

Abstract
PU.1, an Ets family transcription factor, mediates macrophage effector function in inflammation by regulating gene expression. But, the extent and nature of PU.1 function in gene expression has not been genetically determined because ablation of PU.1 gene abolishes macrophage development. Here, we epigenetically suppressed PU.1 by stably expressing PU.1 specific siRNA in macrophages, and determined the effect of PU.1 deficiency on expressions of key inflammatory genes: Toll-like receptor 4 (TLR4), cyclooxygenease-2 (COX-2), and macrophage inflammatory protein-1α (MIP-1α). PU.1-silenced cell lines expressed lower TLR4 mRNA and COX-2 protein, but higher MIP-1α protein, than controls. Over-expression of PU.1 suppressed LPS-induced MIP-1α production. PU.1 occupied proximal and distal cognate sites in the endogenous MIP-1α promoter, but dissociated only from the distal sites in response to lipopolysaccharide, suggesting a novel negative regulatory mechanism by PU.1. Together, our results defined PU.1 function in differentially regulating expressions of TLR4, COX-2 and MIP-1α.
Keywords: PU.1, epigenetic suppression, TLR4, COX-2, MIP-1α, gene regulation, macrophages, inflammation
PU.1, an Ets transcription factor exclusively expressed in myeloid cells, determines the development of macrophages, as shown in PU.1 gene knockout mice that have no macrophage [1]. In macrophages, PU.1 has been shown to induce expressions of various inflammatory genes including Toll like receptor 4 (TLR4) [2], cyclooxygenas-2 (COX-2) [3], and macrophage inflammatory protein-1α (MIP-1α) [4]. But, this function has not been genetically determined due to the close genetic link between PU.1 and macrophage development. Nevertheless, these results suggest that PU.1 is an essential transcription factor in macrophage-mediated innate immunity.
Macrophages are essential cells in innate immunity. They sense pathogens and then initiate inflammatory responses [5], which is conferred, in part, by abundant expression of Toll like receptor 4 (TLR4), a receptor for bacterial endotoxin (lipopolysaccharide: LPS) [6]. Thus, mice harboring defective TLR4 are more susceptible to bacterial infection [7]. LPS binding to TLR4 activates a series of transcription factors such as NF-κB [6], C/EBP-β [8], and PU.1[3], resulting in induction of inflammatory genes including cyclooxygenase-2 (COX-2) [6] and MIP-1α [4]. Although PU.1, activated by LPS, may induce TLR4, it is unclear whether LPS treatment leads to expression of functional TLR4.
COX-2 plays an important role in inflammation by catalyzing the production of prostanoids that have diverse effects on inflammation, such as recruitment of inflammatory cells, increase of vascular permeability, and induction of vasodilation [9–11]. Thus, transcriptional regulation of COX-2 gene expression has been studied extensively, showing that CREB, NF-κB, and C/EBP-β are major transcription factors for COX-2 expression in various cell types [12–15]. In addition, PU.1 was suggested as a macrophage specific factor for COX-2 expression in macrophages [3], but another in vitro study failed to relate PU.1 and COX-2 expression [16]. Therefore, whether or not PU.1 regulates COX-2 expression in macrophages remains controversial.
MIP-1α is a C-C chemokine that binds to G protein coupled receptors CCR3 and CCR5 and recruits diverse inflammatory effector cells including macrophages [17]. A previous report located a PU.1 binding site in the promoter of MIP-1α, along with C/EBP-β. In that study, although over-expression of C/EBP-β supports MIP-1α expression, over-expression of PU.1, however, failed to do so, and elevated PU.1 expression was not correlated with MIP-1α production in spleen focus-forming virus-induced murine erthroleukemias (MEL) cells[4]. Thus, despite the biochemical, in vitro binding capability of PU.1 to the MIP-1α promoter, it remains unknown whether or not PU.1 is functionally involved in MIP-1α expression.
Here, by epigenetically suppressing PU.1 in macrophages, we examined the role of PU.1 in TLR4 expression and in TLR4-meidated COX-2 and MIP-1α expressions. Due to low transfection efficiency in macrophages, it was necessary to generate macrophage cell lines that stably express siRNA specific for PU.1. Our results defined the role of PU.1 in regulating expression of those genes, in which PU.1 functioned as a positive and a negative regulator.
Materials and methods
Cell Culture
A murine macrophage cell line, RAW 264.7 (ATCC, Rockville, MD), was maintained in DMEM (Cellgro) containing 10% fetal bovine serum (FBS) (Hyclone). LPS treatment was described previously [3].
RNAi Plasmid Constructs, plasmids, and transfection
A pair of oligonucleotides designed by a software, targeting at from +115 to +135nt, was inserted into pSUPER.retro.puro plasmid (OligoEngine, Seattle, WA): the forward, 5′-GATCCCCGCCATAGCGATCACTACTGTTCA AGAGACAGTAGTGATCGCTATGGC TTTTTGGAAA-3′ and the reverse, 5′-AGCTTTTCCAAAAAGCCATAGCGATCACTACTGTCTCTTGAACAGTAGTGATCGCTATGGCGGG-3′. Plasmids were purified by Endo-free Maxiprep kit (Qiagen). RAW264.7 transfected with GenePORTER 2 (Gene Therapy Systems, San Diego, CA) was selected and maintained under 2μg/ml of Puromycin (SIGMA). The plasmids encoding PU.1 and C/EBP-β were gifts from Dr. Atchison (University of Pennsylvania) and Dr. Sealy (Vanderbilt University), respectively.
Western Blotting
Total cell lysate were prepared as described previously [3]. Proteins separated by SDS-PAGE were analyzed by appropriate antibodies with enhanced chemoluminescence (ECL plus, Amersham). Antibodies for α-PU.1 (rabbit polyclonal), α-p65 (rabbit polyclonal), αIκBα (rabbit polyclonal), α-actin antibody, and α-Ets-1/2 (goat polyclonal) were obtained from Santa Cruz Biotechnology except α-murine COX-1 and -2 (Cayman Chemical).
RT-PCR
Total RNA was prepared by RNeasy kit per the protocol of the manufacturer (Qiagen). 2μg of RNA was used for cDNA synthesis. Actin cDNA from each sample was used to normalize the samples for differences in PCR efficiency. TLR4 mRNA quantity was determined by using end-point dilution PCR, including three serial 1 to 5 dilutions (1:1, 1:5, 1:25, and 1:125) of RT products for PCR amplification. To avoid genomic DNA contamination, equal amounts of RNA from each sample were PCR amplified without RT reaction. cDNA was amplified with Taq polymerase (Perkin-Elmer) and appropriate primers at 94°C for 40 s, 60°C for 30 s, and 72°C for 40 s for 35 cycles with an initial 4 min denaturation at 95°C and final 10-min extension at 72°C. The primers for TLR4 were 5′-GGAAGTTTCTCTGGACTAACAAGTTTAGA -3′ and 5′-AAATTGTGAGCCACATT GAGTTTC-3′. The primers for β-actin were 5′-AGAGGGAAATCGTGCGTGAC-3′; and 5′-CA ATAGTGATGACCTGGCCGT-3′.
Flow cytometry analysis
One million cells pre-incubated with normal IgG were stained with either phycoerythrin-conjugated anti-TLR4/MD2 or isotypic IgG (Santa Cruz Biotechnology) at 4°C for 45 min in a medium (DMEM medium, 10% newborn calf serum), and analyzed by a FACScan flow cytometer (Becton Dickinson, Mountain View, Calif.) and CellQuest software.
Luciferase assay
Cells were transfected with NF-κB firefly luciferase reporter construct, along with tk-Renilla luciferase construct. Dual luciferase assay was performed per the protocol of the manufacturer (Promega).
Chromatin Immunoprecipitation (ChIP) Assay
Reagents and assay procedure were described previously [18]. DNA was amplified as follows: 94° C 240 s; 30~32 cycles at 94° C 40 s, 54° C 40 s and 72° C 60 s; final elongation at 72° C 10 min. The primers used for the distal sites were 5′-ACACTGGATAACTGCTTACTTT-3′ and 5′-AGTACACTCATAACATTGGT GA-3′, and those for the proximal site 5′-GTGGCCTAGTCACTTTGCG-3′ and 5′-CAGCTCTCAACTCGTGACC-3′. Each experiment was performed at least three times independently.
Results
PU.1 up-regulates TLR4 mRNA expression
To silence PU.1 expression by siRNA in macrophages, to which transfection is poor, we stably transfected RAW264.7 cells with an empty host vector plasmid or a plasmid encoding siRNA specific for the murine PU.1 gene, and the transfected cells were selected under 2μg/ml of Puromycin. As shown in Fig. 1A, two independent PU.1 siRNA cell lines, PU 5.7 and PU 5.9, suppressed PU.1 expression, while a stable transfectant with the empty vector plasmid, PU 4.11, expressed PU.1 similar to the parental cell line, RAW264.7. To examine whether PU.1 siRNA cross-silences other Ets family transcription factors, we measured expressions of Ets- 1 and -2 in PU 5.7, and found no differences in their expressions (Fig. 1B). These results show that our PU.1 siRNA cell lines specifically suppressed PU.1 expression without affecting related proteins.
Fig. 1. PU.1 up-regulates TLR4 mRNA expression.
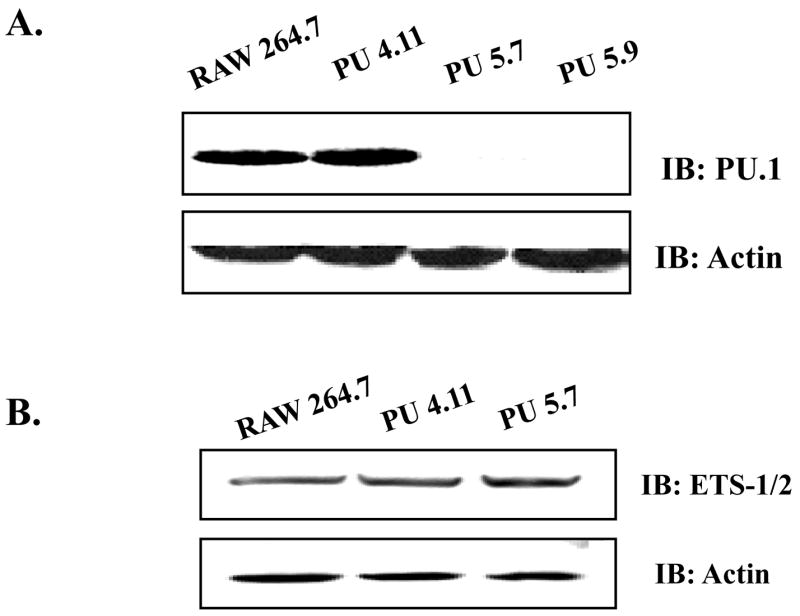
(A) Silencing PU.1 expression in siRNA cell lines, PU 5.7 and PU 5.9, was determined by Western blot with α-PU.1 antibody (top panel) and α-actin antibody for internal controls (bottom panel). (B) Potential cross-silencing was examined in PU 5.7 by Western blot with α-ETS ½ antibodies. (C) TLR4 mRNA expression was examined by semi-quantitative RT-PCR of PU 5.7 (left panel) and PU 5.9 (right panel). Included was PCR for TLR4 without RT reaction (-RT) to exclude genomic DNA contamination. β-actin as internal controls was similarly analyzed.
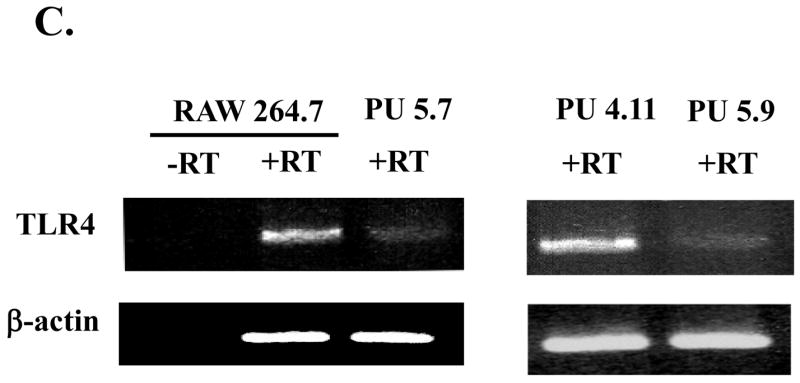
Next, to examine whether PU.1 regulates expression of TLR4 mRNA, we extracted total RNA from the PU.1-silenced cell lines and analyzed it by semi-quantitative RT-PCR. As shown in Fig. 1C, PU 5.7 expressed a low level of TLR4 mRNA expression compared to RAW264.7 (left panel). Similarly, PU 5.9 expressed less TLR4 mRNA than PU 4.11 (right panel). Together, these data indicate that PU.1 positively regulates TLR4 mRNA expression.
Decrease of TLR4 mRNA does not affect TLR4 signaling
Since TLR4 needs to complex with a cofactor, MD2, for its cell surface presentation and responsiveness to LPS [19; 20], we examined whether low TLR4 mRNA expression affects the cell surface expression of TLR4/MD2 and responsiveness to LPS. First, for determination of the cell surface expression of TLR4, the cells were treated with LPS for different periods, stained with phycoerythrin-conjugated TLR4/MD2 antibody, and subsequently analyzed by FACS. As shown in Fig. 2A and B, the surface expression of TLR4/MD2 complex, at a steady state and during LPS treatment, was not significantly different between the parental and PU.1 siRNA cell line, PU 5.7.
Fig. 2. Expression of cell surface TLR4 and NF-κB activation in the PU.1-silenced cell line.


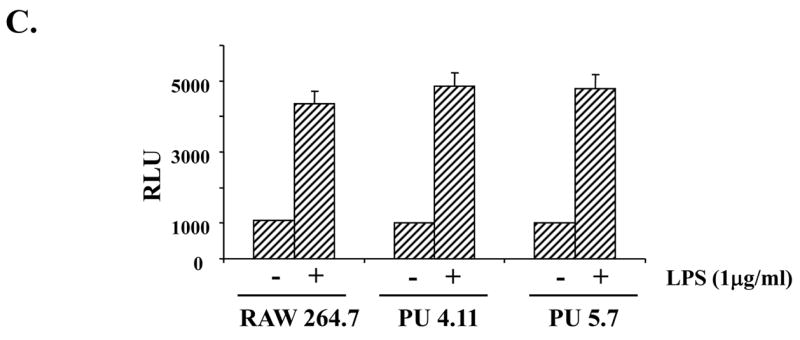
Surface expression of TLR4/MD2 was determined in RAW 264.7 cells (A) and PU 5.7 cells (B) by FACS analyses. The cells differentially treated with LPS (0.1μg/ml) were fixed and stained with α-TLR4/MD2 antibody. The dotted line represents staining with PE-conjugated isotypic IgG and solid line does for with PE-conjugated TLR4/MD2 antibody. (C) NF-κB activity of PU.1-silenced cell line was measured by a reporter assay. The cells transfected with an NF-κB-Luciferase reporter and tk-Renilla constructs were treated with LPS (1 μg/ml) for 4 h. The results were an average of triplicate settings, and experiment was performed three times independently.
Next, in order to determine responsiveness to LPS, the cells were transfected with an NF-κB-Luciferase reporter construct and subsequently treated with LPS for NF-κB activation, an indicative of activated TLR4 signaling. As shown in Fig. 2C, NF-κB activation was not different from controls, indicating intact responsiveness to LPS. Similar results were also obtained in another PU.1-silenced cell line, PU 5.9 (data not shown). Together, these results suggest that although PU.1 deficiency results in a reduction of a steady level of TLR4 mRNA, this low TLR4 mRNA expression is sufficient for maintaining the cell surface presentation and function of TLR4.
PU.1 regulates early COX-2 expression
Previously, we showed that over-expression of PU.1 enhances COX-2 expression elicited by LPS treatment [3], which was, however, not supported by a recent in vitro study conducted in another laboratory [16]. These results prompted us to test whether or not PU.1 regulates COX-2 expression in macrophages by using the PU.1 siRNA cell lines. We treated the cells with LPS for up to 8 h, and analyzed COX-2 expression by Western blotting. As shown in Fig. 3, the PU.1 siRNA cell line, PU 5.7, expressed lower level of COX-2 expression than controls at 2 and 4 h after LPS treatments (compare lanes 2 and 3 to lanes 5 and 6). But, at 8h after LPS treatment, COX-2 expression in PU 5.7 was not different from PU 4.11. On the other hand, COX-1 expression was not different in these two cell lines regardless of LPS treatment (data not shown). We obtained similar results using two additional PU.1-silenced cell lines, PU 5.6 and PU 5.9, (data not shown). Together, these results demonstrate that PU.1 regulates early expression of COX-2 in macrophages.
Fig. 3. PU.1 up-regulatesCOX-2 expression.

COX-2 expression in PU.1-silenced cell lines was determined by Western blot analysis. The cells were treated with LPS (1 μg/ml) for up to 8 h, and total cell lysate was analyzed by immunoblotting for COX-2 (top panels) and actin (bottom panels).
PU.1 down-regulates MIP-1a expression
Since it remains unknown whether or not PU.1 functionally regulates MIP-1α expression, we determined the role of PU.1 in MIP-1α expression. First, since a previous report showed that C/EBP-β is involved in MIP-1α expression [4], we tested whether over-expression of C/EBP-β induces MIP-1α in macrophages. As shown in Fig. 4A, transfection of RAW264.7 with a C/EBP-β expressing plasmid induced MIP-1α (lanes 3 and 4), suggesting that, consistent with the previous report [4], C/EBP-β supports MIP-1α expression. Next, to determine the role of PU.1 in LPS-induced MIP-1α expression, we treated the PU.1 silenced cells, PU 5.7, with LPS for different periods, and measured MIP-1α expression by Western blotting. As shown in Fig. 4B, unlike control (lane 1), PU 5.7 cells expressed MIP-1α without LPS treatment (lane 4), which was further enhanced by LPS treatment (compare lanes 2 and 3 to lanes 5 and 6), suggesting that PU.1 functions as a suppressor of MIP-1α expression. To test this possibility, we transfected RAW 264.7 cells with increasing amounts of a PU.1 expressing plasmid prior to LPS treatment for 4h, and analyzed MIP-1α expression by Western blotting. As shown in Fig. 4C, over-expression of PU.1 suppressed MIP-1α expression. Together, these results suggest that PU.1 is a negative regulator of MIP-1α expression.
Fig. 4. PU.1 negatively regulates MIP-1α expression.
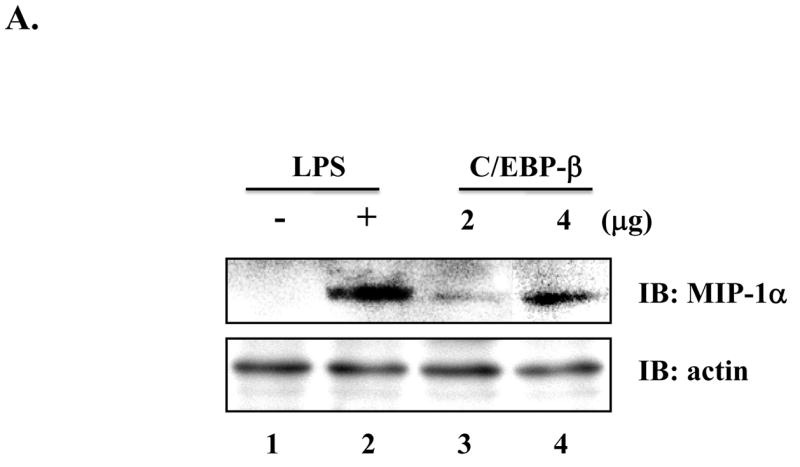
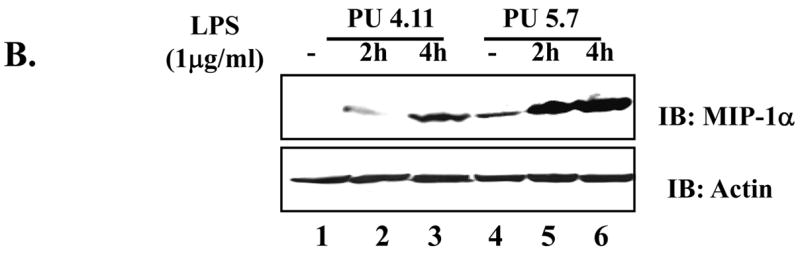

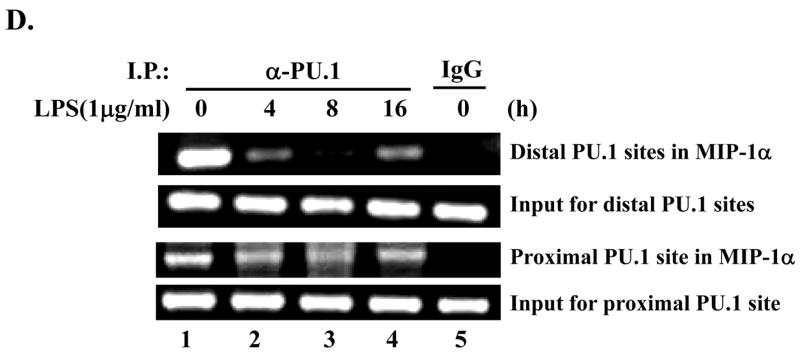
(A) RAW 264.7 cells were transfected with pcDNA3.1 (lanes 1 and 2) or a plasmid encoding C/EBP-β (lanes 3 and 4) for 48h. Transfection was normalized with pcDNA3.1 to 4μg. Total cell lysate was analyzed by Western blot for MIP-1α (top panel) and actin (bottom panel). (B) MIP-1α expression in PU 5.7 following LPS treatment was determined by Western blot analysis. (C) RAW 264.7 cells were transfected with increasing amounts of a PU.1-expressing plasmid, and the transfected cells were treated with LPS for 2h. MIP-1α expression was determined by Western blot analysis. (D) PU.1 binding to the endogenous MIP-1α promoter was analyzed by ChIP assay. PU.1 bound to DNA was immunoprecipitated by α-PU.1 antibody (lanes 1 to 4), and co-precipitated DNA was analyzed by PCR for distal PU.1 sites (top two panels) and proximal site (bottom two panels). Included was isotypic IgG to exclude a nonspecific immunoprecipitation (lane 5).
In order to elucidate a novel suppressive mechanism by PU.1, we examined PU.1 binding to the endogenous MIP-1α promoter in response to LPS treatment. First, we analyzed the murine MIP-1α promoter sequence by the TFSEARCH program (version 1.3, Tokyo University, Japan), which located, within a 1.5kb-long promoter, three c-Ets sites: two sites, from −949 to −959 nt and from −1078 to − 1090 nt, designated as distal binding sites, and one site, from −342 to − 352 nt, as a proximal binding site. Next, to examine whether PU.1 utilizes these sites, we performed chromatin immunoprecipitation (ChIP) assay. RAW cells were treated with LPS for different periods, and fixed to cross-link DNA and bound transcription factors. For precipitation of PU.1 bound DNA, the nuclear fraction was collected, sonicated, and added with α-PU.1 antibody. DNA precipitated with PU.1 was eluted and amplified by the specific sets of primers flanking either the distal binding sites or the proximal binding site. As shown in Fig. 4D, PU.1 bound to these binding sites in an unstimulated condition (lane 1). While constitutively binding to the proximal site regardless of LPS treatment (third panel from the top), PU.1 lost binding to the distal sites at early time points (lanes 2 and 3) but returned to the sites at a late time point after LPS treatment (lane 4). Taken together, these results suggest that differential binding of PU.1 to these sites, in response to LPS, is associated with down-regulation of MIP-1α expression.
Discussion
Bacterial infection is associated with severe sepsis, the 10th leading cause of death in the US [21], which is characterized by an uncontrolled, adverse host immune reaction that is independent of resolving bacterial infection [22]. Given the key role of macrophages in innate immunity, in which PU.1 is a central transcription factor [23], elucidating PU.1 function in regulation of inflammatory gene expression could provide a clue to control excessive inflammation that results in organ dysfunction. But, due to its close link with macrophage development [1], the precise function of PU.1 has not been genetically determined. In this study, we attempted to circumvent this issue by epigenetically suppressing PU.1 expression and to genetically determine PU.1 function in regulating inflammatory gene expression such as TLR4, COX-2, and MIP-1α.
LPS bound TLR4 activates PU.1, and PU.1 has been shown to increase TLR4 transcription [2]. Thus, it is conceivable that LPS treatment increases TLR4 expression and thereof responsiveness to LPS, which, however, remains controversial [24; 25]. Consistent with the published results [2; 24], our data clearly show that PU.1 positively regulated TLR4 gene expression. But, our results show that LPS treatment decreased the level of the cell surface TLR4, and we found no evidence that the level of the cell surface TLR4 was recovered during LPS treatment (data not shown). It seems that a low level of TLR4 mRNA did not affect the level of cell surface TLR4 and its signaling. Thus, our results suggest that LPS treatment does not enhance, rather suppresses, TLR4 function.
COX-2, induced by inflammatory stimuli, regulates inflammation, and aberrant expression of COX-2 is associated with cancer [26]. Numerous studies have shown that COX-2 expression results from a complex interplay among key transcription factors such as NF-κB, C/EBP-β, and PU.1. Since the involvement of PU.1 is controversial [16], we addressed this by using the PU.1-silenced cell lines. Our data show that deficiency of PU.1 resulted in decreased COX-2 protein expression at early, but not at later time point, indicating that the impact of PU.1 on COX-2 expression was time dependent. It is notable that the PU.1-silenced cell lines exhibited intact TLR4 signaling, excluding the possibility that reduced COX-2 expression is due to impaired TLR4 signaling. In conjunction with our previous report showing that PU.1 binds to two different sites in the endogenous COX-2 promoter in response to LPS, we conclude that PU.1 is involved in COX-2 expression.
MIP-1α, rapidly induced by LPS and TNF-α, promotes inflammation by recruiting immune effector cells [17]. Although previous study implicated PU.1 in MIP-1α gene expression [4], neither high expression of PU.1 in MEL cells [4] nor GM-CSF treatment to increase PU.1 expression [27] resulted in MIP-1α expression, leaving the role of PU.1 in MIP-1α expression unknown. Our results suggest that PU.1 suppresses MIP-1α gene expression. Consistent with this, over-expression of PU.1 suppressed MIP-1α expression elicited by LPS treatment. Thus, our results provide explanation why expression of PU.1 failed to induce MIP-1α.
Dual, opposing effects of a transcription factor are not unprecedented. Yin-Yang 1 (YY1) has been documented as a dual regulator of transcription [28; 29]. This function is in part determined by acetylation or deacetylation of YY1, which are mediated by histone acetylases (HATs) and histone deacetylases (HDACs), respectively [28; 29]. Likewise, PU.1 interacts with HATs and HDACs [30; 31]. As yet, it is unknown whether the acetylation status of PU.1 is associated with its dual activity.
In summary, we attempted to define the role of PU.1 in macrophage effector function. To that end, we established PU.1 loss-of-function cell lines by utilizing siRNA. The PU.1-silenced cell lines maintained functional TLR4 signaling, which allowed us to study PU.1 function in inflammatory gene expression following LPS treatment. Our data demonstrated that PU.1 positively regulates TLR4 and COX-2 expression but negatively regulates MIP-1α. Our study also supplies evidence that siRNA approaches in macrophages could provide important tools to decipher functions of PU.1 and other regulatory factors in macrophages.
Acknowledgments
This work was supported by The Department of Veterans Affairs and NIH Grants HL061419, HL075557, and HL066196.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 2.Rehli M, Poltorak A, Schwarzfischer L, Krause SW, Andreesen R, Beutler B. PU.1 and interferon consensus sequence-binding protein regulate the myeloid expression of the human Toll-like receptor 4 gene. J Biol Chem. 2000;275:9773–9781. doi: 10.1074/jbc.275.13.9773. [DOI] [PubMed] [Google Scholar]
- 3.Joo M, Park GY, Wright JG, Blackwell TS, Atchison ML, Christman JW. Transcriptional regulation of the cyclooxygenase-2 gene in macrophages by PU.1. J Biol Chem. 2004;279:6658–6665. doi: 10.1074/jbc.M306267200. [DOI] [PubMed] [Google Scholar]
- 4.Grove M, Plumb M. C/EBP, NF-kappa B, and c-Ets family members and transcriptional regulation of the cell-specific and inducible macrophage inflammatory protein 1 alpha immediate-early gene. Mol Cell Biol. 1993;13:5276–5289. doi: 10.1128/mcb.13.9.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kooguchi K, Hashimoto S, Kobayashi A, Kitamura Y, Kudoh I, Wiener-Kronish J, Sawa T. Role of alveolar macrophages in initiation and regulation of inflammation in Pseudomonas aeruginosa pneumonia. Infect Immun. 1998;66:3164–3169. doi: 10.1128/iai.66.7.3164-3169.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 7.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 8.Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, Nakajima T, Hirano T, Kishimoto T. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 1990;9:1897–1906. doi: 10.1002/j.1460-2075.1990.tb08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fletcher JR. Eicosanoids. Critical agents in the physiological process and cellular injury. Arch Surg. 1993;128:1192–1196. doi: 10.1001/archsurg.1993.01420230020003. [DOI] [PubMed] [Google Scholar]
- 10.Williams JA, Shacter E. Regulation of macrophage cytokine production by prostaglandin E2. Distinct roles of cyclooxygenase-1 and -2. J Biol Chem. 1997;272:25693–25699. doi: 10.1074/jbc.272.41.25693. [DOI] [PubMed] [Google Scholar]
- 11.Williams TJ, Morley J. Prostaglandins as potentiators of increased vascular permeability in inflammation. Nature. 1973;246:215–217. doi: 10.1038/246215a0. [DOI] [PubMed] [Google Scholar]
- 12.Inoue H, Nanayama T, Hara S, Yokoyama C, Tanabe T. The cyclic AMP response element plays an essential role in the expression of the human prostaglandin-endoperoxide synthase 2 gene in differentiated U937 monocytic cells. FEBS Lett. 1994;350:51–54. doi: 10.1016/0014-5793(94)00731-4. [DOI] [PubMed] [Google Scholar]
- 13.Hwang D, Jang BC, Yu G, Boudreau M. Expression of mitogen-inducible cyclooxygenase induced by lipopolysaccharide: mediation through both mitogen-activated protein kinase and NF-kappaB signaling pathways in macrophages. Biochem Pharmacol. 1997;54:87–96. doi: 10.1016/s0006-2952(97)00154-8. [DOI] [PubMed] [Google Scholar]
- 14.Caivano M, Gorgoni B, Cohen P, Poli V. The induction of cyclooxygenase-2 mRNA in macrophages is biphasic and requires both CCAAT enhancer-binding protein beta (C/EBP beta ) and C/EBP delta transcription factors. J Biol Chem. 2001;276:48693–48701. doi: 10.1074/jbc.M108282200. [DOI] [PubMed] [Google Scholar]
- 15.Wadleigh DJ, Reddy ST, Kopp E, Ghosh S, Herschman HR. Transcriptional activation of the cyclooxygenase-2 gene in endotoxin-treated RAW 264.7 macrophages. J Biol Chem. 2000;275:6259–6266. doi: 10.1074/jbc.275.9.6259. [DOI] [PubMed] [Google Scholar]
- 16.Kang YJ, Wingerd BA, Arakawa T, Smith WL. Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. J Immunol. 2006;177:8111–8122. doi: 10.4049/jimmunol.177.11.8111. [DOI] [PubMed] [Google Scholar]
- 17.Menten P, Wuyts A, Van Damme J. Macrophage inflammatory protein-1 Cytokine. Growth Factor Rev. 2002;13:455–481. doi: 10.1016/s1359-6101(02)00045-x. [DOI] [PubMed] [Google Scholar]
- 18.Park GY, Joo M, Pedchenko T, Blackwell TS, Christman JW. Regulation of macrophage cyclooxygenase-2 gene expression by modifications of histone H3. Am J Physiol Lung Cell Mol Physiol. 2004;286:L956–L962. doi: 10.1152/ajplung.00338.2003. [DOI] [PubMed] [Google Scholar]
- 19.Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 20.Kawasaki K, Nogawa H, Nishijima M. Identification of mouse MD-2 residues important for forming the cell surface TLR4-MD-2 complex recognized by anti-TLR4-MD-2 antibodies, and for conferring LPS and taxol responsiveness on mouse TLR4 by alanine-scanning mutagenesis. J Immunol. 2003;170:413–420. doi: 10.4049/jimmunol.170.1.413. [DOI] [PubMed] [Google Scholar]
- 21.Murphy SL. National vital statistics report. Anonymous, National Center for Health Statistics; Hyattsville, Md: 2000. Deaths: final data for 1998. [PubMed] [Google Scholar]
- 22.Wheeler AP, Bernard GR. Treating patients with severe sepsis 1 N. Engl J Med. 1999;340:207–214. doi: 10.1056/NEJM199901213400307. [DOI] [PubMed] [Google Scholar]
- 23.Lloberas J, Soler C, Celada A. The key role of PU.1/SPI-1 in B cells, myeloid cells and macrophages. Immunol Today. 1999;20:184–189. doi: 10.1016/s0167-5699(99)01442-5. [DOI] [PubMed] [Google Scholar]
- 24.Roger T, Miconnet I, Schiesser AL, Kai H, Miyake K, Calandra T. Critical role for Ets, AP-1 and GATA-like transcription factors in regulating mouse Toll-like receptor 4 (Tlr4) gene expression. Biochem J. 2005;387:355–365. doi: 10.1042/BJ20041243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pedchenko TV, Park GY, Joo M, Blackwell TS, Christman JW. Inducible binding of PU.1 and interacting proteins to the Toll-like receptor 4 promoter during endotoxemia. Am J Physiol Lung Cell Mol Physiol. 2005;289:L429–L437. doi: 10.1152/ajplung.00046.2005. [DOI] [PubMed] [Google Scholar]
- 26.FitzGerald GA. COX-2 and beyond: Approaches to prostaglandin inhibition in human disease. Nat Rev Drug Discov. 2003;2:879–890. doi: 10.1038/nrd1225. [DOI] [PubMed] [Google Scholar]
- 27.Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 28.Shi Y, Lee JS, Galvin KM. Everything you have ever wanted to know about Yin Yang 1…. Biochim Biophys Acta. 1997;1332:F49–F66. doi: 10.1016/s0304-419x(96)00044-3. [DOI] [PubMed] [Google Scholar]
- 29.Thomas MJ, Seto E. Unlocking the mechanisms of transcription factor YY1: are chromatin modifying enzymes the key? Gene. 1999;236:197–208. doi: 10.1016/s0378-1119(99)00261-9. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto H, Kihara-Negishi F, Yamada T, Hashimoto Y, Oikawa T. Physical and functional interactions between the transcription factor PU.1 and the coactivator CBP. Oncogene. 1999;18:1495–1501. doi: 10.1038/sj.onc.1202427. [DOI] [PubMed] [Google Scholar]
- 31.Kihara-Negishi F, Yamamoto H, Suzuki M, Yamada T, Sakurai T, Tamura T, Oikawa T. In vivo complex formation of PU.1 with HDAC1 associated with PU.1-mediated transcriptional repression. Oncogene. 2001;20:6039–6047. doi: 10.1038/sj.onc.1204756. [DOI] [PubMed] [Google Scholar]