

Abstract
We report the generation and characterization of a diiron(III) intermediate formed during reaction with dioxygen of the reduced hydroxylase component of toluene/o-xylene monooxygenase from Pseudomonas sp. OX1. The decay rate of this species is accelerated upon mixing with phenol, a substrate for this system. Under steady state conditions, hydrogen peroxide was generated in the absence of substrate. The oxidized hydroxylase also decomposed hydrogen peroxide to liberate dioxygen in the absence of reducing equivalents. This activity suggests that dioxygen activation may be reversible. The linear free energy relationship determined from hydroxylation of para substituted phenols under steady state turnover has a negative slope. A value of ρ < 0 is consistent with electrophilic attack by the oxidizing intermediate on the aromatic substrates. The results from these steady and pre-steady state experiments provide compelling evidence that the diiron(III) intermediate is the active oxidant in ToMO and a peroxodiiron(III) transient, despite differences between its optical and Mössbauer spectroscopic parameters and those of other peroxodiiron(III) centers.
Keywords: toluene monooxygenase, carboxylate-bridged diiron proteins, oxygen activation, hydrocarbon oxidation
Introduction
Dioxygen activation at structurally homologous active sites in metalloproteins often leads to transient species having similar spectroscopic properties. For example, heme prosthetic groups bind dioxygen in their ferrous forms to generate η1-peroxoiron(III) intermediates.1,2 Protonation of the distal oxygen atom in this intermediate facilitates O–O bond cleavage and formation of an (oxo)iron(IV)–porphyrin cation radical (Scheme 1). The reactivity of the heme iron unit is modulated by the proximal ligand trans to the O2-binding site on the metal center and by the surrounding protein matrix, dictating dioxygen binding or activation depending on the requisite enzyme function. Similar phenomena occur for the non-heme carboxylate-bridged diiron (CBDI) protein superfamily, in which a reduced, diiron(II), form reacts with dioxygen (Scheme 1).3,4 The choice of non-bridging ligands at the dimetallic center, histidine vs aspartate or glutamate, and the availability of an open coordination site determine whether O2 will bind reversibly or be activated for substrate oxidation. For dioxygen activation, the diiron(II) form of the protein typically reacts with dioxygen to form a peroxodiiron(III) center. In the hydroxylase component of methane monooxygenase (MMOH), either homolytic5 or heterolytic6 cleavage of the O–O bond in this species occurs to yield a high-valent di(μ-oxo)diiron(IV) core, designated Q. In the R2 subunit of ribonucleotide reductase (RNR-R2), the peroxo intermediate carries out a one-electron oxidation of W48 to form a mixed-valent diiron(III,IV) species, designated X.7 The peroxodiiron(III) units in MMOH and RNR-R2 are the only ones in the CBDI enzyme superfamily that evolve to form iron(IV) intermediates.
Scheme 1.
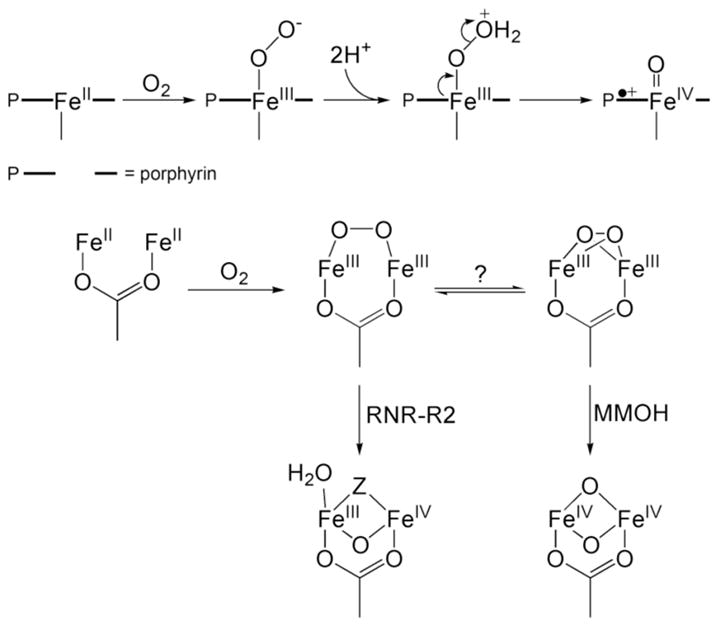
Dioxygen Activation at Iron-Heme and CBDI Centers
The primary coordination spheres of stearyol-ACP Δ9D desaturase,8 rubrerythrin,9 and the hydroxylase component of toluene/o-xylene monooxygenase (ToMOH)10,11 bear a strong resemblance to those of MMOH and RNR-R2. No intermediates have previously been reported for reaction of the reduced forms of these enzymes with dioxygen. Addition of the substrate-carrier protein conjugate to reduced Δ9D prior to reaction with dioxygen allowed such an intermediate to be generated, however, the Mössbauer, rR, and optical spectroscopic parameters of which are indicative of a μ-1,2-peroxodiiron(III) species.12,13
We previously reported a reaction of an I100W mutant of ToMOH from Pseudomonas sp. OX1 that, in its reduced form ToMOHred, reacts with O2 to provide a transient mixed-valent diiron(III,IV)–W• chromophore that was characterized by various spectroscopic methods.14,15 Of particular interest was the identification of a diiron(III) intermediate, formed prior to the diiron(III,IV)–W• species (λmax 500 nm), having Mössbauer parameters and optical absorption features different from those of other peroxodiiron(III) transients.4,16 Its isomer shift lies within the range of known peroxide-bridged synthetic diiron(III) complexes, but the quadrupole splitting differs by more than 0.3 mm/s.16–18 No transient optical absorption band, other than that of the deprotonated tryptophanyl radical signal at 500 nm, were apparent during reaction of the reduced ToMOH I100W/regulatory protein (ToMOD) complex with O2.
In the present article we describe the results of experiments, including single- and double-mixing rapid-freeze quench Mössbauer studies, to determine whether this same diiron(III) species is generated in native ToMOH. As will be demonstrated, such an intermediate does indeed accumulate over a relatively long time scale of 1 sec, and reacts with substrate as examined by double-mixing methods in which the oxygenated species was first generated and then exposed to buffer containing phenol. To gain further insight into the nature of this intermediate, steady-state experiments were conducted to determine (i) whether hydrogen peroxide evolves during catalytic turnover; (ii) whether a peroxide shunt pathway to this species is accessible in this system; and (iii) the reactivity profile of the diiron(III) center in comparison to those of other CBDI enzymes and synthetic complexes. The results suggest that this intermediate contains a {O22−} fragment bound to a diiron(III) center, but with a previously unknown coordination geometry and/or protonation state.
Experimental Methods
General Considerations
Plasmids containing the genes for the Pseudomonas sp. OX1 ToMO component proteins were kindly supplied by the laboratory of Professor Alberto Di Donato, Naples, Italy. Expression and preparation of ToMOH and ToMOD were carried out as described elsewhere10 with the following changes. For 57Fe enrichment, ToMOH was expressed in media (10 g tryptone, 10 g NaCl, and 2 g Cassamino acids per liter of ddH2O) supplemented with 40 μM 57FeCl3. The solution of 57FeCl3 was prepared by dissolving enriched metal powder (96.7% isotopic purity, Advanced Materials Technologies Ltd., Nes-Ziona, Israel) in concentrated hydrochloric acid. Ion exchange buffers contained 57FeCl3 (150 μM) instead of Mohr’s salt. Specific activities for phenol for the isolated components were greater than 1200 mU/mg. The iron content of the hydroxylase was 4.2 ± 0.3 Fe:H dimer as measured by the ferrozine assay.19 HPLC experiments were performed with a Vydac protein & peptide C18 column connected to an Agilent 1200 series instrument equipped with a multi-wavelength detector. Instrumentation for optical and Mössbauer spectroscopy are as reported elsewhere.14,20 All other reagents were purchased from Aldrich Chemical Company and used as received.
Rapid-Freeze Quench Sample Preparation for Mössbauer Spectroscopy
Samples of 57Fe-enriched ToMOH and ToMOD were reduced and dialyzed as described previously.14,15 For single-mixing rapid-freeze quench (RFQ) experiments, a solution of ToMOH:2ToMOD (690 μM) in 25 mM MOPS buffer, pH 7.0, was rapidly combined with an equal volume of O2-saturated buffer at 4.0 °C. The oxygenated protein was allowed to react for a specific period of time, ranging from 34 ms to 15 min, after which the protein mixtures were quenched in isopentane at −140 °C. Samples were packed into Mössbauer sample cups and stored in liquid nitrogen. For double-mixing experiments, oxygenated buffer and the reduced protein solution (510 μM ToMOH, 1.02 mM ToMOD) were first mixed with an equal volume of O2 saturated buffer and allowed to react for ~ 170 ms. This oxygenated protein solution was then mixed with a half volume of buffer containing phenol (4.6 mM in 25 mM MOPS buffer, pH 7.0), aged for times varying from 25 ms to 30 min, and quenched in isopentane at −140 °C. A sample of the 0.17-s aged oxygenated protein without phenol was collected as a reference. The dilution factors for single- and double-mixing samples were two and three, respectively. The product of phenol oxidation was determined and quantified in double-mixing RFQ Mössbauer samples, for which the oxygenated protein was allowed to react with substrate for at least 5 s in the following manner. Ascorbic acid (20 μL, 22 mM in ddH2O) was added to an aliquot (50 μL) of a thawed Mössbauer sample. This mixture was allowed to stand for 5 min, after which TFA (30 μL, 50% in ddH2O) was added. Samples were centrifuged and the supernatant was loaded onto a Vydac column. HPLC conditions for separation of hydroxylated products from phenol were 0% buffer B for 7 min, 0% to 40% B for 1 min (linear gradient), 40% to 100% for 7 min (linear gradient), and 100% B for 3 min (A: 1% acetonitrile, 98.8% ddH2O, 0.2% TFA; B: 49.9% acetonitrile, 49.9% ddH2O, 0.2% TFA). The optical absorption at 280 nm was monitored with time for all samples. Retention times for catechol, resorcinol, and hydroquinone were determined under these conditions. Mössbauer measurements were carried out at 4.2 K with an applied magnetic field parallel to the γ-irradiation using spectrometers described previously.20 Analyses of the Mössbauer spectra were performed with the program WMOSS (WEB Research). The zero velocity refers to the centroid of a room-temperature spectrum of a metallic Fe foil.
Assay for Hydrogen Peroxide Release in the Steady-State
A colorimetric assay employing potassium thiocyanate and ferrous ammonium sulfate was used to detect hydrogen peroxide.21 Each reaction solution contained 1 μM ToMOH, 2 μM ToMOC, 4 μM ToMOD, 0.1 μM ToMOF, and 1 mM NADH in 500 μL of Tris/HCl buffer, pH 7.5, at 25 °C. ToMOC and ToMOF are the Rieske protein and NADH oxidoreductase components of the toluene/o-xylene monooxygenase (ToMO) system, respectively. In samples containing phenol as a substrate, the concentration was 4 mM. Reactions were initiated by addition of NADH, quenched after a specified reaction time with 100 μL of 0.4 M trichloroacetic acid, and centrifuged at 13,500 × g for 5 min. After centrifugation, aliquots of solutions of potassium thiocyanate (100 μL, 2.5 M) and ferrous ammonium sulfate (200 μL, 10 mM) in ddH2O were added to the supernatant (500 μL). The mixture was shaken vigorously and allowed to stand for five min before the absorbance at 480 nm was measured. Solutions of hydrogen peroxide were standardized by measuring the absorbance at 240 nm.22 These solutions were treated as described for the enzyme reaction mixtures to generate a standard curve. The consumption of NADH to generate hydrogen peroxide was determined by monitoring the absorbance decrease at 340 nm, which corresponds to the depletion of NADH (ε340 = 6220 M−1cm−1), for five min, quenching the reaction, and measuring the concentration of hydrogen peroxide present. To monitor the absorbance change at 340 nm, the initial NADH concentration in the assay solution was reduced from 1 mM to 0.2 mM.
An Attempt to Initiate Catalysis by a Peroxide Shunt Pathway
HPLC analysis of reaction mixtures was conducted following steady-state experiments as above in which NADH and ToMOF were omitted and hydrogen peroxide was added as a reduced dioxygen source. Assay mixtures (0.5 mL) contained 15 μM ToMOH, 30 μM ToMOC, 40 μM ToMOD, 20 mM hydrogen peroxide, and 2 mM phenol in 0.1 M Tris/HCl buffer, pH 7.5. ToMOH was the last component added to each mixture. These solutions were stirred and allowed to react at 25 °C for specified times ranging from 30 s to 12 min. Samples were quenched with 100 μL of 0.4 M trichloroacetic acid, centrifuged, and loaded onto the HPLC column as described above. A series of experiments was also performed in which the concentrations of hydrogen peroxide produced were assayed as outlined above except that, prior to measuring the absorbance, the sample was diluted 15-fold with ddH2O. Solutions of hydrogen peroxide, which were standardized by measuring the absorbance at 240 nm, were used to calibrate HPLC peak integrals. Dioxygen release from reaction mixtures containing ToMOH and ToMOD was assayed by the alkaline pyrogallol method.23 Anaerobic assay mixtures for O2 generation contained 100 μM ToMOH and 400 μM ToMOD in 200 μL of 25 mM MOPS, pH 7.5, to which hydrogen peroxide (0.6 mL, 30% solution) was added. Control samples were also prepared in which the protein solution or hydrogen peroxide was substituted with buffer.
Hammett Studies
The steady-state activity of ToMOH using as substrates the following para-substituted phenols, cresol, phenol, fluorophenol, chlorophenol, and nitrophenol, were determined by directly quantifying the catechol products by an HPLC protocol described above. Assay mixtures comprising 0.3 μM ToMOH, 15 μM ToMOD, 2 μM ToMOC, 40 nM ToMOF, and 1 mM substrate in 0.1 M Tris/HCl, pH 7.5, were incubated at 25 °C. Steady-state turnover was initiated by addition of NADH to a final concentration of 1 mM. Aliquots (100 μL) were removed at 10-s intervals, quenched with TFA (10 μL), vortexed for 5 s, and centrifuged at 5000 rpm for 5 min to pellet the denatured protein. Each aliquot was then frozen in liquid nitrogen and stored at −78 °C. For p-chloro and p-nitrophenol, samples were taken at 30-s instead of 10-s intervals. The supernatant was analyzed by HPLC as mentioned previously. The absorbance was monitored at 280 nm for all substrates except for nitrophenol, the samples of which were monitored at 465 nm. Standard curves were generated for catechol products with the appropriate substituent in the 4-position. 4-Fluorocatechol was synthesized according to a literature procedure24 and characterized by 1H-NMR spectroscopy. The solid catechols were vacuum-sublimed and stored in the dark prior to use.
Results
Formation of a Diiron(III) Intermediate by Oxygenation of ToMOHred
Because the diiron(III) intermediate identified in the ToMOH I100W variant is diamagnetic and exhibits no apparent optical absorption bands,14,15 Mössbauer spectroscopy was used to seek spectroscopic evidence for the possible accumulation of the same diiron(III) intermediate in the reaction of reduced wild-type ToMOH with O2. The reaction was freeze-quenched at various reaction times (0.07 s, 0.14 s, 0.44 s, 2 s, and 32 s) by the RFQ method (Experimental Methods) and the resulting samples were characterized by Mössbauer spectroscopy. Figure 1 displays the 4.2-K Mössbauer spectrum of the diiron(II) ToMOH before reaction with O2 (A). Also presented are spectra of several freeze-quenched samples (B–D) that clearly reveal the sequence of events that occur during the reaction. By comparison with the spectrum of the reduced ToMOH reactant (A), the spectrum of a sample freeze-quenched at 0.14 s reaction time (B) shows ~ 45% reduction in intensity of the quadrupole doublet arising from the reduced, diiron(II) ToMOH and the appearance of a sharp quadrupole doublet (red line in B) with parameters (δ= 0.55 ± 0.02 mm/s, ΔEQ = 0.67 ± 0.03 mm/s, and line width = 0.27 mm/s) that are identical to those of the diiron(III) intermediate found in ToMOH I100W. A high-field experiment confirms that this doublet arises from a diamagnetic system (Figure S1). These observations establish that the diiron(II) sites in wild-type ToMOH indeed react rapidly with O2 to generate a diiron(III) intermediate that is the same as the one in ToMOH I100W. Accumulation of this intermediate reaches a maximum corresponding to 44% of the total Fe absorption and remains unchanged for several seconds (compare spectra B and C) before decaying directly to the diiron(III) product (orange line in D). No other intermediates were detected.
Figure 1.
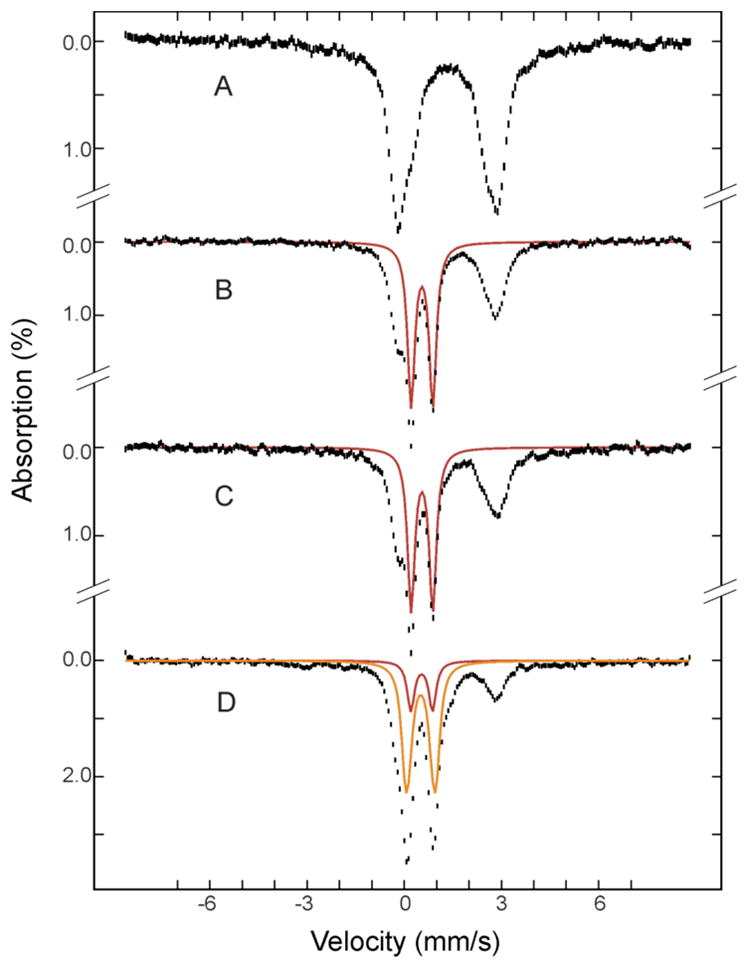
Mössbauer spectra of freeze-quenched samples from the reaction of reduced ToMOH:2ToMOD mixtures with O2. The samples were frozen before mixing (A) and 0.14 s (B), 2 s (C), and 32 s (D) after mixing. The spectra (vertical bars) are collected at 4.2 K in a 50 mT field applied parallel to the γ-ray beam. The red and orange lines are simulated spectra of the diiron(III) intermediate and diiron(III) product, respectively. The spectrum of the diiron(III) intermediate (red) was modeled with a single quadrupole doublet with δ = 0.55 mm/s, ΔEQ = 0.67 mm/s and line width = 0.27 mm/s. The spectrum of the diiron(III) product (orange) was simulated as a superposition of two equal intensity quadrupole doublets with parameters δ = 0.52 mm/s, ΔEQ = 0.73 mm/s and line width = 0.32 for doublet 1 and δ = 0.52 mm/s, ΔEQ = 0.97 mm/s and line width = 0.28 mm/s for doublet 2. They are plotted at the following absorption intensities: red, 44%, 41% and 13% in B, C, and D; orange, 41% in D. Unreacted diiron(II) protein accounts for ~ 56% of total Fe absorption at maximal accumulation of the intermediate (B). These parameters and intensities are the results of a global analysis of the entire set of the Mössbauer spectra including those that are not depicted here.
Kinetics of the Reaction of ToMOHred with O2
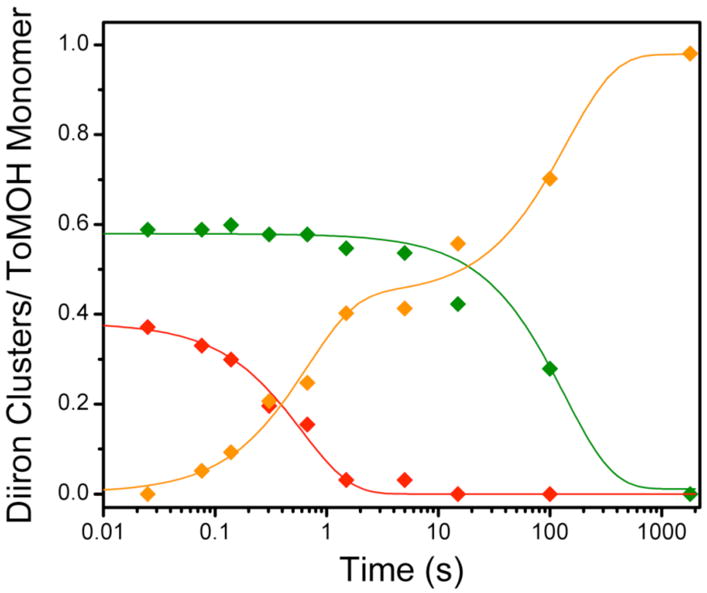
To obtain a quantitative estimate of the time evolution of Fe species generated during the oxidation of reduced ToMOH, it is necessary to deconvolute the Mössbauer spectra into various spectral components corresponding to the different Fe species. Although δ values for the diiron(III) intermediate (0.55 mm/s) and product (0.52 mm/s) are similar, different ΔEQ values for the intermediate (0.67 mm/s) and product (0.85 mm/s) allow the spectra of these two species to be readily deconvoluted (Figure 2). A global analysis of the entire set of the Mössbauer spectra, including those for time points not depicted, yields characteristic parameters reported above as well as percentages of Fe absorption for the species detected in the freeze-quenched samples. Based on the Fe and protein contents determined for the freeze-quenched samples, 3.5 Fe atoms/ToMOH dimer, these percentages can be converted into amounts of diiron clusters per ToMOH monomer. The results are presented in Figure 3 (diamonds). Analysis of these data indicate that the diiron(III) intermediate (red diamonds) forms rapidly with a rate constant of ~ 26 s−1 and accumulates to a maximum of 0.4 clusters/ToMOH monomer (or 0.8 clusters/dimer). It decays slowly at a rate of 0.045 s−1 to generate the diiron(III) product (orange diamonds and line). Data for the reduced ToMOH (green diamonds and line) reveal two populations for the diiron(II) sites that are distinguishable by their rates of reaction with O2. One population (0.4 cluster/ToMOH monomer) reacts rapidly (26 s−1) to form the diiron(III) intermediate, while the remaining diiron(II) sites react slowly at a rate of ~ 0.02 s−1.
Figure 2.
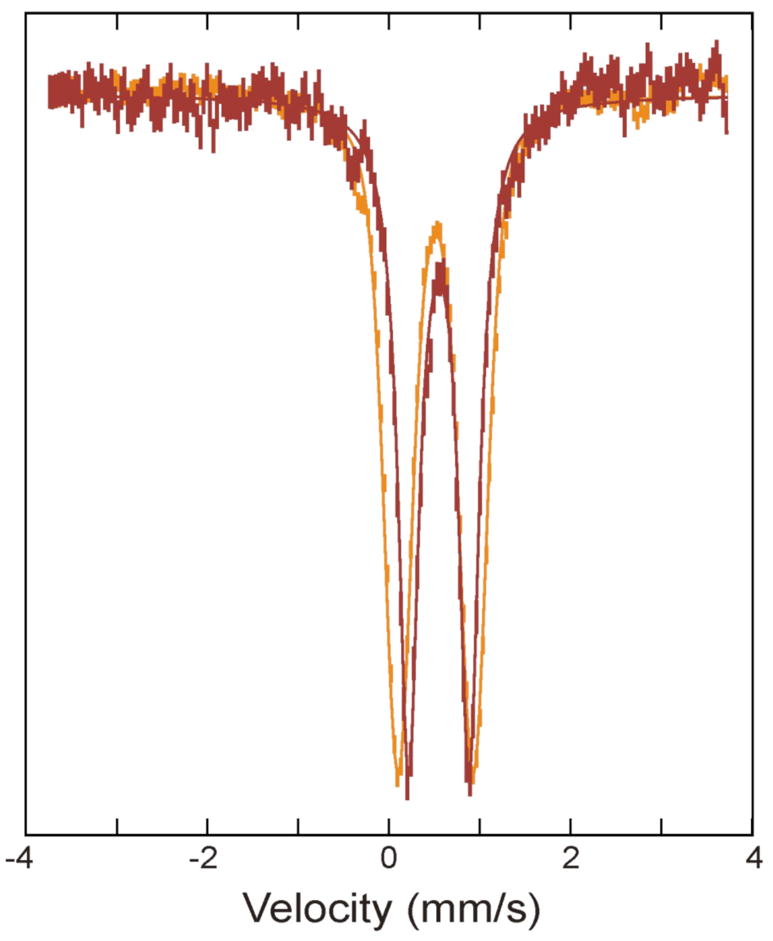
Overlay of Mössbauer spectra of the diiron(III) intermediate (red) and product (orange). The spectrum for the intermediate (red vertical bars) was prepared by removing the 56% diiron(II) contribution from the raw spectrum of the 0.14 s freeze-quenched sample (Figure 1B), and the product spectrum (orange vertical bars) was the spectrum of as-purified oxidized ToMOH. For comparison the two spectra are scaled to matching intensities. The solid lines are theoretical spectra simulated with parameters of the Fe species reported in the caption of Figure 1.
Figure 3.
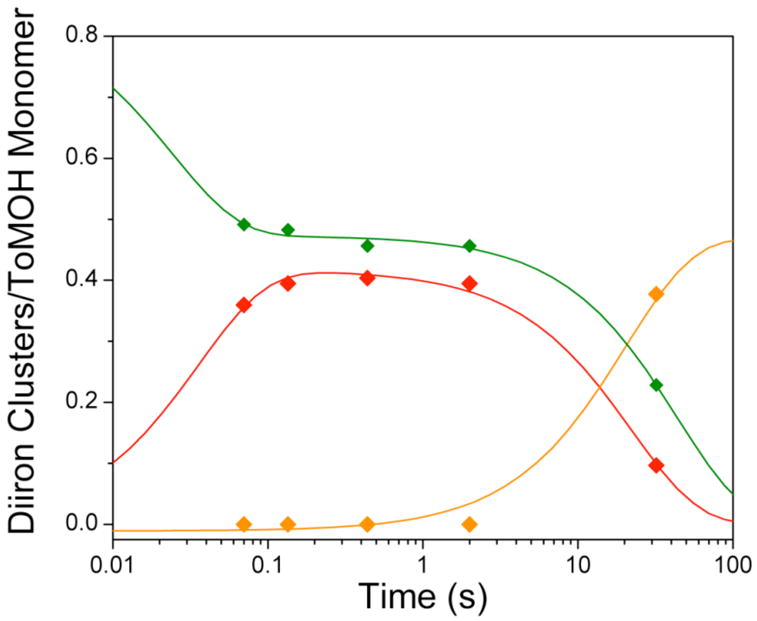
Speciation plot for reaction of diiron(II) ToMOH:2ToMOD with dioxygen. Reduced diiron(II) protein (
 ) reacts with dioxygen at ~ 26 s−1 to form a diiron(III) transient species (
) reacts with dioxygen at ~ 26 s−1 to form a diiron(III) transient species (
 ). Only ~45% of diiron(II) clusters give rise to this intermediate. The diiron(III) transient subsequently decays to the oxidized resting state (
). Only ~45% of diiron(II) clusters give rise to this intermediate. The diiron(III) transient subsequently decays to the oxidized resting state (
 ) at ~ 0.045 s−1. The unreacted diiron(II) centers decay slowly at 0.02 s−1 to generate several ill-defined ferric species, the production of which is not depicted. Solid lines are one- or two-exponential fits to the data.
) at ~ 0.045 s−1. The unreacted diiron(II) centers decay slowly at 0.02 s−1 to generate several ill-defined ferric species, the production of which is not depicted. Solid lines are one- or two-exponential fits to the data.
Effect of Phenol on Decay of the Diiron(III) Intermediate
The reactivity of the diiron(III) intermediate with phenol as a substrate was probed by using the double-mixing RFQ technique and Mössbauer spectroscopy. Reduced ToMOH was allowed to react with O2 for 0.17 s to afford a maximum accumulation of the diiron(III) intermediate prior to the introduction of the substrate phenol. The oxygenated protein mixture was then mixed with a buffer solution containing phenol. The reaction was allowed to proceed for various times and then quenched at −140 °C (Experimental Methods). Samples were collected for Mössbauer investigations. The resulting Mössbauer spectra for some of the time points are presented in Figure 4. As in the single-mixing experiments, the spectrum of the 0.17-s aged oxygenated protein shows that ~ 40% of the diiron(II) sites evolve to the diiron(III) intermediate (Figure 4, spectrum A, red line). After mixing with phenol, the quadrupole doublet corresponding to this species decreases its intensity almost immediately and becomes nearly undetectable at 5 s (Figure 4, B–D, red lines). This behavior is in stark contrast to the relatively stable accumulation of this species observed in the absence of phenol. The diiron(III) product is also detected almost immediately after mixing with phenol and grows into a dominant species at 5 s (Figure 4, B–D, orange lines). The unreacted diiron(II) protein, however, remains relatively stable (compare the absorption peak at 3 mm/s in spectra A to D) and starts to decay several seconds after mixing with phenol, behavior similar to that observed in the absence of phenol. As in the single mixing study, a global analysis to deconvolute the entire set of spectra into spectral components was performed and kinetic data for the Fe species generated after mixing with phenol were thereby obtained. The data are depicted in Figure 5 (diamonds) together with results of kinetic modeling of the formation and decay of the diiron species (color lines). The decay rate constant for the diiron(III) intermediate increases by more than 40-fold, from 0.045 s−1 to 2.0 s−1, in the presence of phenol. Because the amount of unreacted diiron(II) sites remains constant during the decay phase of this transient, the decay of the diiron(III) intermediate (Figure 5, red line) parallels the early formation of the diiron(III) product (Figure 5, orange line), both in rate constant (2.0 s−1) and in the extent of accumulation (0.4 clusters/ToMOH monomer). Decay of the unreacted diiron(II) species, with a rate constant of ~ 0.01 s−1, accounts for the second phase production of the diiron(III) product. No other intermediate was detected. Mössbauer parameters of the diiron(III) intermediate differ in the presence and absence of phenol (Figure 6); in the presence of phenol, this species was modeled by a quadrupole doublet with parameters (δ = 0.52 ± 0.02 mm/s and ΔEQ = 0.62 ± 0.03 mm/s) that are slightly different from those (δ = 0.55 mm/s and ΔEQ = 0.67 mm/s) observed in the absence of this substrate. Also, the absorption lines of this doublet are broader by 0.03 mm/s and 0.06 mm/s for the low-energy and high-energy line, respectively, in samples containing phenol (Figure 6). These differences, although minor, suggest that phenol perturbs the diiron center of the intermediate prior to reacting with it.
Figure 4.
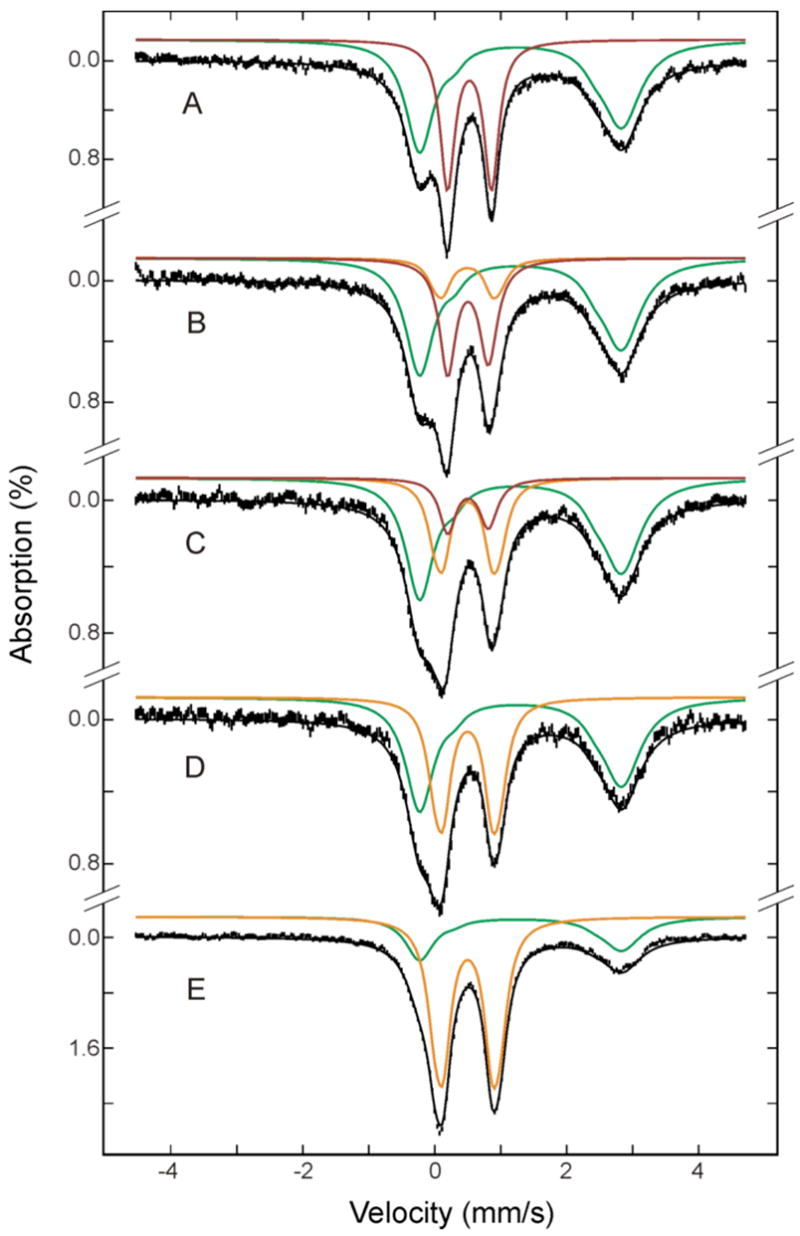
Mössbauer spectra of double-mixing RFQ samples for reaction of the diiron(III) intermediate with buffer containing phenol. Reduced protein was allowed to react with O2 for 0.17 s to generate the intermediate before mixing with phenol (A). After mixing with phenol, the reaction mixture was freeze-quenched at 0.14 s (B), 0.67 s (C), 5 s (D) and 100 s (E). The spectra (vertical bars) are collected at 4.2 K in a 50 mT applied field parallel to the γ-ray beam. The green, red and orange lines are simulated spectra, respectively, of the unreacted diiron(II) protein, diiron(III) intermediate and diiron(III) product. The spectra of the diiron(III) intermediate in the absence of phenol (red in A) and diiron(III) product (orange) were simulated using the parameters reported in the caption of Figure 1. The spectrum of the diiron(III) intermediate was perturbed slightly by the presence of phenol (see the following Figure 6) and was simulated with the altered parameters reported in text (red lines in B-E). The spectrum of the unreacted diiron(II) was modeled as a superposition of two quadrupole doublets with an intensity ratio of 10 to 1, and the following parameters: δ= 1.32 mm/s, ΔEQ = 3.06 mm/s for the intense doublet, and δ = 1.39 mm/s and ΔEQ = 2.13 mm/s for the minor doublet. The simulated spectra are plotted at the following absorption intensities: green, 61%, 60%, 58%, 57% and 29%; red, 39%, 30%, 14%, 0% and 0%; orange, 0%, 12%, 28%, 43% and 71% in A, B, C, D and E, respectively. The black lines overlaid with the spectra are composite spectra including all the Fe species mentioned above.
Figure 5.
Speciation plot for reaction of the diiron(III) intermediate with phenol. The intermediate (
) decays at 2 s−1 to give rise to the early formation phase of diiron(III) product (
). Diiron(II) clusters (
) that do not traverse the intermediate oxidize more slowly, with a rate constant of ~0.01 s−1 to product. Solid lines represent one- or two-exponential fits to the data.
Figure 6.
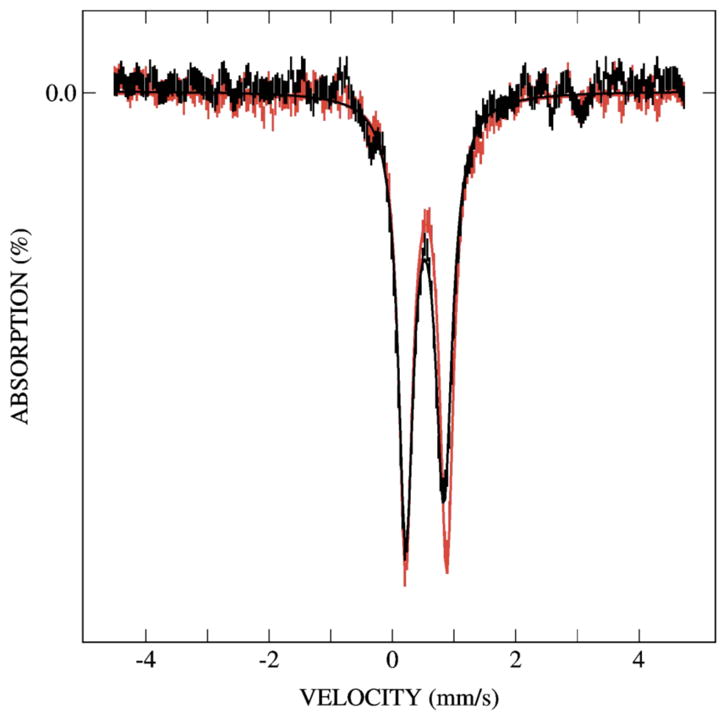
Overlay of Mössbauer spectra of the diiron(III) intermediate in the absence (red) and presence (black) of phenol. These spectra were prepared by removing the unreacted diiron(II) contributions from the spectrum shown in Figure 4A and spectrum of a sample freeze-quenched at 70 ms after mixing with phenol. Addition of phenol results in smaller values for δ and ΔEQ as compared to the intermediate generated in the absence of substrate (see text for parameters). Absorption lines also broaden in the presence of phenol by 0.03 and 0.06 mm/s, for low- and high-energy lines, respectively.
The acceleration of the decay of the diiron(III) transient by phenol provides strong evidence that this species is responsible for the substrate oxidation. Indeed, catechol was detected and determined by HPLC analyses to be the only observed aromatic product in the double-mixing samples at reaction times equal to and longer than 5 s. The concentration of this hydroxylation product in the samples was 145 ±1 μM, in good agreement with the estimated intermediate concentration, ~ 135 μM, accumulated before mixing with phenol. The intermediate concentration was estimated from the result of the Mössbauer analysis (40% Fe absorption for the intermediate before mixing with phenol) and protein and Fe contents determined for the double mixing samples (170 μM protein and 3.92 Fe/ToMOH dimer). A broad optical absorption band centered at 580 nm, consistent with that of an iron-catecholate charge transfer transition, was observed in optical spectra of samples prepared by double-mixing (Figure 7). Coordination of catechol to the diiron center, however, appears to cause only minor perturbations to the diiron site, since the Mössbauer spectrum of the final product, the oxidized protein (Figure 4E, orange line), could be simulated with quadrupole doublets identical to those used to model the Mössbauer absorption of the as-purified diiron(III) ToMOH (Figure 2, orange spectrum).
Figure 7.
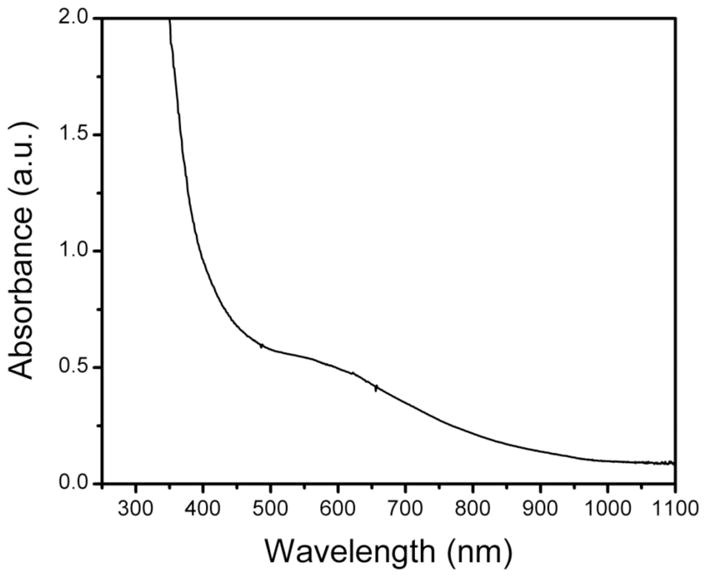
Optical spectrum of a sample following an RFQ double-mixing Mössbauer experiment. The broad absorption band is centered at ~ 580 nm.
Release of Hydrogen Peroxide by ToMO under Steady-State Conditions
Under steady-state conditions, hydrogen peroxide was evolved in the absence of phenol (Figure 8). Its rate of formation could be modeled with a hyperbolic function, suggesting that the enzyme system may be inactivated in the presence of hydrogen peroxide, as reported for phenol hydroxylase from Pseudomonas sp. CF600.25 The efficiency of the NADH oxidase reaction, defined as the ratio of the decrease in NADH concentration to the increase in hydrogen peroxide concentration, was 51(4)%. Addition of substrate to the reaction mixture retarded peroxide formation, implying that phenol reacts either with a peroxodiiron(III) center or a precursor of such a transient.
Figure 8.
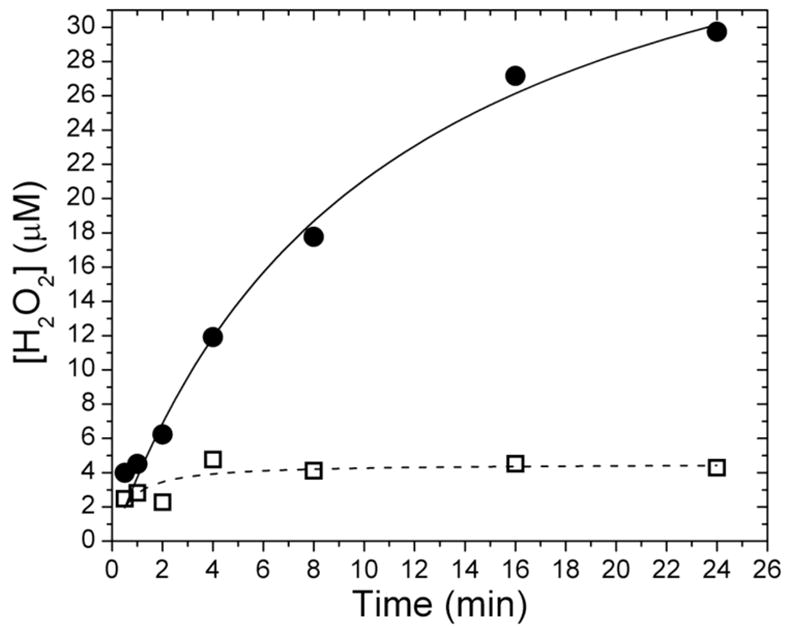
Concentration of hydrogen peroxide evolved under steady-state conditions with () and without (•) phenol. Hydrogen peroxide is formed slowly as NADH is consumed by the enzyme system. The addition of substrate inhibits peroxide formation. Data were fit with hyperbolic functions.
Peroxide Shunt Pathway and Catalase Activity of ToMOHox
Hydroxylation products were not detected in samples containing hydrogen peroxide, ToMOHox, ToMOD, and ToMOC. Inclusion of ToMOC in the reaction mixtures was to assure that protein component interactions would not affect our inability to access a peroxide shunt pathway. We noticed during the course of the experiments that hydrogen peroxide was rapidly consumed. Its concentration was monitored in acid-quenched samples by integrating the peak eluting at ~ 3.3 min in HPLC traces recorded at 240 nm (Figure 9) and by the colorimetric method used to assay for hydrogen peroxide release. Samples in which the hydroxylase was omitted showed no appreciable decrease in H2O2 concentration.
Figure 9.
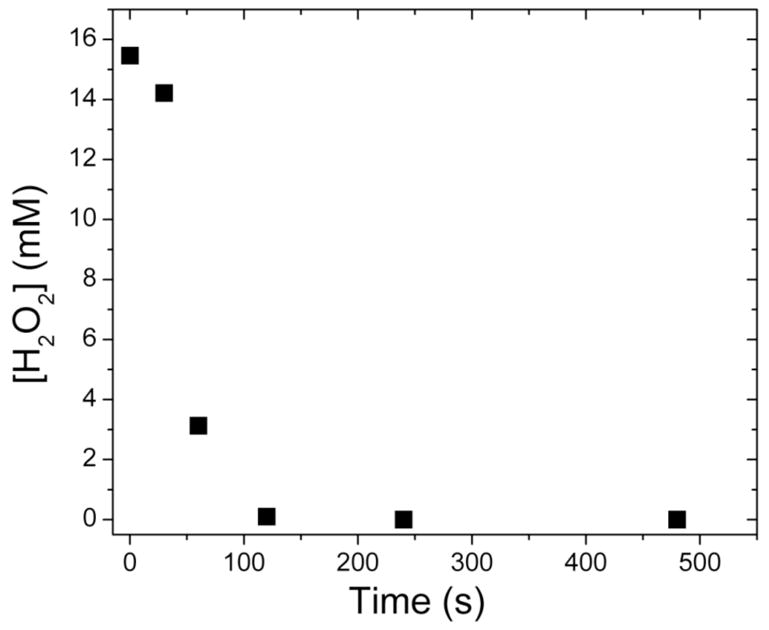
Change in the concentration of hydrogen peroxide with time after mixing with ToMOH–ToMOD mixtures. Hydrogen peroxide is rapidly consumed and evolved dioxygen in a manner independent of the presence of substrate. A peroxide shunt pathway was not accessed because catechol was not observed in assay solutions that contained phenol.
Experiments utilizing only ToMOHox and ToMOD revealed that dioxygen was rapidly evolved upon addition of aliquots of hydrogen peroxide. The evolved gas from these reaction mixtures oxidized the pale pink solution of alkaline pyrogallol to a brown color. Addition of hydrogen peroxide to vials containing buffer, or addition of buffer to protein solutions, did not result in oxidation of pyrogallol.
Hammett Relationship for Substrate Oxidation by ToMO
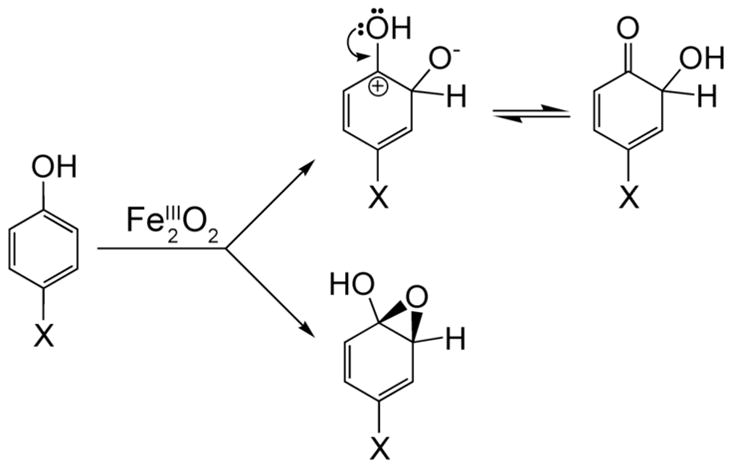
Initial reaction rates were used to generate Hammett plots for the oxidation of phenolic substrates by ToMO (Figure 10). Plots of the substituent constants σm and σp+ against the logarithm of the ratio of the initial rates for the substituted phenols and phenol were fit with a linear equation. Reaction constants, ρ, of -1.3 and -1.7 for σm and σp+ were determined for this system, consistent with electrophilic attack on the aromatic substrate. The rate data were plotted against two substituent constants because of the hydroxylation mechanism proposed for the homologous T4MO system. In that system, NIH shift experiments suggested that the substrate is attacked either at the site of oxidation (σm), to yield a substrate cation, or at the para and meta positions (σp+), to form a transient arene epoxide (Scheme 2). There is appreciable scatter in the plots, however, with R2 ~ 0.81 in both cases. Scaling the initial reaction velocities to account for the percentage of deprotonated substrate does not significantly improve the quality of the fit.
Figure 10.
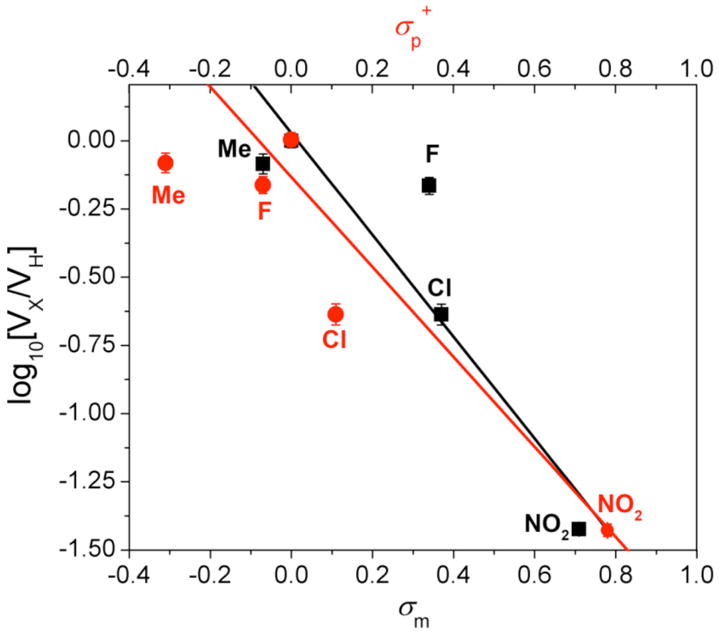
Hammett plots for oxidation of p-substituted phenols by ToMO. The values of ρ were −1.3 and −1.7 for σm (■) and σ+ (
 ) respectively. A negative reaction constant is indicative of electrophilic reactions. The goodness of fit as measured by R2 for either σ value is 0.81.
) respectively. A negative reaction constant is indicative of electrophilic reactions. The goodness of fit as measured by R2 for either σ value is 0.81.
Scheme 2.
Proposed Pathways for Substrate Hydroxylation
Discussion
Mechanism of Dioxygen Activation by ToMOH
The reduced diiron(II) forms of the CBDI enzymes are reactive toward dioxygen, activating the molecule to yield intermediates with Fe–O bonds that react with substrates.3,12,26–29 The first intermediate observed spectroscopically in the systems characterized thus far is a peroxodiiron(III) adduct, with well-defined spectroscopic parameters, including a near-IR optical absorption band.4 Despite the structural homology of the active sites of ToMOH and MMOH,10 the first intermediate observed upon oxygenation of ToMOHred is a diiron(III) transient for which no such absorption feature could be detected. The Mössbauer spectroscopic parameters, δ and ΔEQ, for this intermediate are 0.55 and 0.67 mm/s, respectively, which are both lower than those of peroxodiiron(III) centers in related enzymes and model complexes. Despite the spectroscopic differences, we tentatively assign the diiron(III) transient formed after oxygenation of reduced ToMOH as a peroxodiiron(III) center. This assignment is based on the following observations: (i) the reactivity of the transient is similar to that reported for peroxodiiron(III) intermediates in other systems and requires that the regulatory protein ToMOD be present in order to form; (ii) hydrogen peroxide is liberated during steady-state turnover in the absence of substrate; and (iii) by analogy to dioxygen activation in other CBDI systems, in which the diiron(III) intermediates are all peroxo species. The observed spectroscopic differences from peroxo intermediates in other systems may arise from a weak or red-shifted absorption band in the optical spectrum, photodecomposition, additional ligands coordinated to the diiron center, or an alternative geometry or protonation state of the peroxide moiety.
The {O22−} fragments in the peroxodiiron(III) intermediates in RNR-R2,30,31 Δ9D,12 and Ft32,33 are proposed to bind in a cis-μ-1,2- fashion and that in MMOH has alternatively been given the same assignment34–36 or that of a μ–η2, η2 side-on peroxide.5,28,37 In the former geometry, the out-of-plane and in-plane π* dioxygen orbitals interact with dyz and dz2 orbitals, respectively, on the iron atoms. The out-of-plane orbitals form π–bonds and the in-plane orbital gives rise to σ–bonds between peroxide and the dimetallic unit. In the case of iron(III), π-bonding dominates the interaction because the metal orbitals are half-filled.38 The characteristic low energy absorption feature centered at ~ 700 nm in μ-1,2-peroxodiiron(III) species arises from ππ* to dyz transitions, according to one formalism.38 The strength of this π–bonding interaction dictates therefore the energy and intensity of this transition as well as the O–O bond strength. In systems where high-valent oxygenated diiron species are observed, π–bonding is proposed to dominate the interaction between the {O22−} fragment and the diiron center because cleavage of the consequent weaker O–O is more facile. The extinction coefficients for the peroxodiiron(III) intermediate in MMOH, RNR-R2, Δ9D, and Ft follow the expected trend therefore, with those reported for this transient in RNR-R2 and MMOH being greater than those for the same species in Δ9D and Ft. If π-bonding in the ToMOH intermediate is weak, the energy and intensity of this charge transfer are predicted to decrease. The absorption maximum will therefore be red-shifted, with a smaller extinction coefficient than that of similar diiron(III) species. Weaker π-bonding would favor peroxide release, disfavor formation of high-valent intermediates, and reduce the electron density on the iron atoms. The Mössbauer parameters are indicators of the symmetry of the electric field gradient around (ΔEQ), and effective charge or oxidation state (δ) of, the iron atoms. Reducing the electron density therefore should lower δ relative to systems with stronger π-bonds. Longer Fe–O bonds may increase the symmetry, translating into smaller ΔEQ values. The Fe–O–O bond angle, which determines in part the Fe–Fe distance, strongly influences the π-bonding contribution from peroxide fragment, with smaller angles favoring σ-only bonding. If the absorption band is therefore weak and low in energy in ToMOHperoxo, we predict that the Fe–Fe distance in this intermediate will be shorter than that observed in peroxo-adducts of other CBDI systems.
Another possibility is that the peroxodiiron(III) species might decompose upon irradiation, as reported previously for a cis-μ-1,2-peroxodiiron(III) complex38 and for the superoxodiiron(III) intermediate in myo-inositol oxygenase.26 Reaction of reduced ToMOH I100W and ToMOD with dioxygen quantitatively converts the diiron(III) intermediate to the mixed-valent diiron(III,IV)–W• couple.14,15 The calculated extinction coefficient at 500 nm of the radical agrees with the accumulation of ToMOHperoxo, judging by Mössbauer spectroscopy. The photodecomposition rate of the diiron(III) transient, therefore, must be slower than ~ 1 s−1 to permit quantitative formation of the diiron(III,IV)–W• species. Given an estimated formation rate constant of 26 s−1 for this intermediate from Mössbauer studies, we predict that this transient should accumulate to adequate levels to permit detection of any optical features. We can therefore exclude photodecomposition as an explanation for the lack of an optical absorption band.
The nature, number, and protonation state of the bridging ligands in dioxygen adducts of CBDI model complexes influence the spectroscopic features of the characterized cis-μ-1,2-peroxodiiron(III) intermediates. Model compounds with oxo,39,40 hydroxo,40 or alkoxo17,18,41,42 bridges opposite the bound peroxide have absorption maxima, extinction coefficients, and Mössbauer parameters that cover a broad range of values (Table 1).16–18,40,43 A number of these compounds have values for δ within range of the present 0.52 mm/s value, although all the ΔEQ parameters are ~ 1.0 mm/s. Amino acid side chains in the active site pocket of ToMOH, such as E243, which shifts between the Mn(II)-reconstituted and diiron(III) forms,10,11 could be incorporated as additional oxygen atom bridges, thereby modulating the absorption and Mössbauer spectra of ToMOHperoxo. Any absorption band arising from this intermediate would either have to be obscured by end absorption from the protein optical band at 280 nm or lie in the near-IR region, beyond the range of our diode array instrument detector.
Table 1.
Mössbauer and Optical Spectroscopic Parameters for μ-1,2-Peroxodiiron(III) Complexes
| Donor Set | Bridging Ligands | λmax [nm] | ε[M−1cm−1] | δ[mm/s] | ΔEQ [mm/s] | Ref. |
|---|---|---|---|---|---|---|
| N3O3 | hydroxo | 644 | 3000 | 0.50 | 1.31 | 40 |
| N3O3a | oxo | 577 | 1500 | 0.50 | 1.46 | 40 |
| N3O3a | alkoxo
carboxylato |
665 | 2300 | 0.57 | 1.44 | 17 |
| N3O3 | alkoxo
carboxylato |
710 | 1500 | 0.58 | 1.17 | 17 |
| N3O3 | alkoxo
carboxylato |
500 – 800 | 1700 | 0.65
0.58 |
1.70
0.70 |
16 |
| N3O3 | dicarboxylato | 694 | 2650 | 0.66 | 1.40 | 18 |
| N4O2* | oxo | 494, 648, 846 | 1100, 1200, 230 | 0.54 | 1.68 | 43 |
These compounds were characterized by Mössbauer and optical spectroscopy only.
Finally, the geometry or protonation state of the peroxide fragment in ToMOHperoxo may differ from those of other μ-1,2-peroxodiiron(III) intermediates. Diiron(III) centers containing 1,1-peroxo-, 1,1-hydroperoxo-, or η1: η2-peroxo bridges have all been proposed for transition states linking the reduced diiron(II) forms to diiron(III) and high-valent intermediates in MMOH and RNR-R2.5,37,38,44 If the intermediate is protonated, such proton transfers could not be rate-determining within the limits of RFQ methods because we find no solvent kinetic isotope effect for either its formation or decay in single-mixing Mössbauer studies.45 Hydroperoxy intermediates in cytochromes P450 and heme iron synthetic complexes are proposed to be electrophilic oxidants, reacting with electron-rich substrates.46 MMOHperoxo and the peroxo intermediate in RNR-R2 are also capable of oxidizing electron-rich substrates, such as aromatic side chain residues,27,47 alkenes,29 and ethers.28 The reactivity profile of the intermediate reported here is similar to those of hydroperoxoiron(III) species in heme systems, MMOHperoxo, and the peroxo intermediate in RNR-R2. ToMOH can epoxidize or hydroxylate halogenated alkenes and a number of aromatic compounds, preferring electron-rich substrates over poorer ones.48,49 A preliminary cryoreduction study of the ToMOH intermediate gives rise to an EPR-active species, which will allow us to probe its structure by 17O-ENDOR spectroscopy in future work.50
ToMOH is unique among BMMs studied to date because it can serve as a catalase in the absence of reducing equivalents and as an NADH oxidase in conjunction with the other components of the enzyme system. The catalase activity of ToMOHox suggests that dioxygen activation by ToMOHred may be reversible, if protons are lost prior to ET. Dioxygen binds reversibly to diiron(II) clusters in respiratory proteins, such as hemerythrin, but has not been reported for CBDI enzymes that catalyze substrate oxidation. RFQ Mössbauer spectra of samples from reaction of dioxygen with ToMOHred and ToMOD demonstrate that ToMOHperoxo decays to form only the oxidized diiron(III) center. The catalase reaction was conducted with more than a 20-fold excess of hydrogen peroxide, which could drive the reaction in the reverse direction. Hydrogen peroxide could react with ToMOHox, liberating two water molecules and generating ToMOHperoxo (Scheme 3). Oxidation of the peroxide fragment by the diiron(III) center evolves dioxygen with formation of ToMOHred. Binding of a second molecule of hydrogen peroxide, possibly in a μ-1,1 fashion, to the diiron(II) center would allow for protonation of the distal oxygen atom followed by cleavage of the O–O bond. This binding mode for a peroxide fragment to a diiron(II) center has been previously proposed during reaction of synthetic peroxodiiron(III) complexes with the diiron(II) starting compound.42 The oxo-bridged diiron(III) center formed after O–O bond cleavage can react with water to form ToMOHox or with hydrogen peroxide to generate ToMOHperoxo.
Scheme 3.
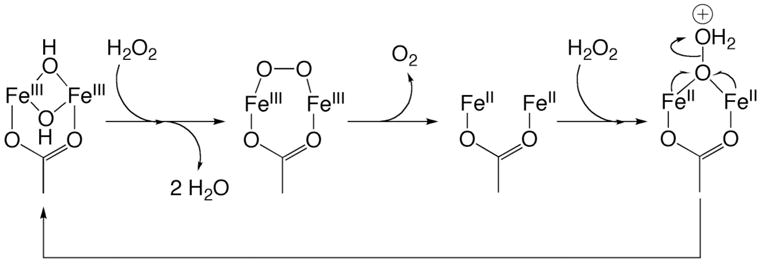
Reaction of ToMOHox with Hydrogen Peroxide
The ToMO enzyme system catalyses the two-electron reduction of dioxygen to hydrogen peroxide with an efficiency of 51%. Reductive quenching of the diiron(III) intermediate in the absence of substrate could explain this poor efficiency. Electron transfer from reduced ToMOC to ToMOHperoxo prior to dissociation of peroxide may allow for complete reduction of dioxygen to water, as occurs in MMOH.19 The combined rate for dissociation of ToMOD, and binding and ET from reduced ToMOC to ToMOHperoxo, would have to exceed the decay rate of the transient, for which the rate constant is 0.05 s−1. The catalytic rate of phenol hydroxylation is 20-fold greater than the decay rate of ToMOHperoxo.51 The rate of protein complex formation and ET is faster therefore than that of ToMOHperoxo decay, suggesting that reductive quenching of ToMOHperoxo is kinetically feasible. The reduction of ToMOHperoxo could give rise to a peroxodiiron(II) species, similar to that proposed in Scheme 3.
Substrate Hydroxylation by ToMOH
The rate constant for decay of the ToMOH diiron(III) intermediate is sensitive to phenol, increasing by more than thirty-fold, from 0.05 s−1 to 1.7 s−1, in its excess. The reaction is quantitative and forms catechol. The Mössbauer spectrum of ToMOHperoxo differs with phenol present, in contrast to that of the diiron(III) end product in double-mixing samples, which can be modeled with parameters identical to those in the absence of catechol (Figures 2 and 4). Presumably, phenol accesses the CBDI active site via the channel and binds to the diiron center before undergoing oxidation. Coordination of phenol to a CBDI center in this manner would direct hydroxylation at the ortho position. The high regioselectivity of phenol hydroxylation by ToMO parallels that of model complexes in which the substrates are tethered to ligands bound to iron and where oxidization occurs regiospecifically at the closest accessible position.18,52,53
Substrate coordination might potentiate reaction pathways that are otherwise unavailable in its absence. The choice of phenol as substrate for our experiments was determined because of the weak regioselectivity exhibited by the enzyme system for toluene. ToMO oxidizes phenol exclusively to catechol, but oxidation of toluene yields a mixture of cresols.51 To ensure that the Mössbauer spectrum of the diiron(III) product could be readily distinguished from that of the diiron(III) intermediate, phenol was therefore chosen as substrate.
A possible detailed mechanism for phenol hydroxylation is shown in Scheme 4. Dioxygen activation involves formation a peroxodiiron(III) intermediate, which we arbitrarily depict with a μ-1,2- geometry. Phenol next coordinates to one iron(III) center atom, after which there is an electrophilic attack of the {O22−} fragment on the arene π-system. The resulting arene epoxide ring opens, and rearomatization leads to oxidized diiron(III) center with bound product. In the absence of substrate, the intermediate slowly liberates hydrogen peroxide to return the resting diiron(III) state, ToMOHox. For reaction with toluene, a similar mechanism is expected but with substrate orientation being affected by residues within the active site pocket rather than by coordination to the diiron center. No EPR active species were observed in samples from the double-mixing studies. We previously reported the reaction of the diiron(III) intermediate to produce a neutral tryptophanyl radical centered at the mutagenic site (I100W).14,15 Oxidation of phenol to catechol did not produce a similar substrate-based radical with an appreciable lifetime. Substrate proximity to the diiron center may direct the reaction through radical pathways at longer distances and two-electron processes at shorter distances.
Scheme 4.
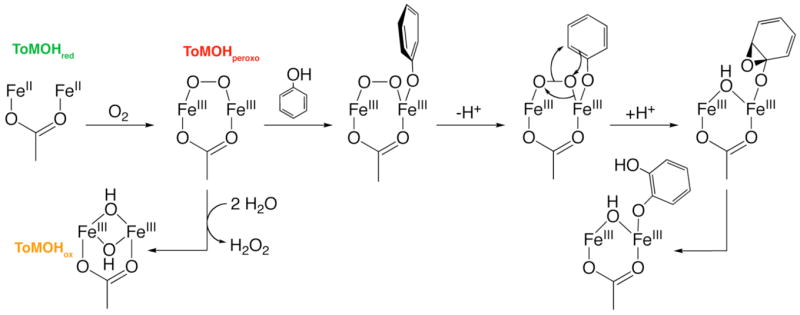
Substrate Hydroxylation and Dioxygen Activation by ToMOH
Linear free energy relationships for steady-state hydroxylation of substrate by ToMO have negative slopes (Fig. 10), indicative of electrophilic attack on the aryl ring with substrate oxidation being at least partially rate-limiting. Activation of an aromatic substrate by donation from a hydroxyl group predisposes the system to electrophilic attack. As such, hydroxylation could proceed by a mechanism different from that that used for less activated arenes, such as toluene and benzene. Hammett studies of the homologous T4MO enzyme system employing substituted benzenes as substrates, however, are consistent with an electrophilic attack on the π-ring.54 The results of NIH shift experiments using T4MO suggested that hydroxylation involves formation of a transient arene epoxide that opens reversibly, or attack at a specific carbon atom, to yield a substrate carbocation.55 The intermediate in ToMOH is therefore electrophilic, demonstrating a preference for two-electron oxidation of electron-rich substrates, such as alkenes and arenes, or one-electron abstraction from a nearby redox active residue, as occurred in the I100W variant.
Slow binding of substrate in the active site pocket will retard the steady-state hydroxylation rate and bring about deviations from a linear Hammett relationship. Mutation of two glutamate residues, E214 and E284, on the protein surface near the channel opening and far from the active site enhance the rate of steady-state hydroxylation of 4-nitrophenol.49 More than 60% of this substrate is deprotonated at pH 7.5. Removal of negative charge at the opening of the substrate access channel could eliminate repulsion with deprotonated carboxylate side chains of E214 and E284, increasing the ease of substrate access to the active site. Correcting the observed hydroxylation rate of substrates in the Hammett study for the fraction of substrate that is protonated at pH 7.5 did not improve the quality of the fit. Other factors, such as the energy cost in bringing a charged species into a buried, neutral active site pocket in ToMOH, may also hinder access and affect the oxidation of these electron-poor aromatic compounds.
A portion of the product remains bound to the diiron center, as evidenced by the appearance of a broad absorption band at 580 nm. Phenol, the substrate used here, is also a reaction product of benzene hydroxylation by ToMOH. Phenol, like catechol, can also bind to the diiron center and does so, as evidenced by the growth of an absorption band centered at 495 nm during titration of ToMOHox with phenol (data not shown). A similar absorption band was reported in a mutant of RNR-R2 in which F208 is hydroxylated and binds to the diiron center.27 Product release in this pre-steady-state system may be slow because the installed hydroxyl group allows coordination to the diiron center. Dissociation and egress of products is proposed to be the rate-determining step in steady-state hydroxylation by MMOH.56 Coordination of products to the diiron center after hydroxylation, and the effects of these events on the reaction rate, are probably common to the hydroxylation mechanisms of BMMs. In ToMOH, a linear Hammett relationship provides strong evidence that substrate hydroxylation limits the reaction rate, implying that product dissociation is at least partially rate-limiting.
Quantitation of catechol formed in double-mixing samples was possible only after treatment of the diiron(III) protein with ascorbic acid. Under steady-state conditions, reduction of product-bound ToMOHox facilitates its release to restart a catalytic cycle. Reduction-facilitated product release has been proposed previously for MMOH.19,57 Such a mechanism, whereby product release is enhanced by reduction of the diiron center would require that diffusion of product out of the active site occur more rapidly than subsequent steps involving dioxygen activation, hydrocarbon diffusion into the active site, and hydroxylation. Previously steady-state oxidation of benzene by ToMO results in the initial accumulation of phenol, and phenol is only hydroxylated to catechol at low benzene concentrations.51 The accumulation of phenol is therefore evidence that the rate of product dissociation exceeds that of reduction of the oxidized hydroxylase and subsequent dioxygen activation.
ToMOD enhanced binding of phenol to the diiron center by a factor of two, as measured by optical titration of the oxidized hydroxylase with phenol.45 This result suggests that hydroxylase-regulatory protein interactions perturb the diiron center, possibly by reorienting residues to increase the active site volume and allow substrate access. The flexibility of the active site to facilitate changes in the volume of the substrate-binding cavity was reported previously for other BMMs. For example, the volume is increased by partial unwinding of helix E in MMOHox upon binding of 6-bromohexanol.58 Analogous helical rearrangements were also noted in the crystal structure of the PHH–PHM complex.59 The regulatory protein in these systems may therefore effect structural changes to allow for dioxygen activation and substrate access to, and/or product egress from, the diiron center.
Implications for the CBDI Protein Family
Half-sites reactivity, whereby two equivalent active sites alternate in traversing opposite halves of the catalytic cycle, has been proposed in RNR-R2,60 Δ9D,12 and MMOH.19 The maximum observed accumulation of the peroxodiiron(III) intermediate in ToMOH is consistently recorded to be ~ 45%, with the remaining protein present as unreactive diiron(II) starting material. Such reproducible yields of the intermediate generated upon oxygenation of reduced wild type and mutant I100W14,15 hydroxylases suggests that dioxygen activation in this system only occurs at one active site per dimer. This conserved feature of CBDI family members may reflect allosteric changes transmitted through the protein framework as amino acid side chains shift with the transition from the reduced enzyme to intermediate and oxidized forms.
The protein framework can modulate the mechanism of dioxygen activation and the reactivity of those intermediates, as evidenced by the ability of MMOH and RNR-R2 to form high-valent species, of Δ9D and Ft to form only peroxo-bridged intermediates, and of ToMOH to form a putative peroxodiiron(III) transient with different spectroscopic characteristics. DFT methods have proposed that compression of the Fe–Fe vector by the protein matrix aids in the formation of Q.5 Subtle changes afforded by the orientation of subunits or complex formation with other components may be responsible for tuning the different reactivities of BMMs.
Conclusions
The reactive intermediate in ToMOH that hydroxylates arenes is a diiron(III) species, which we tentatively assign as a peroxo adduct. This intermediate is an electrophile, similar to μ-1,2-peroxodiiron(III) transients in RNR-R2 and MMOH. ToMOH is distinct, however, because it generates hydrogen peroxide during steady-state turnover in the absence of substrate and acts as a catalase, consuming hydrogen peroxide to liberate dioxygen. Our results suggest that the dioxygen activation step(s) may be reversible in this system, which has not been previously reported for any BMM. The Mössbauer parameters and optical characteristics of ToMOHperoxo differentiate it from other μ-1,2- peroxodiiron(III) centers in biology. Further characterization of this intermediate by ENDOR, XAS, and X-ray crystallography will provide invaluable insight into factors that govern dioxygen activation at CBDI centers in biology.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants GM32134 (to SJL) and GM 47295 (to BHH). LJM was supported in part by a fellowship from the Martin Society. SUPPORTING INFORMATION. Field-dependent Mössbauer spectra of the diiron(III) intermediate. This material is available free of charge via the Internet at http://pubs.acs.org.
ABBREVIATIONS
- CBDI
carboxylate-bridged diiron
- Δ 9D
stearoyl-ACP Δ9D desaturase
- Ft
ferritin
- MMO
methane monooxygenase enzyme system
- MMOH
hydroxylase component of MMO
- MMOR
NADH oxidoreductase component of MMO
- MMOB
regulatory component of MMOH
- RFQ
rapid-freeze quench
- RNR-R2
Class I ribonucleotide reductase R2-subunit
- rR
resonance Raman
- ToMO
toluene/o-xylene monooxygenase system
- ToMOH
hydroxylase component of ToMO
- ToMOD
regulatory component of ToMO
- ToMOC
Rieske protein component of ToMO
- ToMOF
NADH oxidoreductase component of ToMO
References
- 1.Nam W. Acc Chem Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]
- 2.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 3.Kurtz DJ., Jr J Biol Inorg Chem. 1997;2:159–167. [Google Scholar]
- 4.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew Chem, Int Ed. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 5.Rinaldo D, Philipp DM, Lippard SJ, Friesner RA. J Am Chem Soc. 2007;129:3135–3147. doi: 10.1021/ja0654074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SK, Lipscomb JD. Biochemistry. 1999;38:4423–4432. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- 7.Sturgeon BE, Burdi D, Chen S, Huynh BH, Edmondson DE, Stubbe J, Hoffman BM. J Am Chem Soc. 1996;118:7551–7557. [Google Scholar]
- 8.Lindqvist Y, Huang W, Schneider G, Shanklin J. EMBO J. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- 9.Jin S, Kurtz DJ, Jr, Liu Z-J, Rose J, Wang B-C. J Am Chem Soc. 2002;124:9845–9855. doi: 10.1021/ja026587u. [DOI] [PubMed] [Google Scholar]
- 10.Sazinsky MH, Bard J, Di Donato A, Lippard SJ. J Biol Chem. 2004;279:30600–30610. doi: 10.1074/jbc.M400710200. [DOI] [PubMed] [Google Scholar]
- 11.McCormick MS, Sazinsky MH, Condon KL, Lippard SJ. J Am Chem Soc. 2006;128:15108–15110. doi: 10.1021/ja064837r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664–14671. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- 13.Broadwater JA, Achim C, Münck E, Fox BG. Biochemistry. 1999;38:12197–12204. doi: 10.1021/bi9914199. [DOI] [PubMed] [Google Scholar]
- 14.Murray LJ, García-Serres R, Naik S, Huynh BH, Lippard SJ. J Am Chem Soc. 2006;128:7458–7459. doi: 10.1021/ja062762l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray LJ, García-Serres R, Davydov R, McCormick MS, Naik S, Hoffman BM, Huynh BH, Lippard SJ. manuscript in preparation. [Google Scholar]
- 16.Kim K, Lippard SJ. J Am Chem Soc. 1996;118:4914–4915. [Google Scholar]
- 17.Ookubo T, Sugimoto H, Nagayama T, Masuda H, Sato T, Tanaka K, Maeda Y, Okawa H, Hayashi Y, Uehara A, Suzuki M. J Am Chem Soc. 1996;118:701–702. [Google Scholar]
- 18.Yamashita M, Furutachi H, Tosha T, Fujinami S, Saito W, Maeda Y, Takahashi K, Tanaka K, Kitagawa T, Suzuki M. J Am Chem Soc. 2007;129:2–3. doi: 10.1021/ja063987z. [DOI] [PubMed] [Google Scholar]
- 19.Gassner GT, Lippard SJ. Biochemistry. 1999;38:12768–12785. doi: 10.1021/bi990841m. [DOI] [PubMed] [Google Scholar]
- 20.Ravi N, Bollinger JM, Jr, Huynh BH, Edmondson DE, Stubbe J. J Am Chem Soc. 1994;116:8007–8014. [Google Scholar]
- 21.Hildebrandt AG, Roots I. Arch Biochem Biophys. 1975;171:385–397. doi: 10.1016/0003-9861(75)90047-8. method b. [DOI] [PubMed] [Google Scholar]
- 22.Keller RJ, Halmes NC, Hinson JA, Pumford NR. Chem Res Toxicol. 1993;6:430–433. doi: 10.1021/tx00034a007. [DOI] [PubMed] [Google Scholar]
- 23.Chufán EE, Verani CN, Puiu SC, Rentschler E, Schatzschneider U, Incarvito C, Rheingold AL, Karlin KD. Inorg Chem. 2007;46:3017–3026. doi: 10.1021/ic061686k. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 24.Corse J, Ingraham LL. J Org Chem. 1951;16:1345–1348. [Google Scholar]
- 25.Cadieux E, Vrajmasu V, Achim C, Powlowski J, Münck E. Biochemistry. 2002;41:10680–10691. doi: 10.1021/bi025901u. [DOI] [PubMed] [Google Scholar]
- 26.Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr Proc Natl Acad Sci USA. 2006;103:6130–6135. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baldwin J, Voegtli WC, Khidekel N, Moënne-Loccoz P, Krebs C, Pereira AS, Ley BA, Huynh BH, Loehr TM, Riggs-Gelasco PJ, Rosenzweig AC, Bollinger JM., Jr J Am Chem Soc. 2001;123:7017–7030. doi: 10.1021/ja002114g. [DOI] [PubMed] [Google Scholar]
- 28.Beauvais LG, Lippard SJ. J Am Chem Soc. 2005;127:7370–7378. doi: 10.1021/ja050865i. [DOI] [PubMed] [Google Scholar]
- 29.Valentine AM, Stahl SS, Lippard SJ. J Am Chem Soc. 1999;121:3876–3887. [Google Scholar]
- 30.Moënne-Loccoz P, Baldwin J, Ley BA, Loehr TM, Bollinger JM., Jr Biochemistry. 1998;37:14659–14663. doi: 10.1021/bi981838q. [DOI] [PubMed] [Google Scholar]
- 31.Yun D, García-Serres R, Chicalese BM, An YH, Huynh BH, Bollinger JM., Jr Biochemistry. 2007;46:1925–1932. doi: 10.1021/bi061717n. [DOI] [PubMed] [Google Scholar]
- 32.Hwang J, Krebs C, Huynh BH, Edmondson DE, Theil EC, Penner-Hahn JE. Science. 2000;287:122–125. doi: 10.1126/science.287.5450.122. [DOI] [PubMed] [Google Scholar]
- 33.Moënne-Loccoz P, Krebs C, Herlihy K, Edmondson DE, Theil EC, Huynh BH, Loehr TM. Biochemistry. 1999;38:5290–5295. doi: 10.1021/bi990095l. [DOI] [PubMed] [Google Scholar]
- 34.Que L, Jr, Dong Y. Acc Chem Res. 1996;29:190–196. [Google Scholar]
- 35.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625–2658. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 36.Valentine AM, Lippard SJ. J Chem Soc, Dalton Trans. 1997;1997:3925–3931. [Google Scholar]
- 37.Gherman BF, Baik MH, Lippard SJ, Friesner RA. J Am Chem Soc. 2004;126:2978–2990. doi: 10.1021/ja036506+. [DOI] [PubMed] [Google Scholar]
- 38.Brunold TC, Tamura N, Kitajima N, Moro-oka Y, Solomon EI. J Am Chem Soc. 1998;120:5674–5690. [Google Scholar]
- 39.Shan X, Que L., Jr Proc Natl Acad Sci USA. 2005;102:5340–5345. doi: 10.1073/pnas.0409640102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Furutachi H, Fujinami S, Nagatomo S, Maeda Y, Watanabe Y, Kitagawa T, Suzuki M. J Am Chem Soc. 2005;127:826–827. doi: 10.1021/ja045594a. [DOI] [PubMed] [Google Scholar]
- 41.Feig AL, Lippard SJ. J Am Chem Soc. 1994;116:8410–8411. [Google Scholar]
- 42.Feig AL, Becker M, Schindler S, van Eldik R, Lippard SJ. Inorg Chem. 1996;35:2590–2601. doi: 10.1021/ic951242g. [DOI] [PubMed] [Google Scholar]
- 43.Dong Y, Zang Y, Shu L, Wilkinson EC, Que L., Jr J Am Chem Soc. 1997;119:12683–12684. [Google Scholar]
- 44.Bollinger JM, Jr, Tong WH, Ravi N, Huynh BH, Edmondson DE, Stubbe J. J Am Chem Soc. 1994;116:8015–8023. [Google Scholar]
- 45.Murray LJ, Naik S, Ortillo DO, Huynh BH, Lippard SJ. unpublished results. [Google Scholar]
- 46.Nam W, Ryu YO, Song WJ. J Biol Inorg Chem. 2004;9:654–660. doi: 10.1007/s00775-004-0577-5. [DOI] [PubMed] [Google Scholar]
- 47.Ormö M, deMaré F, Regnström K, Åberg A, Sahlin M, Ling J, Loehr TM, Sanders-Loehr J, Sjöberg BM. J Biol Chem. 1992;267:8711–8714. [PubMed] [Google Scholar]
- 48.Ryoo D, Shim H, Canada K, Barbieri P, Wood TK. Nat Biotechnol. 2000;18:775–778. doi: 10.1038/77344. [DOI] [PubMed] [Google Scholar]
- 49.Vardar G, Ryu K, Wood TK. J Biotechnol. 2005;115:145–156. doi: 10.1016/j.jbiotec.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 50.Murray LJ, Davydov R, Naik S, Huynh BH, Hoffman BM, Lippard SJ. unpublished results. [Google Scholar]
- 51.Cafaro V, Izzo V, Scognamiglio R, Notomista E, Capasso P, Casbarra A, Pucci P, Di Donato A. Appl Environ Microbiol. 2004;70:2211–2219. doi: 10.1128/AEM.70.4.2211-2219.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carson EC, Lippard SJ. Inorg Chem. 2006;45:828–836. doi: 10.1021/ic051471v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carson EC, Lippard SJ. Inorg Chem. 2006;45:837–848. doi: 10.1021/ic051476s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mitchell KH, Studts JM, Fox BG. Biochemistry. 2002;41:3176–3188. doi: 10.1021/bi012036p. [DOI] [PubMed] [Google Scholar]
- 55.Mitchell KH, Rogge CE, Gierahn T, Fox BG. Proc Natl Acad Sci USA. 2003;100:3784–3789. doi: 10.1073/pnas.0636619100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lipscomb JD. Annu Rev Microbiol. 1994;48:371–399. doi: 10.1146/annurev.mi.48.100194.002103. [DOI] [PubMed] [Google Scholar]
- 57.Lee SK, Nesheim JC, Lipscomb JD. J Biol Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- 58.Sazinsky MH, Lippard SJ. J Am Chem Soc. 2005;127:5814–5825. doi: 10.1021/ja044099b. [DOI] [PubMed] [Google Scholar]
- 59.Sazinsky MH, Dunten PW, McCormick MS, Di Donato A, Lippard SJ. Biochemistry. 2006;45:15392–15404. doi: 10.1021/bi0618969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sjöberg BM, Karlsson M, Jörnvall H. J Biol Chem. 1987;262:9736–9743. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.