

Abstract
Short practical syntheses for five deuterium labeled derivatives of dimethylallyl diphosphate (DMAPP) useful for enzymological studies are reported. These include the preparation of the C1-labeled derivatives (R)-[1-2H]3-methylbut-2-enyl diphosphate ((R)-[1-2H]1-OPP) and (S)-[1-2H]3-methylbut-2-enyl diphosphate ((S)-[1-2H]1-OPP), the C2-labeled derivative [2-2H]3-methylbut-2-enyl diphosphate ([2-2H]1-OPP), and the methyl-labeled derivatives (E)-[4,4,4-2H3]3-methylbut-2-enyl diphosphate ((E)-[4,4,4-2H3]1-OPP) and (Z)-[4,4,4-2H3]3-methyl-but-2-enyl diphosphate ((Z)-[4,4,4-2H3]1-OPP).
Isoprenoid compounds constitute a large diverse class of “small molecule” natural products with over 35,000 individual known metabolites. The vast majority of these compounds are ultimately derived from two fundamental isoprenoid building blocks, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP, 1-OPP). DMAPP is the electrophilic primer required to initiate the 1’-4 (head-to-tail) chain elongation reactions for the biosynthesis of polyisoprenoid compounds,1 which are generated by successive alkylations of IPP by a growing allylic diphosphate chain.2,3 DMAPP is also the substrate for biosynthesis of monoterpenes with non-head-to-tail skeletons4 and a variety of metabolites where the dimethylallyl moiety is attached to non-isoprenoid fragments.1
The prenyltransfer enzymes that catalyze electrophilic alkylation reactions with DMAPP are typically highly stereoselective. Because many enzymes do not tolerate substitutions that substantially increase their steric bulk of the substrates, the stereochemistries of these reactions are best studied with isotopically labeled derivatives of the normal substrates. We now report short practical routes for the stereoselective synthesis of deuterated derivatives of DMAPP, which collectively place label at each of the carbons bearing hydrogen atoms.
The syntheses of (R)-[1-2H]3-methyl-2-butenyl diphosphate ((R)-[1-2H]1-OPP) and (S)-[1-2H]3-methyl-2-butenyl diphosphate ((S)-[1-2H]1-OPP) are outlined in Scheme 1. [1-2H]-3-Methyl-2-butenal ([1-2H]3)5 was reduced with BITIP catalysts derived from (S)- and (R)-BINOL and Bu3SnH6 to give (R)-[1-2H]1-OH and (S)-[1-2H]1-OPP, respectively. The 1H NMR spectra of (R)- and (S)-[1-2H]1-OH were similar to that reported for the S enantiomer obtained by a fermenting yeast reduction of [1-2H]3.7 A portion of each alcohol was converted to the corresponding Mosher ester.8,9 The C1 protons in the 1H NMR spectra of the diastereomeric esters from (R)- and (S)-[1-2H]1-OH and (R)-(-)-Mosher’s chloride were cleanly resolved, giving peaks at 4.77 and 4.82 ppm, respectively. A comparison of peak intensities indicated that the enantiomeric ratios for (R)- and (S)-[1-2H]1-OH were 96/4 and 94.5/5.5, respectively. Diphosphates (R)- and (S)-[1-2H]1-OPP were synthesized from corresponding chiral alcohols by treatment with trichloroacetonitrile and inorganic phosphate,10 a procedure that does not alter the absolute stereochemistry of C1.
Scheme 1. Synthesis of (R)- and (S)-[1-2H]1-OPP.
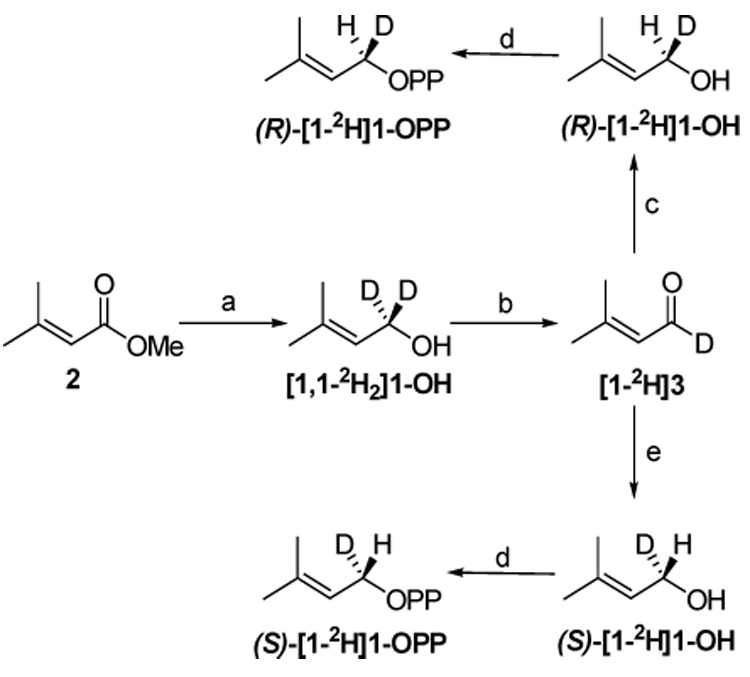
(a) LiAlD4, rt, 2 h, 86%; (b) PCC, CH2Cl2, rt, 3 h, 78%; (c) S-BITIP, Bu3SnH, Et2O, −20 °C, 24 h, 70%; (d) bis-triethylammonium phosphate (TEAP), CCl3CN, CH3CN, rt, 15 min, 30%; (e) R-BITIP, Bu3SnH, Et2O, −20 °C, 24 h, 69%.
[2-2H]3-methyl-2-butenyl diphosphate ([2-2H]1-OPP) was prepared from ethyl acetoacetate (4) as shown in Scheme 2. Deuterium was introduced at the C2 position of the β-ketoester by two exchanges with D2O, to give ~96% exchange of the two methylene hydrogens. The labeled keto ester was then converted to enol phosphate [2-2H]5 followed by treatment with lithium dimethyl cuprate to give [2-2H]2 11 with a 2H content of 93%. The allylic ester was treated with DIBAL to give [2-2H]1-OH. The allylic alcohol was converted into the corresponding diphosphate by the well-established two step halogenation/phosphorylation procedure for synthesis of allylic diphosphates.12
Scheme 2. Synthesis of [2-2H]1-OH.

(a) D2O, rt, 24 h, 2 times, 92%; (b) NaH, ClPO(OEt)2, Et2O, 0 °C, 2 h, 89 %; (c) Me2CuLi, Et2O, −47 °C, 2 h, 93%; (d) DIBAL, Et2O, −70 0 °C, 3 h, 84%; (e) PBr3, Et2O, 2 h, 81%; (f) Tris(tetra-n-butylammonium) hydrogen diphosphate, CH3CN, 34%.
Syntheses of (E)-[4-2H3]3-methyl-2-butenyl diphosphate ((E)-[4,4,4-2H3]1-OPP) and (Z)-[4-2H3]3-methyl-2-butenyl diphosphate ((Z)-[4,4,4-2H3]1-OPP) are shown in Scheme 3. Although both compounds have been prepared previously,13,14 the route we report is an efficient divergent synthesis that yields both stereoisomers. Benzene thiol was added to acetylenic ester 7 to give a 95% yield of a ~3:1 mixture of (E)- and (Z)-8, which were readily separated by column chromatography. The geometries of (E)- and (Z)-8 were assigned from the chemical shift of the methyl 15 and olefinic protons.16 In the 1H NMR spectrum of the major isomer (E)-8, the olefinic proton and methyl protons gave peaks at 5.26 ppm and 2.43 ppm, respectively. For (Z)-8, the corresponding resonances were observed at 5.85 ppm and 1.81 ppm, respectively. Treatment of the isomeric thioenol ethers with CD3MgBr in presence of CuI gave corresponding α,β-unsaturated esters. Treatment of (E)- and (Z)-[4,4,4-2H3]4 with LAH gave allylic alcohols (E)- and (Z)-[4,4,4-2H3]1-OH, respectively. The corresponding diphosphates were prepared by halogenation/phosphorylation.12
Scheme 3. Synthesis of (E)- and (Z)-[4,4,4-2H3]1-OPP.
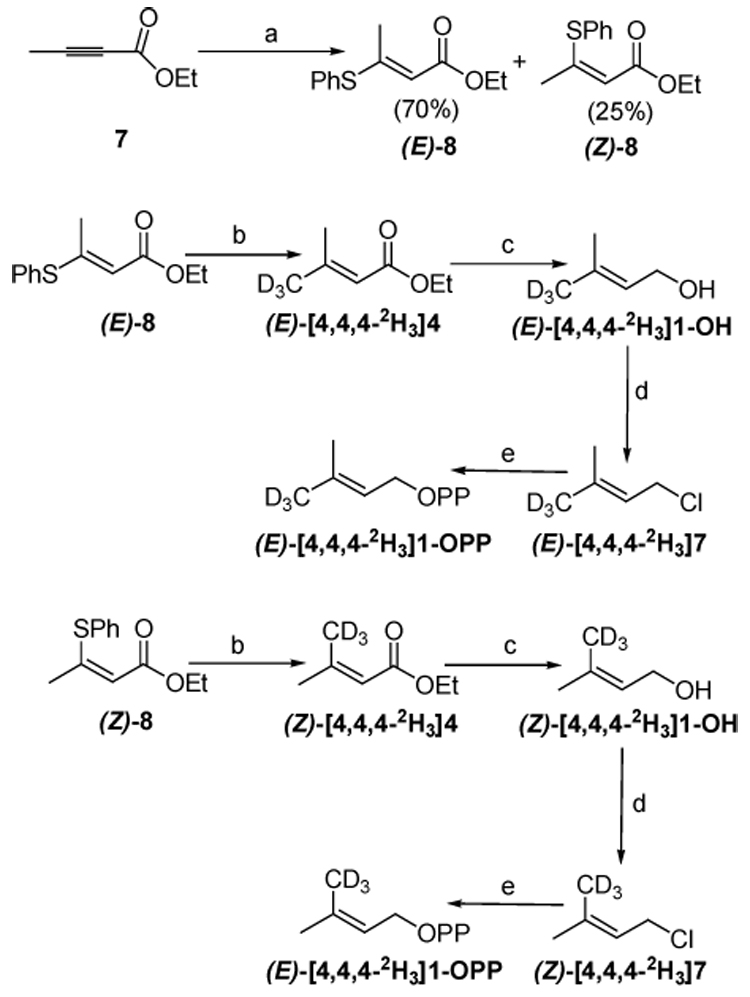
(a) PhSH, NaOH, EtOH, rt, 4 h; (b) CD3MgBr, CuI, THF, −65 °C, 3 h, 81%/87%; (c) LiAlH4, Et2O, 0 °C, 2.5 h, 82%/75%; (d) NCS, CH2Cl2, DMS, 78%/82%; (e) Tris(tetra-n-butylammonium) hydrogen diphosphate, CH3CN, 60%/63%.
In summary, we report short practical syntheses for five labeled derivatives of DMAPP where deuterium is stereospecifically incorporated at specific protonated carbons in the molecule.
Experimental Section
(R)- and (S)- [1-2H]3-Methyl-2-buten-1-ol ((R)- and (S)-[1-2H]1-OH)
A mixture of (S)-BINOL (1.14 g, 4.0 mmol), Ti(O-iPr)4 (1.2 mL, 1.2 mmol), CF3COOH (1.2 mL of 0.5 M in CH2Cl2) and oven-dried 4 Å molecular sieves (8 g), in ether was heated at reflux for 1 h. The solution was cooled to room temperature, and [1-2H]3 (1.02 g, 12 mmol) was added. The mixture was stirred for 5 min and cooled to −78 °C before Bu3SnH (4.19g, 14.4 mmol) was added. The mixture was stirred for 10 min and then placed in a −20 °C freezer for 24 h. The usual workup 6 and flash chromatography on silica gel with 3% ether in pentane gave (R)-[1-2H]1-OH as a colorless oil; 0.73 g (70%; >99 % 2H); 1H NMR (CDCl3), δ 1.67 (s, 3H) 1.74 (s, 3H), 4.10 (d, J = 6.9 Hz, 1H), 5.40 (d, J = 7.2 Hz, 1H); 13C NMR (CDCl3) δ 18.0, 25.9, 59.2, 123.7, 136.7; Mass spectrum m/z (rel intensity) 87 (28), 72 (100), 54 (20), 49 (25), 42 (23); HRMS (EI) calcd for C5H9 2HO (M+) 87.0793, found 87.0796.
Following the same procedure with (R)-BINOL gave (S)-[1-2H]1-OH 7 as a colorless oil; 0.72 g (69 % yield; >99 % 2H); 1H NMR (CDCl3) δ 1.67 (s, 3H) 1.73 (s, 3H), 4.10 (d, J = 6.9 Hz, 1H), 5.39 (d, J = 7.2 Hz, 1H); 13C NMR (CDCl3) δ 18.0, 25.9, 59.1, 123.7, 136.6; Mass spectrum m/z (rel intensity) 87 (29), 72 (100), 54 (18), 42 (23); HRMS (EI) calcd for C5H9 2HO (M+) 87.0793, found 87.0799.
Ethyl [2-2H]3-methyl-2-butenoate ([2-2H]2)
To a solution of lithium dimethylcuprate (40 mmol, 2 eq) in ether, cooled to −47 °C, was added enol phosphate [2-2H]5 (5.3 g, 20.0 mmol), and the mixture was stirred at −47 °C. After 2 h the mixture was poured into an ice cold mixture of 50% aqueous NH4Cl and concd NH4OH (5:1) and the aqueous phase was extracted with ethyl ether. The combined ether extracts were washed with brine, dried over MgSO4, and concentrated. Flash chromatography on silica gel (CH2Cl2) gave 2.13 g (93% yield, 93% 2H) of a colorless oil; 1H NMR (CDCl3) δ 1.27 (t, J= 7.2 Hz, 3H,), 1.88 (s, 3H), 2.16 (s, 3H), 4.13 (2H, q, J = 7.2 Hz); 13C NMR (CDCl3) δ 14.5, 20.3, 27.5, 59.6, 116.3, 156.5, 166.9. Mass spectrum m/z (rel intensity) 129 (4), 72 (18), 57 (16), 43 (100); HRMS (EI) calcd for C7H11 2HO2 (M+) 129.0899, found 129.0920.
Ethyl (E)- and (Z)-[4-2H3]3-methyl-2-butenoate ((E)- and (Z)-[4,4,4-2H3]4)
To a stirred suspension of CuI (5.1 g, 0.03 mol) in dry THF at −63 °C was added CD3MgI (67 mL of 1 M, 0.07 mol) in ether. After stirring for 15 min at −63 °C, a solution of E-8 (3.00 g, 0.014 mol) in dry THF was added and stirring was continued for 3 h. The mixture was then poured into sat NH4Cl and stirred for 15 min at room temp. The aqueous layer was extracted with ether. The combined organic layers were washed with 5% NaOH, water and brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was chromatographed over silica (9:1 hexane/ether) to give 1.43 g (81 % yield, >99 % 2H3) of a colorless oil; 1H NMR (CDCl3) δ 1.27 (t, J = 7.2 Hz, 3H), 2.17 (d, J = 1.2 Hz, 3H), 4.15 (q, J = 7.2 Hz, 2H), 5.67 (d, J = 1.2 Hz, 1H); 13C NMR (CDCl3) δ 14.5, 20.3, 59.6, 116.3, 156.6, 166.9; HRMS (CI) [M+1]+ calculated for C7H9 2H3O2 132.1100, found 132.1108.
Following the same procedure, 3.00 g (0.014 mol) of (Z)-8 gave (Z)-[4,4,4-2H3]4 as a colorless oil; 1.54 g (87% yield, >99 % 2H3); 1H NMR 14,15 (CDCl3) δ 1.28 (t, J = 7.2 Hz, 3H), 1.89 (d, J = 1.0 Hz, 3H), 4.15 (t, J = 7.2 Hz, 2H), 5.68 (d, J = 1 Hz, 1H); 13C NMR (CDCl3) δ 14.5, 27.5, 59.6, 116.4, 156.7, 166.9; HRMS (CI) [M+1]+ calculated for C7H9 2H3O2 132.1100, found 132.1107.
Supplementary Material
Supporting Information Available: General experimental details, characterization data for all compounds (except (R)- and (S)-[1-2H]1-OH, [2-2H]2, (E)- and (Z)-[4,4,4-2H3]4) and 1H and 13C or 31P spectra for (R)- and (S)-[1-2H]1-OH, (R)- and (S)-[1-2H]1-OPP, [2-2H]5, [2-2H]2, [2-2H]1-OH, [2-2H]1-OPP, (E)-8, (Z)-8, and (E)-[4,4,4-2H3]4. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
This study was supported by NIH Grant GM 21328.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Poulter CD. Acc. Chem. Res. 1990;23:70. [Google Scholar]
- 2.Kellogg BA, Poulter CD. Curr. Opin. Chem. Biol. 1997;4:570–578. doi: 10.1016/s1367-5931(97)80054-3. [DOI] [PubMed] [Google Scholar]
- 3.Koyama T, Ogura K. In: Comprehensive Natural Products Chemistry: Isoprenoids. Cane DE, editor. Vol. 2. Oxford: Elsevier Science; 1998. p. 73. [Google Scholar]
- 4.Rivera SB, Swedlund BD, King GJ, Bell RN, Hussey CE, Shattuck-Eidens DM, Wrobel WM, Peiser GD, Poulter CD. Proc. Natl. Acad. Sci. U.S.A. 2001;98:4373. doi: 10.1073/pnas.071543598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vassilikogiannakis G, Chronakis N, Orfanopoulos M. J. Am. Chem. Soc. 1998;120:9911. [Google Scholar]
- 6.Keck GE, Krishnamurthy D. J. Org. Chem. 1996;61:7638. doi: 10.1021/jo961593s. [DOI] [PubMed] [Google Scholar]
- 7.King CHR, Poulter CD. J. Am. Chem. SOC. 1982;104:1413. [Google Scholar]
- 8.Dale JA, Dull DL, Mosher HS. J. Org. Chem. 1969;34:2543. [Google Scholar]
- 9.Gao Y, Hanson RM, Klunder JM, KO SY, Masamune H, Sharpless KB. J. Am. Chem. Soc. 1987;109:5765. [Google Scholar]
- 10.Keller RK, Thompson R. J. Chromatogr. 1993;645:161. doi: 10.1016/0021-9673(93)80630-q. [DOI] [PubMed] [Google Scholar]
- 11.Sum FM, Weiler L. Tetrahedron. 1981;37:303. [Google Scholar]
- 12.Davisson VJ, Woodside AB, Neal TR, Stremler KE, Muehlbacher M, Poulter CD. J. Org. Chem. 1986;51:4768. [Google Scholar]
- 13.Mohanty SS, Uebelhart P, Eugster CH. Helv. Chim. Acta. 2000;83:2036. [Google Scholar]
- 14.Shibuya M, Chou HM, Fountoulakis M, Hassam S, Kim SU, Kobayashi K, Otsuka H, Rogalska E, Cassady JM, Floss HG. J. Am. Chem. Soc. 1990;112:297. [Google Scholar]
- 15.Hill RK, Yan S, Arfin SM. J. Am. Chem. Soc. 1973;95:7857. doi: 10.1021/ja00804a048. [DOI] [PubMed] [Google Scholar]
- 16.Mori K, Mori H. Tetrahedron. 1987;43:4097. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: General experimental details, characterization data for all compounds (except (R)- and (S)-[1-2H]1-OH, [2-2H]2, (E)- and (Z)-[4,4,4-2H3]4) and 1H and 13C or 31P spectra for (R)- and (S)-[1-2H]1-OH, (R)- and (S)-[1-2H]1-OPP, [2-2H]5, [2-2H]2, [2-2H]1-OH, [2-2H]1-OPP, (E)-8, (Z)-8, and (E)-[4,4,4-2H3]4. This material is available free of charge via the Internet at http://pubs.acs.org.