered by LPS, and we hypothesized would be an ideal candidate for testing in the rabbit model of shock described herein.
Figure 1.

Anti-rabCD14 blocks LPS-induced TNF release and LPS binding to CD14-bearing cells in vitro. (A) Heparinized rabbit blood was preincubated with anti-rabCD14 10 μg/ml (▵), 3 μg/ml (▴), 1 μg/ml (○), isotype-matched IgG 10 μg/ml (■), or no addition (•) followed by stimulation with LPS for 4 hr at 37°C. TNF released into the plasma was measured by using the WEHI clone 13 cytolytic assay. (B) Rabbit peritoneal exudate macrophages (PEM) or transfected CHO cells expressing rabbit CD14 as cell surface protein (CHO-rabCD14) were left untreated (-0-), exposed to FITC-LPS, or pretreated with anti-rabCD14 followed by exposure to FITC-LPS and then examined on the fluorescence-activated cell sorter. In contrast to the blocking effect of anti-CD14, an isotype control mAb was without effect.
Effects of Anti-CD14 or Anti-TNF mAb on LPS-Induced Injury in Rabbits.
To avoid acute systemic effects of high doses of LPS here we used an animal model in which three sequential doses of LPS were administered over a 24-hr period. This protocol resulted in severe organ injury and/or death in the 24 hr after the last LPS injection; no deaths were recorded during the first 24 hr of the experiment although various measures of sublethal injury were recorded during the first 24 hr including decreased MAP, leukopenia, and systemic cytokine release. The rationale for this model derives from a variety of studies that suggest septic shock in man is the result of the cumulative effects of mediators released after multiple exposures to bacteria or bacterial products such as LPS (4). We speculated that the model used in these studies provided a unique opportunity to investigate the utility of mAb blockade of CD14 or of a single mediator TNF when the treatment was administered prophylactically or after some exposure to LPS. Data of 48-hr survival and a summary of organ injury evaluated from histopathology are contained in Table 1.
Table 1.
Incidence of 48-hr survival and organ injury
| Anti-rabCD14
|
Anti-rabTNF
|
Controls
|
||||
|---|---|---|---|---|---|---|
| Prophylactic treatment | Delayed treatment | Prophylactic treatment | Delayed treatment | LPS ± control mAb | No LPS | |
| 48-hr survival | 100%* (13/13) | 100%* (26/26) | 100%* (11/11) | 67% (8/12) | 68% (21/31) | 100%* (15/15) |
| Renal cortical necrosis | 0%† (0/13) | 0%† (0/26) | 64% (7/11) | 58% (7/12) | 84% (26/31) | 0%† (0/15) |
| Pulmonary injury | 0%† (0/13) | 0%† (0/26) | 27% (3/11) | 33% (4/12) | 61% (19/31) | 0%† (0/15) |
P < 0.05 compared with untreated group (LPS ± control mAb) using Fisher Exact test.
P < 0.001 compared with untreated group (LPS ± control mAb) using Fisher Exact test.
LPS injections (5 μg/kg) at 0, 5, and 24 hr induce lethality by 48 hr in approximately one-third of the untreated animals (10/31). In this model, deaths do not occur acutely but were noted to occur between 24 and 48 hr; multiple organ injury and lethality requires all three LPS injections. The experimental group termed untreated includes animals receiving LPS alone (n = 22), prophylactic isotype-matched mAb treatment (5 mg/kg i.v. at t = −1, +4, and +23 hr; n = 4), and delayed isotype-matched mAb treatment (5 mg/kg i.v. at t = +4 and +23 hr; n = 5). The isotype-matched treatment groups behaved identically to animals receiving LPS alone providing justification for inclusion in the untreated group. In contrast to the ≈30% mortality in the untreated group, no deaths were recorded in the various anti-rabCD14 treatment groups during the 48-hr experimental period (39/39) and, remarkably, no dependency on the time of administration of anti-rabCD14 was noted. Specifically, we noted 100% survival in animals treated at −1 hr (prophylactic treatment; 13/13), at +4 hr (delayed treatment; 10/10), at +8 hr (delayed treatment; 6/6), at +12 hr (delayed treatment; 5/5), and even after +23 hr (delayed treatment; 5/5).
Animals treated with anti-rabTNF demonstrated protection against 48-hr lethality when the antibody was given prophylactically. In contrast, no protection was noted in the group in which anti-rabTNF injection was delayed until +4 hr (Table 1). These data provide a clear difference between the effects of anti-rabCD14 and anti-TNF mAb treatment in this animal model. On one hand, anti-CD14 will blockade production of all cellular mediators induced by LPS when a CD14-dependent pathway is involved whereas anti-TNF treatment will only prevent events related to the deleterious effects of TNF on cells.
After the initial LPS injection, MAP decreased gradually following LPS treatment to <80% of control values (Fig. 2A). In contrast, control groups (no LPS injections) and the group receiving anti-rabCD14 prophylactically remained normotensive throughout the 48-hr experiment. Delayed administration of anti-rabCD14 did not significantly improve recovery from hypotension that was induced by the first dose of LPS.
Figure 2.

Anti-rabCD14 prevents hypotension (A) and attenuates leukopenia (B) in rabbits that received three sequential i.v. injections of LPS (5 μg/kg) at t = 0, 5, and 24 hr (filled symbols). Animals that received LPS were either untreated (•), prophylactically treated with anti-rabCD14 (■, t = −1, 4, and 23 hr), or received anti-rabCD14-delayed treatment (▴, t = 4 and 23 hr). Control animals that did not receive LPS (○) were either untreated (no antibody) or received antibody prophylactic treatment or delayed treatment. (B Inset) WBC counts over 24–48 hr.
In untreated animals, a severe leukopenia, primarily a neutropenia, occurs within 1 hr after the initial LPS injection (Fig. 2B). Initial WBC counts (7,750 ± 300 cells/mm3) declined to a nadir (1,000 ± 90 cells/mm3) by 4 hr; then, a progressive leukocytosis (12,800 ± 2,000 cells/mm3) was noted at 24 hr. The third injection of LPS at t = 24 hr resulted in a similar pattern of leukopenia followed by a return of cells to the circulation in the next 24-hr period. Prophylactic treatment with anti-rabCD14 attenuated the neutropenia seen in the first 24-hr period in this model. In contrast to untreated animals, WBC counts in the anti-rabCD14 prophylactic treatment rabbits declined to a nadir (4,270 ± 400 cells/mm3) by 3 hr but were significantly higher when compared with the untreated animals (P < 0.0001). Moreover, WBC counts in the anti-rabCD14 prophylactic treatment group remained stable during the next 24-hr period. In the anti-rabCD14 delayed treatment group, the pattern of leukopenia (900 ± 100 cells/mm3) and leukocytosis (15,000 ± 200 cells/mm3) was not significantly different when compared with the untreated animals in the first 24 hr. The delayed treatment with anti-rabCD14 did prevent a second precipitous fall in WBC counts after the final LPS injection. In control animals, the WBC counts and differentials remained normal throughout the length of the experiments. The observation that anti-rabCD14 treatment attenuated LPS-induced leukopenia does not discriminate between mechanisms involving direct effects of LPS on leukocyte adhesion or secondary effects of LPS-induced release of mediators that occur via CD14-dependent mechanisms.
One of the hallmarks of the innate immune response to LPS is the rapid systemic release of cytokines. Here we show that LPS induces a pronounced elevation of plasma TNF levels within 1–2 hr after the initial LPS injection in the untreated groups (Fig. 3A). No TNF was detected after the second LPS injection at 5 hr. However, we observed a second although attenuated release of TNF 1–2 hr after the final (t = 24 hr) LPS injection. Measurement of IL-8 revealed a similar pattern with cytokine production detected after the initial and final LPS injections but not after the second injection at 5 hr (Fig. 3B). The failure to observe release of TNF and IL-8 after the t = 5-hr LPS injection presumably results from the development of LPS tolerance (26). Prophylactic treatment with anti-rabCD14 mAb prevented elevations of TNF and IL-8 observed after the initial and final LPS injections. Delayed treatment with anti-rabCD14 (after the first release of cytokines had occurred) prevented the 25–26 hr second wave cytokine peak after the final LPS injection.
Figure 3.

Anti-rabCD14 inhibits release of TNF (A) and IL-8 (B) into serum of rabbits receiving three sequential i.v. injections of LPS (5 μg/kg) at t = 0, 5, and 24 hr (filled symbols). As described in the Fig. 2 legend, animals that received LPS were either untreated (•), prophylactically treated with anti-rabCD14 (■, t = −1, 4, and 23 hr), or received anti-rabCD14 delayed treatment (▴, t = 4 and 23 hr). Control animals that did not receive LPS (○) were either untreated (no antibody) or received antibody prophylactic treatment or delayed treatment.
Septic shock is characterized by multi-organ failure resulting from fibrin deposition, inadequate organ perfusion as well as other mechanisms most likely involving activated leukocytes. The appearance of renal cortical necrosis with sequential LPS injections 24 hr apart has been well described previously in rabbits and is characterized by petechial hemorrhages and microthrombi similar to those noted in disseminated intravascular coagulation (27). Renal cortical necrosis was observed in 84% (26/31) of the untreated group (Fig. 4). Histological examination revealed substantial fibrin deposition in the renal glomerular capillaries and tubular necrosis. We also recorded evidence of pulmonary damage marked by hemorrhage, consolidation, and fibrin deposition in 61% (19/31) of the animals in the untreated groups (Fig. 5). In preliminary studies, we determined that renal cortical necrosis occurred in animals injected with LPS at 0 and 24 hr and did not require the second LPS injection administered at 5 hr. In contrast, the pulmonary injury recorded here was only observed in animals receiving all three injections of LPS. These latter data are consistent with the concept that septic shock and multi-organ failure results from the cumulative effects of multiple exposures to endotoxin. Other tissues including liver and spleen also revealed the presence of micro-thrombi (data not shown). In marked contrast, no salient pathological changes are seen in the kidney (Fig. 4), lung (Fig. 5), or other tissues such as spleen, liver, or adrenal glands (not shown) on gross or histological examination in any of the anti-rabCD14 treatment groups regardless of whether the antibody was administered as a prophylactic or delayed treatment.
Figure 4.
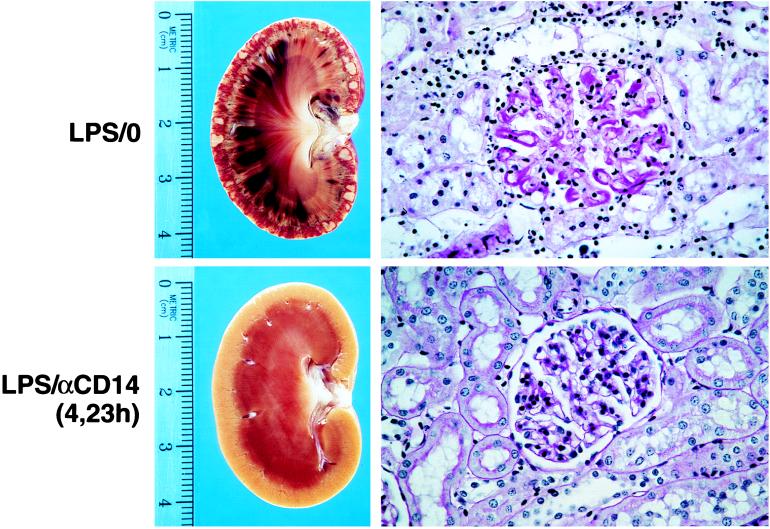
Anti-rabCD14 protects from renal injury induced by three sequential i.v. injections of LPS (5 μg/kg at t = 0, 5, and 24 hr). The upper panels show representative lesions from an untreated rabbit (that received LPS but no antibody: LPS/0). The hemisectioned kidney shows advanced cortical necrosis, and the periodic acid/Schiff reagent-stained histologic section shows marked glomerular fibrin deposition and tubular necrosis. The lower panels are representative of the anti-rabCD14-treated groups (LPS:αCD14) showing a hemisectioned kidney and histologic section from a rabbit that received LPS injections and antibody delayed treatment at t = 4 and 23 hr. Kidneys from rabbits that received anti-rabCD14 antibody (either prophylactic or delayed treatment) were essentially normal, with changes limited to mild edema and occasional inflammatory infiltrates (X400).
Figure 5.
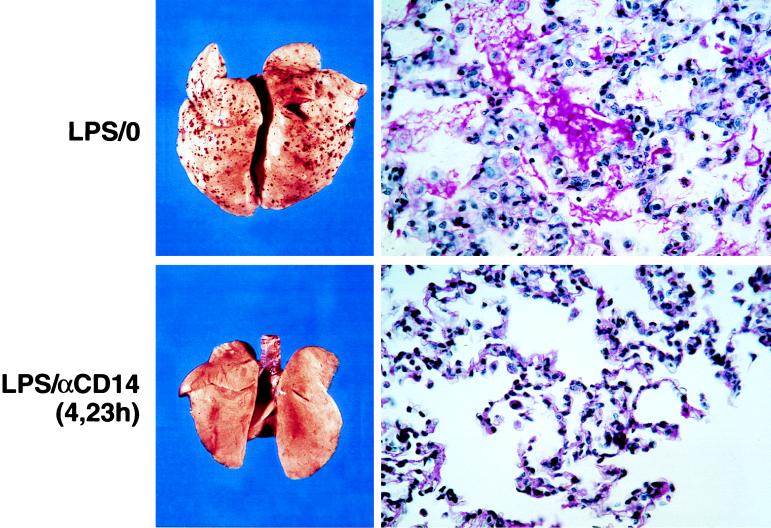
Anti-rabCD14 protects from lung injury induced by three sequential i.v. injections of LPS (5 μg/kg at t = 0, 5, and 24 hr). The upper panels show representative lesions from an untreated rabbit (that received LPS but no antibody: LPS/0). Petechial hemorrhages and edema were evident on gross examination, and periodic acid/Schiff reagent-stained histologic sections show sero-fibrinous exudate and inflammatory infiltrates in the alveoli. The lower panels are representative of the anti-rabCD14 treated groups (LPS:αCD14) showing lung from an animal that received LPS injections and anti-rabCD14 delayed treatment at t = 4 and 23 hr. The lungs from anti-rabCD14-treated animals (prophylactic or delayed treatment) were grossly normal without edema or petechiae, and microscopic changes were limited to increased number of leukocytes observed in the alveolar capillaries (X400).
In this endotoxemia model, the first LPS injection induces a large cytokine response but in the absence of further stimulation does not lead to organ injury or death. The second (5 hr) LPS injection, although inducing essentially no further cytokine release, apparently potentiates injury induced by the 24-hr injection. In a group of 5 rabbits, we observed no lethality and no renal or lung injury 48 hr after two sequential 10 μg of LPS doses at 0 and 5 hr. The 24 hr LPS challenge induces a substantially diminished cytokine response but triggers organ injury and death. The protection afforded by anti-CD14 mAb in this model can be attributed to blocking of the priming effect of the first dose of LPS (antibody pretreatment) or to blocking of the triggering action of the 24-hr dose (delayed antibody treatment). Importantly, as noted below, when we compared anti-CD14 and anti-TNF, only the former antibody provided protection when administered as delayed antibody treatment.
We also compared the effects of anti-rabCD14 treatment protocols by using anti-rabbit TNF mAb. Either prophylactic or delayed treatment with anti-rabTNF did not provide the same degree of protection as seen with the anti-rabCD14-treated animals (Table 1). Pretreatment with anti-rabTNF neutralized TNF and did confer survival benefit (11/11) but only gave partial protection from renal cortical damage (7/11) and pulmonary injury (3/11). Delayed treatment with anti-rabTNF at +4 hr failed to provide the survival benefit observed with (8/12) and did not prevent renal cortical necrosis (7/12) or pulmonary injury (4/12). Moreover, 48-hr survivors with renal cortical necrosis would not be expected to survive long-term given the nearly complete destruction of the kidney caused by this lesion.
CONCLUSION
The severe physiologic injury and organ failure that often accompanies GN sepsis is generally acknowledged to be a result of repeated exposure to LPS. Recognition of LPS by the innate immune system results in the release of a multiplicity of mediators, and while some mediators appear to play a more prominent role than others on producing injury, disappointing results from numerous clinical trials have shown that therapeutic agents targeting single mediators do not protect from injury (28). Even when such therapies may provide benefit, it is usually confined to either prophylactic or very early stage treatments. There is now a plethora of data suggesting an essential role for CD14 in recognition of LPS by cells and more importantly in triggering cell activation processes. It is generally agreed that CD14 almost certainly acts in concert with additional, but as yet unidentified, cellular components to initiate a transmembrane signaling cascade. However, these additional components are not part of a redundant system because blockade of LPS binding to CD14 under physiological conditions is sufficient to prevent the many cellular responses triggered by LPS.
Unfortunately, treatment of septic patients developing symptoms predictive of shock and multi-organ failure often does not occur until there are already some pathophysiologic manifestations apparent. It is possible that other agents that block the pleotropic effects of LPS such as bactericidal-permeability increasing factor might afford protection in the model noted herein. This remains to be evaluated experimentally. Most importantly the model used here suggests that there may be a therapeutic window available for treatment since we have demonstrated clear benefit with respect to organ injury and survival in animals receiving delayed treatment with anti-rabCD14. Whether this contention will be born out in the setting of septic shock in man will only be known after direct testing of anti-CD14 mAb therapy in the clinic.
Acknowledgments
We thank Susan Spray, Doug Brandt, and Greg Campbell with the Department of Animal Resources for their assistance during the surgical procedures and Elizabeth Chastain for her help in preparing the manuscript. This is publication number 11657-IMM from the Department of Immunology of The Scripps Research Institute. This work was supported by grants from the National Institutes of Health: AI15136 and GM08172 and from ICOS Corporation (Seattle, WA).
ABBREVIATIONS
- LPS
lipopolysaccharide
- TNF
tumor necrosis factor
- GN
Gram-negative
- LBP
LPS-binding protein
- MAP
mean arterial pressure
- WBC
white blood cell
- FITC
fluorescein isothiocyanate
- CHO
Chinese hampster ovary
- rab
rabbit
References
- 1.Bone R C. Chest. 1991;100:802–808. doi: 10.1378/chest.100.3.802. [DOI] [PubMed] [Google Scholar]
- 2.Wolff S M, Bennett J V. N Engl J Med. 1974;291:733–734. doi: 10.1056/NEJM197410032911411. [DOI] [PubMed] [Google Scholar]
- 3.Stone R. Science. 1994;64:365–367. doi: 10.1126/science.8153620. [DOI] [PubMed] [Google Scholar]
- 4.Danner R L, Elin R J, Hosseini J M, Wesley R A, Reilly J M, Parillo J E. Chest. 1991;99:169–175. doi: 10.1378/chest.99.1.169. [DOI] [PubMed] [Google Scholar]
- 5.van Deventer S J H, Buller H R, ten Cate J W, Sturk A, Pauw W. Lancet. 1988;1:605–609. doi: 10.1016/s0140-6736(88)91412-2. [DOI] [PubMed] [Google Scholar]
- 6.Ziegler E J, McCutchan J A, Fierer J, Glauser M P, Sadoff J C, Douglas H, Braude A I. N Engl J Med. 1982;307:1125–1130. doi: 10.1056/NEJM198211113072001. [DOI] [PubMed] [Google Scholar]
- 7.Kreger B E, Craven D E, McCabe W R. Am J Med. 1980;68:344–355. doi: 10.1016/0002-9343(80)90102-3. [DOI] [PubMed] [Google Scholar]
- 8.Hazinski M F, Iberti T J, MacIntyre N R, Parker M M, Tribett D, Prion S, Chmel H. Am J Crit Care. 1993;2:224–237. [PubMed] [Google Scholar]
- 9.Evans G F, Snyder Y M, Butler L D, Zuckerman S H. Circ Shock. 1989;29:279–290. [PubMed] [Google Scholar]
- 10.Ramadori G, Van Damme J, Rieder H, Meyer zum Buschendelde K-H. Eur J Immunol. 1988;18:1259–1264. doi: 10.1002/eji.1830180817. [DOI] [PubMed] [Google Scholar]
- 11.Sgadari C, Angiolillo A L, Cherney B W, Pike S E, Farber J M, Koniaris L G, Vanguri P, Burd P R, Sheikh N, Gupta G, et al. Proc Natl Acad Sci USA. 1996;93:13791–13796. doi: 10.1073/pnas.93.24.13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baue A R. Shock. 1995;4:39–40. doi: 10.1097/00024382-199507000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Ulevitch R J, Tobias P S. Annu Rev Immunol. 1995;13:437–457. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 14.Haziot A, Ferrero E, Lin X Y, Stewart C L, Goyert S M. In: CD14-Deficient Mice Are Exquisitely Insensitive to the Effects of LPS. Levin J, Alving C R, Munford R S, Redl H, editors. New York: Wiley; 1995. pp. 349–351. [PubMed] [Google Scholar]
- 15.Haziot A, Ferrero E, Kontgen F, Hijiya N, Yamamoto S, Silver J, Stewart C L, Goyert S M. Immunity. 1996;4:407–414. doi: 10.1016/s1074-7613(00)80254-x. [DOI] [PubMed] [Google Scholar]
- 16.Jack R S, Fan X, Bernheiden M, Rune G, Ehlers M, Weber A, Kirsch G, Mentel R, Furll B, Freudenberg M, et al. Nature (London) 1997;389:742–745. doi: 10.1038/39622. [DOI] [PubMed] [Google Scholar]
- 17.Grau G E, Maennel D N. Nat Med. 1997;3:1193–1195. doi: 10.1038/nm1197-1193. [DOI] [PubMed] [Google Scholar]
- 18.Deitch E A. Shock. 1998;9:1–11. doi: 10.1097/00024382-199801000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Wichterman K A, Baue A E, Chaudry I H. J Surg Res. 1980;29:189–201. doi: 10.1016/0022-4804(80)90037-2. [DOI] [PubMed] [Google Scholar]
- 20.Mathison J C, Wolfson E, Ulevitch R J. J Clin Invest. 1988;81:1925–1937. doi: 10.1172/JCI113540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeForge L E, Remick D G. Immunol Invest. 1991;20:89–97. doi: 10.3109/08820139109054928. [DOI] [PubMed] [Google Scholar]
- 22.Desch C E, Kovach N L, Present W, Broyles C, Harlan J M. Lymphokine Res. 1989;8:141–146. [PubMed] [Google Scholar]
- 23.Espevik T, Nissen-Meyer J. J Immunol Methods. 1986;95:99–105. doi: 10.1016/0022-1759(86)90322-4. [DOI] [PubMed] [Google Scholar]
- 24.Bebbington C R, Hentschel C C G. In: The Use of Vectors Based on Gene Amplification for the Expression of Cloned Genes in Mammalian Cells. Glover D, editor. New York: Academic; 1987. pp. 163–188. [Google Scholar]
- 25.Kravchenko V V, Steinemann S, Kline L, Feng L, Ulevitch R J. Shock. 1996;5:194–201. doi: 10.1097/00024382-199603000-00005. [DOI] [PubMed] [Google Scholar]
- 26.Mathison J C, Virca G D, Wolfson N, Tobias P S, Glaser K, Ulevitch R J. J Clin Invest. 1990;85:1108–1118. doi: 10.1172/JCI114542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raij L, Keane W F, Michael A F. Kidney Int. 1977;12:91–95. doi: 10.1038/ki.1977.85. [DOI] [PubMed] [Google Scholar]
- 28.Dellinger, R. P., Opal, S. M., Rotrosen, D., Suffredini, A. D. & Zimmerman, J. L. (1997) Chest 744–753.