

Abstract
The posterior cingulate cortex (PCC) is the brain region displaying the earliest sign of energy hypometabolism in patients with amnestic mild cognitive impairment (MCI) who develop Alzheimer’s disease (AD). In particular, the activity of the mitochondrial respiratory enzyme cytochrome oxidase (C.O.) is selectively inhibited within the PCC in AD. The present study is the first experimental analysis designed to model in animals the localized cortical C.O. inhibition found as the earliest metabolic sign of early-stage AD in human neuroimaging studies. Rats were used to model local inhibition of C.O. by direct injection of the C.O. inhibitor sodium azide into the PCC. Learning and memory were examined in a spatial holeboard task and brains were analyzed using quantitative histochemical, morphological and biochemical techniques. Behavioral results showed that sodium azide-treated rats were impaired in their memory of the baited pattern in probe trials as compared to their training scores before treatment, without non-specific behavioral differences. Brain analyses showed that C.O. inhibition was specific to the PCC, and sodium azide increased lipid peroxidation, gliosis and neuron loss, and lead to a network functional disconnection between the PCC and interconnected hippocampal regions. It was concluded that impaired memory by local C.O. inhibition in the PCC may serve to model in animals a metabolic lesion similar to that found in patients with amnestic MCI and early-stage AD. This model may be useful as an in vivo testing platform to investigate neuroprotective strategies to prevent or reduce the amnestic effects produced by posterior cingulate energy hypometabolism.
Keywords: animal model, cytochrome oxidase, posterior cingulate, sodium azide, energy metabolism, oxidative stress, TBARS, amnestic mild cognitive impairment, Alzheimer’s disease
INTRODUCTION
Minoshima et al (1994, 1997) were the first to report that metabolic energy reduction in the posterior cingulate cortex (PCC) is the first sign of brain hypometabolism very early in Alzheimer’s disease (AD). Neuroimaging evidence for an early metabolic lesion in PCC has also been found in asymptomatic subjects at genetic risk for AD, such as subjects homozygotic for the E4 allele of the apolipoprotein E gene (Reiman et al., 1996, Small et al., 2000), and in patients with amnestic mild cognitive impairment (MCI) who later develop AD (Mosconi, 2005). Moreover, recent diffusion tensor imaging showed that the cingulum fibers that connect the PCC with hippocampal regions have reduced integrity in both MCI and AD patients (Zhang et al., 2007). This is consistent with a disruption in the functional correlations of a network of brain regions involving the PCC and interconnected medial temporal regions in MCI patients at risk for AD (Sorg et al., 2007).
Human studies support a role of the PCC in spatial orientation and memory, which are affected in amnestic MCI and early-stage AD. Indeed, PET and MRI studies showed that the severity of AD symptoms is correlated with hypometabolism in PCC but not temporal regions (Ishii et al., 1997; Hirono et al., 1998; Alsop et al., 2000). Cammalleri et al. (1996) described a patient with a tumor in the PCC who was unable to navigate in its surroundings due to the inability to use landmarks for route finding. Functional studies in healthy subjects also provide evidence for a role of the PCC in visually-guided behavior relating to aspects of navigation and memory. A spatial attention task showed fMRI activation in the PCC that was strongly correlated with the speed of target detection when spatial cues were present (Mesulam et al., 2001). The PCC was also active when subjects were asked to remember visual landmarks and their movements along a previously learned route (Berthoz, 1997). Similarly, Maguire et al. (1997) found that the PCC was involved during tasks that required learning a room that contained salient objects and empty rooms only distinguished by their shape, as well as remembering environments recently learned and those very familiar. In particular, PET studies showed that the PCC is critically involved in a network for memory retrieval (Nyberg et al., 1996; Cabeza et al., 1997).
Animal studies also suggest a role of the PCC in spatial navigation and memory. Single-cell recordings show that specific sensory stimuli can activate cingulate neurons that are sensitive to the direction the animal is facing (Chen et al., 1994). Furthermore, the PCC may specifically be involved during the late stages of learning. In an active avoidance task, discriminative neuronal activity (cells firing to a stimulus that predicts shock) occurs late in learning in the PCC (Gabriel, 1993). This is supported by studies showing that lesioning this area before task acquisition impaired rats only during the late stages of acquisition (Bussey et al., 1996; Bussey et al., 1997). Lesioning the PCC with NMDA impaired rats’ performance in the radial arm maze, water maze, and an object-in-place task, whereby objects are switched in location in the second trial (Vann and Aggleton, 2002). Aspiration of the PCC produced deficits in the water maze and matching-to-place tasks (Harker and Whishaw, 2002; Sutherland et al., 1988; Whishaw et al., 2001).
The evidence from human and animal studies indicates that the PCC is involved in spatial navigation, perhaps with a bigger contribution during the stages of memory retrieval. Human functional neuroimaging shows that this area is the earliest affected metabolically in patients with AD (Minoshima et al., 1997) and in patients with MCI who later develop AD (Mosconi, 2005). Importantly, Valla et al. (2001) found that the underlying biochemical defect for PCC hypometabolism in AD brains was a selective activity reduction of the mitochondrial enzyme cytochrome oxidase (C.O.). This respiratory enzyme is responsible for the activation of oxygen for aerobic energy metabolism, and it provides an index of neuronal metabolic energy capacity because it is essential for ATP production in mitochondria (Wong-Riley et al., 1998). The energy hypometabolism in the PCC of AD patients is specifically linked to decrement in C.O. activity, it is not found in age-matched control brains, and it is not secondary to decrement in other mitochondrial enzymes or the number of amyloid plaques or neurofibrillary tangles in PCC (Valla et al., 2001). Furthermore, C.O. activity in the superficial layers of PCC in AD brains showed a progressive reduction tightly linked to disease duration, which was not observed in other cortical regions such as the motor cortex (Valla et al., 2001).
There is a need to model in animals PCC hypometabolism similar to that linked to amnestic MCI and early-stage AD to be able to investigate neuroprotective strategies to prevent or reduce the amnestic effects produced by PCC hypometabolism. The goal of this study was to determine whether an amnestic mild impairment could be induced in rats by partial inhibition of C.O. activity in the PCC, and whether this local metabolic lesion would lead to network functional dissociation between the PCC and interconnected hippocampal regions implicated in the progression of AD (Zhang et al., 2007; Sorg et al., 2007). PCC hypometabolism was produced by sodium azide, a mitochondrial toxin that selectively inhibits C.O. activity by blocking the transfer of electrons to oxygen (Bennett et al.,1992). The objective was to model the specific metabolic lesion linked to early-stage AD by inhibiting C.O. activity in the PCC, not just damage this region by other means in a non-specific manner. This study tested this hypothesis by locally inhibiting C.O. activity in the PCC after rats were trained in a spatial food search task and examining its effects on memory retrieval and regional brain metabolism.
MATERIALS and METHODS
Behavioral Training
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas at Austin, and conform to all NIH and USDA guidelines. All experiments were conducted in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. Subjects for the memory impairment study were 25 male Sprague-Dawley rats (Harlan, Houston, TX) weighing 225–250 grams upon arrival and single-housed on a 12 h light-dark cycle. After 1–2 days of acclimation, they were placed on a food restriction procedure and weighed daily to ensure that their weight did not drop below 85% of free-feeding rats of a similar age. They were fed after daily behavioral training. Water was freely available. Rats were handled daily for 5 min on each of the 4–5 days before habituation began. On the two days preceding habituation, rats were given two sucrose pellets (45 mg, Noyes, Lancaster, NH) in their home cage. Two animals were excluded from the experiment. One was removed because it was not eating upon arrival to the research facility and another died during surgery. The remaining rats were trained in a baited holeboard task (Table 1).
Table 1.
Behavioral Protocol in Holeboard Maze
| Day | Procedure | Configuration |
|---|---|---|
| 1–2 | habituation | all holes baited |
| 3–10 | training | 4 holes baited |
| 11 | surgery | |
| 12 | probe | no baited holes |
| 13–16 | retraining | 4 holes baited |
Holeboard training was conducted inside a Med Associates open field chamber 43.2 cm2 (St. Albans, VT) surrounded by clear plexiglass walls 30.5 cm high. Inside the chamber was a holeboard floor insert with sixteen equidistant-spaced holes (3.2 cm2) in a 4 X 4 array and an underlying food tray. The holes were 4.45 cm apart and the outer holes were 8.57 cm from the chamber wall. The bottom insert consisted of a bottom tray, a screen, and a top tray that were positioned underneath the holeboard floor. The bottom insert contained 16 holes with the same dimensions as the holeboard floor. The wire screen was positioned over the holes of the bottom tray which each contained five inaccessible sucrose pellets to mask potential odor cues emanating from the food in the baited holes. Thus, the rats could not use olfactory cues to discriminate between baited and unbaited holes. The task floor was placed 2 cm over the food tray. Infrared photobeams located just under the surface of the top floor detected entries into the holes. The testing room was dimly lit (33 lux) and contained large spatial cues. The chambers were wiped with a diluted solution of Bio-Clean (Stanbio Laboratories, Boerne, TX) and the task floors were rotated between subjects to discourage the use of the previous animal’s markings as a navigational strategy.
Med Associates computer software recorded the latency to complete each trial, novel hole entries, repeat entries and total entries, and organized the data by task and non-task. These parameters were used to calculate reference memory. Reference memory was defined as the ratio of the number of visits and revisits to the baited holes divided by the total number of visits to baited and nonbaited holes.
Rats were run in cohorts of 4–5 rats, resulting in a total number of five cohorts. During habituation, the animals were placed in the holeboard apparatus with all sixteen holes baited with one sucrose pellet. A habituation trial began with the first nosepoke and ended after all pellets were consumed or after 5 min elapsed. Trials continued until each rat consumed at least 15 pellets within a trial; this criterion was achieved in an average of four trials.
After 1–2 days of habituation, spatial learning training began. Four holes were baited in a consistent pattern throughout the training procedure (Figure 1). A trial began with the first nosepoke and ended after 5 min or after all 4 pellets were consumed. Each rat received 5 trials per day for a total of 8 days. Subjects remained in the behavioral room until all the rats in their cohort completed 5 trials. The average intertrial interval was 10 min.
Figure 1.

Schematic drawing of the holeboard. Solid circles correspond to baited holes and open circles to unbaited holes.
Subjects were divided into two groups (sodium azide n = 11 and control n = 12) matched on their pretreatment acquisition performance which was calculated by averaging their reference memory scores for the 8-day training period. On the 9th day, surgery and PCC injections were performed. On the day following surgery, they received one unbaited probe trial to test for memory retrieval. Thereafter, rats were retrained daily for 4 days to determine if the local C.O. inhibition of PCC was specific for memory retrieval or would interfere with task re-acquisition. Retraining was identical to training with the exception of the rats receiving 4 trials per day instead of 5 trials. Fewer trials and days were used during retraining to avoid ceiling effects on learning that could mask group differences.
Surgery
Subjects were anesthetized using a vaporizing machine (E-Z Anesthesia, Euthanex Corp, Palmer, PA). Animals were induced in a host cage filled with 97% breathing air and 3% isoflurane (Aerrane, Baxter Pharmaceutical Products, Deerfield, IL). After the rat was fully anesthetized (~ 3 min), they were placed on a surgical bed maintained at 37°C with their nose in a cone connected to the anesthesia machine. During the surgical procedure, animals were monitored for breathing and anesthesia state, and the concentration of isoflurane was adjusted accordingly.
Rats were placed in the flat skull position in a Stoelting stereotaxic frame. The head was shaved, disinfected with Nolvasan (Wyeth, Madison, NJ), and Lacrilube (Allergan, Irvine, CA) was applied over the eyes to prevent drying. A 2 cm mid-sagittal incision was made over the scalp and a retractor was inserted into the skin to expose the area. The periosteum was removed by scraping with cotton swabs. Bone wax (Ethicon, Somerville, NJ) was used to stop bleeding, if necessary.
After drilling 0.8 mm bilateral holes over the PCC, a 30-G dental needle was slowly inserted. The needle was attached by tubing to Hamilton syringes that were placed in a microinjection pump which allowed for precise and slow infusions. Four bilateral injections of sodium azide (3 M sodium azide dissolved in phosphate buffer, pH = 7.4, 0.5 μL per injection) were made, alternating left and right hemispheres.
A 3M concentration was selected since it was previously determined that a 3M intrastriatal injection of sodium azide significantly increased lactate and decreased ATP concentrations after 3 hours (Brouillet et al., 1994). Each injection was made over a 10 min period in the PCC within the following atlas coordinates (from Bregma, in mm): A/P −2.8 to −3.3; M/L ±0.2 to 0.4; D/V −1.2 to −1.3 (Paxinos and Watson, 1997). The needle was left in place for a few minutes after each injection and removed very slowly. Three control animals received phosphate buffer injections in the PCC (vehicle). The remaining control rats (n = 9) received the same procedure without drilling or injection (sham). All control animals were under anesthesia for the same amount of time as sodium azide-injected rats. As there was no difference between vehicle and sham control groups in reference memory performance in the probe (t(10) = 0.753, p = 0.469) and retraining trials (t(10) = 1.518, p = 0.160), these rats were treated as a single control group for subsequent analysis.
At the end of surgery, the skin was sutured using 9 mm wound clips (Reflex, CellPoint Scientific, Gaithersburg, MD) and treated with a topical antibiotic containing pramoxine hydrochloride (Neosporin). A single 5 mg/kg injection of carprofen (Rimadyl, Pfizer) was administered intramuscularly as a post-operative NSAID analgesic. The animal was removed from the anesthesia system and allowed to waken, 5–10 min on average. Upon recovery, the rat was moved to its home cage.
Brain Processing
At the end of the last day of retraining, brains were removed by rapid decapitation, frozen in isopentane, and stored in an ultra-low freezer at −40°C. Euthanasia was done rapidly by decapitation without the use of anesthetics to avoid their effect on brain enzymatic activity. Each brain was sectioned at 40 μm in a −17°C cryostat; sections were collected onto glass slides in two series and kept frozen at −40°C until processed. One series was used for histochemical analysis of C.O. activity. The adjacent series was Nissl-stained with cresyl violet for analysis of cell bodies and cell counting.
Quantitative C.O. histochemistry metabolic mapping (Gonzalez-Lima and Cada, 1998b) was used to analyze brain regional effects due to PCC injection. Experimental tissue slides were processed in batches for enzyme histochemistry and each batch included two standard slides. An additional 15 untreated Sprague-Dawley male rats were used to prepare brain homogenates for tissue standards for quantitative C.O. histochemistry. Standards slides contained tissue sections of varying thickness (10, 20, 40, 60, and 80 μm) with known C.O. activity and were cut from frozen rat brain homogenate. The C.O. activity of the standards was determined in a spectrophotometric experiment. The standards allowed for quantification and calibration among staining batches (Gonzalez-Lima and Cada, 1998b; Gonzalez-Lima and Jones, 1994).
Chemicals were purchased from Sigma. Quantitative C.O. histochemistry was performed as previously described (Gonzalez-Lima and Cada, 1998b; Gonzalez-Lima and Jones, 1994). Briefly, frozen sections were first incubated at 4°C for 5 min in a 0.1 M sodium phosphate buffer (PBS, pH 7.6) solution containing 0.5% glutaraldehyde and 9% sucrose, to facilitate adherence to the slides. This was followed by three baths of 9% sucrose in PBS (5 min each) at graded temperatures in order to bring the tissue from 4°C to room temperature and to remove red blood cells. Next, the slides were placed in a preincubation bath of 50 mM Tris buffer that included 0.55 g cobalt chloride, 200 g sucrose, 10 mL dimethyl sulfoxide, and 774 mL of 0.1 M hydrochloric acid for 10 min. Following a 5 min rinse in PBS, sections were incubated for 60 min at 37°C in an oxygen-saturated solution containing 0.35 g 3,3′-diaminobenzidine tetrahydrochloride, 52.5 mg cytochrome c, 14 mg catalase, 35 g sucrose, and 1.75 mL dimethyl sulfoxide in 700 mL PBS. The reaction was stopped by fixing the tissue in 9% sucrose PBS-buffered formalin (4%) for 30 min. Sections were dehydrated in baths of increasing concentrations of ethanol (30, 50, 70, 95, 95, 100, and 100%, 5 min each), cleared in xylene (3 times, 5 min each), and coverslipped with Permount.
C.O. activity was quantified by optical densitometry using Java software (Jandel Scientific) as detailed in Gonzalez-Lima and Cada (1998b). Briefly, a Kodak calibration strip with various gray levels of known optical densities (O.D.) was imaged and subsequent measurements were automatically reported to a spreadsheet in O.D. Tissue sections were placed on a DC-powered lightbox, captured with a Javelin analog CCD camera, and digitized with a Targa-M8 frame-grabber. The field of view was approximately 0.75 × 1.0 cm. For each area of interest, a researcher blind to group assignment measured O.D. bilaterally across three adjacent brain sections per subject. A standard brain atlas (Paxinos and Watson, 1997) and our C.O. histochemical brain atlas (Gonzalez-Lima and Cada, 1998a) was used to delineate the regions. For each brain region, the group mean was calculated averaging each median value per subject. The subareas of the PCC we sampled for C.O. activity are shown in Figure 2.
Figure 2.
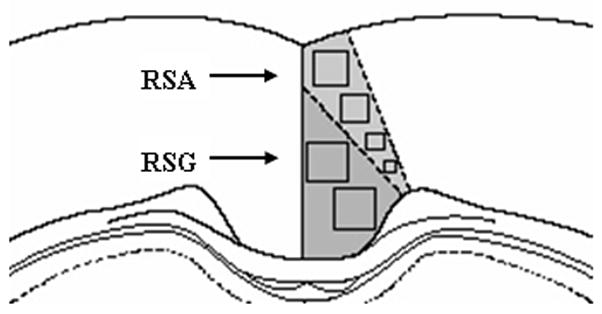
Coronal brain diagram around Bregma −2.8 mm indicating the sampled areas of the PCC measured in the C.O. activity image analysis.
The PCC was defined as the posterior half of the medial limbic cortex located above the caudal half of the corpus callosum, between Bregma −1.8 and −5.3 mm (Paxinos and Watson, 1997). The posterior cingulate includes Zilles’ retrosplenial agranular (RSA) and retrosplenial granular (RSG) cortices, rostral to the splenium of the corpus callosum (Bussey et al., 1997; Paxinos and Watson, 1997). Because we did not investigate the area caudal to the splenium of the corpus callosum, the term “posterior cingulate” cortex (PCC) was used to describe this cortical region rather than the name “retrosplenial” cortex. Anatomical connectivity studies show that the PCC is connected with motor, visual, and subicular cortices, and the anterior thalamus (Berthoz, 1997; Ohnishi et al., 2006), which were areas also examined for C.O. activity in this study.
For each standard, ten measurements per thickness per batch were measured by image analysis. Using the mean O.D. values and C.O. activity of the tissue standards, O.D. values for each area of interest were converted to units of C.O. activity. The linearity of each batch was verified by regression equations (r2 > .90) and calibration curves were calculated for each batch so that increased tissue thickness in the standards was linearly correlated with an increase in histochemically-revealed C.O. activity (Gonzalez-Lima and Cada, 1998b). This curve predicted C.O. activity from the O.D. measures of the experimental tissues within each batch. The use of individual batch standards and accompanying regression curves aided in the control of interbatch variability. C.O. activity is reported as μmol/min/g of tissue.
Sodium azide is a specific inhibitor of C.O., and since the volume of drug injected was small (0.5 μL) and not likely to diffuse substantially, C.O. histochemistry also estimated the location of injection. Metabolic inhibition might affect other brain regions that connect with the PCC. Thus, we examined the activity in areas with major efferent and/or afferent connections to the PCC. Of the regions examined, the anterior thalamic nuclei and the subiculum are the most heavily connected to the PCC (Finch et al., 1984; van Groen and Wyss, 1992; van Groen and Wyss, 2003). The PCC is a component of the Papez circuit, an anatomical network connecting many limbic structures that are also implicated in spatial behavior (Harker and Whishaw, 2002; Mello e Souza et al., 1999; van Groen et al., 2004). Therefore, we also measured activity in this circuit which involves the following regions (projection fibers are in brackets): cingulate cortex [cingulum] → entorhinal & perirhinal cortices [perforant pathway] → hippocampus proper [alveus] → subiculum [fornix] → mammillary bodies [mamillothalamic tract] → anterior thalamic nuclei [anterior thalamic radiation] → cingulate cortex.
The local neurotoxic effects of sodium azide were confirmed morphometrically by determining neuronal and glial densities in the PCC. Nissl staining was used to count cell bodies, obtain stereological estimates of PCC cell density, and determine if the changes in C.O. activity were accompanied by neurodegeneration. For each subject, we stained the alternate slide to the one that showed the lowest C.O. activity in the PCC. Frozen brain sections were delipidized in a series of baths: 95% ethanol, 70% ethanol, distilled water, and 0.05 M sodium acetate buffer (pH 4) (2.5 min each). This was followed by staining in 0.1% cresyl violet in sodium acetate buffer at 45°C for 4 min, differentiated in 70% and 95% ethanol, and dehydrated in sequential baths of 95%, 100% and 100% ethanol (5 min each). Slides were then cleared in two washes of xylene (5 min each) and coverslipped with Permount.
Cell counts were measured by an experimenter who was blind to subjects’ group assignment. Cell density (cells/volume) in the PCC was estimated from Nissl-stained sections of vehicle control (n = 3), sham control (n = 3), and sodium azide (n = 6) subjects using the optical dissector stereological method. The imaging setting consisted of a Labophot-2 binocular bright field microscope (Nikon Corporation, Tokyo, Japan) connected to a DVC-340 scan camera, DVCView 3.3 imaging software (DVC Company, Austin, TX), a Microcode II digital readout (Boeckeler Instruments, Inc., Tucson, AZ) and a PC computer. Cell density was estimated in three different PCC areas: 1) superficial layers II/III of the dorsal PCC (RSA), 2) deep layers V/VI of the dorsal PCC (RSA), and 3) layers III/IV of the ventral PCC (RSG). Differential cell count was obtained based on morphological criteria for brain cells. Neurons were identified by large ellipsoidal/pyramidal cell body, low nucleus/cytoplasm ratio, heterochromatic nucleus, and visible nucleolus; while glial cells appearance consisted of a small cell body, high nucleus/cytoplasm size ratio, and hyperchromatic nucleus. Cells (except those on top of the sections) were identified at 50X through the thickness of a section and were counted when they first came into focus within an unbiased counting frame (Harding et al., 1994). One sample per PCC area was obtained from each of three sections per subject within a randomly determined cerebral hemisphere. PCC cell density per area was calculated as Nvcells = ΣQ−/Σv(frame), where ΣQ− is the sum of cell counts per sample, and Σv(frame) is the area of the unbiased counting frame (0.0396 mm2, adjusted for 140X magnification) multiplied by the length of the analyzed region (d). The length was calculated as d = (No. of sections − 1) x No. of section series x section thickness.
We also tested whether neurotoxicity of C.O. inhibition from sodium azide may be mediated by an increase in oxidative stress by measuring lipid peroxidation in brain tissue after administration of sodium azide in vitro. Rat brain homogenates were exposed to different concentrations of sodium azide and lipid peroxidation was quantified using the thiobarbituric acid reactive substances (TBARS) assay as previously reported (Zhang et al., 2006). The range of sodium azide concentrations was based on the final concentration (100 mM) of 3M sodium azide after distribution in the PCC volume, which was estimated in one of the subjects from C.O. stained sections by means of the Cavalieri method (VPCC = 29.4 mm3). Adult Sprague-Dawley male rats (5 months old) were decapitated, and their brains were quickly extracted and homogenized in a glass-glass homogenizer (20–25 strokes). Homogenates were aliquoted in 1.5 mL Eppendorf tubes, slowly frozen in isopentane at −40°C and stored until further use. Freeze-thaw cycles were avoided. On the day of the experiment, homogenates were diluted at 12.5% (w/v) in PBS (pH 7.4 at room temperature), vortexed, and kept in ice. Homogenates were combined with sodium azide [0 mM (n = 8), 5 mM (n = 6), 25 mM (n = 2), 50 mM (n = 4) or 100 mM (n = 4)] and incubated at 37°C in the dark for 1 hr in closed 1.5 mL Eppendorf tubes. After incubation, the proteins were precipitated by the addition of 25% trichloroacetic acid (50/50, v/v). Samples were then vigorously vortexed and centrifuged at 10,000 rpm for 30 min at room temperature. Supernatants were combined with 1% thiobarbituric acid in 0.3% NaOH (50/50 v/v) and incubated in glass test tubes at 90°C for 40 min. Samples were then maintained at room temperature for 5 min and the absorbances were measured in a Shimadzu spectrophotometer at λ = 532 nm using tetraethoxypropane as an external standard (ε = 34.5 mM−1 min−1). The levels of lipid peroxidation were expressed as TBARS units (μmol/L).
Statistical Analysis
All statistical testing was carried out using the SPSS 11.5 software package (SPSS, Inc., Chicago, IL). Differences were considered statistically significant at the two-tailed p < .05 level for all tests. Repeated-measures analysis of variance (ANOVA) containing one within-subject variable (training day) and one between-subject variable (group) was used to verify learning performance before surgery. Matching subjects to treatment group based on their training performance prevented pre-treatment group differences that might influence their post-treatment performance.
The effects of PCC hypometabolism on reference memory during the probe task were analyzed using Student’s t test. Post-training scores were evaluated with repeated-measures ANOVA followed by pair-wise comparisons using simple and repeated contrasts. Student’s t tests assessed group differences in the following measures of holeboard performance: total nosepokes; initial nosepokes to baited and unbaited holes; repeat nosepokes to baited and unbaited holes; total nosepokes to baited and unbaited holes; inter-nosepoke interval between baited and unbaited holes; and latency to complete the probe.
Group differences in brain C.O. data and cell counts in the Nissl-stained slides were examined using Student’s t tests. Furthermore, within each group, the functional intercorrelations between the PCC and other brain regions were analyzed by computing inter-regional activity correlations (Nair and Gonzalez-Lima, 1999). Activity values for both control and sodium azide-treated groups were analyzed with Pearson’s correlation coefficients.
To investigate the possibility of network changes in addition to direct pairwise PCC intercorrelations we performed a multivariate stepwise discriminant analysis on each subgroup of regions, as used by McIntosh and Gonzalez-Lima (1994). This permits to identify regions which might differentiate the experimental and control groups based not on mean C.O. differences, but on network covariance differences. Since a discriminant function is a weighted linear combination of variables, a stepwise discriminant analysis can identify regions which are influential in combination, but which may not show individual mean differences (McIntosh & Gonzalez-Lima, 1994). The analyses were conducted using a forward stepwise procedure with the minimization of Wilk’s Lambda (a measure of residual variance) as the criteria for deriving the discriminant equation.
For the TBARS experiment, comparisons were made using one-way ANOVA followed by Dunnett’s test for multiple comparisons, which compares the mean of each experimental group with the mean of the control group.
RESULTS
Behavior
Overview
The behavioral results showed that although the groups did not differ in reference memory in the probe test, a significant within-subject difference was found. Only sodium azide-treated rats were impaired in their memory of the baited pattern in the probe trial as compared to their training scores before treatment. This was not due to a difference in general activity since the time to complete the task and the total number of nose pokes was not different in sodium azide-treated rats than in control rats.
Acquisition
The reference memory scores during acquisition are similar to what has been shown in another study using a similar training paradigm consisting of 4 trials per day for 7 days (Lannert and Hoyer, 1998). All rats significantly improved reference memory of the baited holeboard over the 8 days of training, F(7, 147) = 23.80, p < .001 (Figure 3). However, there was no effect of group (F(1, 21) = 0.00, p > .05) before treatment. This was expected since the animals were matched into treatment groups based on their similar pre-treatment training performance.
Figure 3.
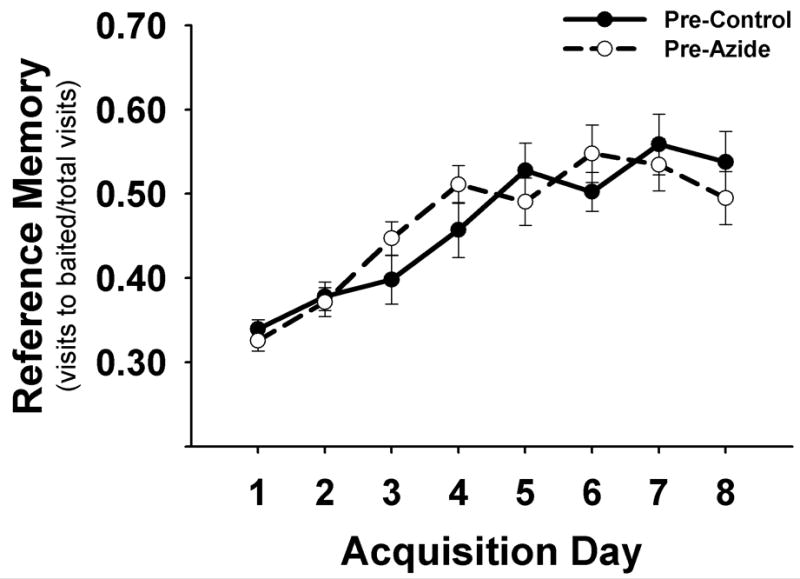
Pre-treatment acquisition performance in the holeboard. Means ± S.E.M. of reference memory scores of the baited pattern are plotted by day (5 trials/day). Rats showing similar learning performance were matched into two pre-treatment groups.
Memory probe
During the unbaited probe trial, the reference memory of sodium azide-injected animals was less than that of control animals (0.27 versus 0.37, respectively, Figure 4) (t(21) = −1.671, p > .05). After comparing each subject’s probe scores to their maximum training score, the mean group difference approached significance (F(1, 21) = 4.10, p = .056). When probe scores were compared to training averages within each group, only the rats treated with sodium azide showed significant impaired memory for the baited pattern in the probe trial (Figure 4; t(10) = 5.395, p < .001). The control group did not show a significant decline in memory (t(11) = 1.877, p > .05).
Figure 4.
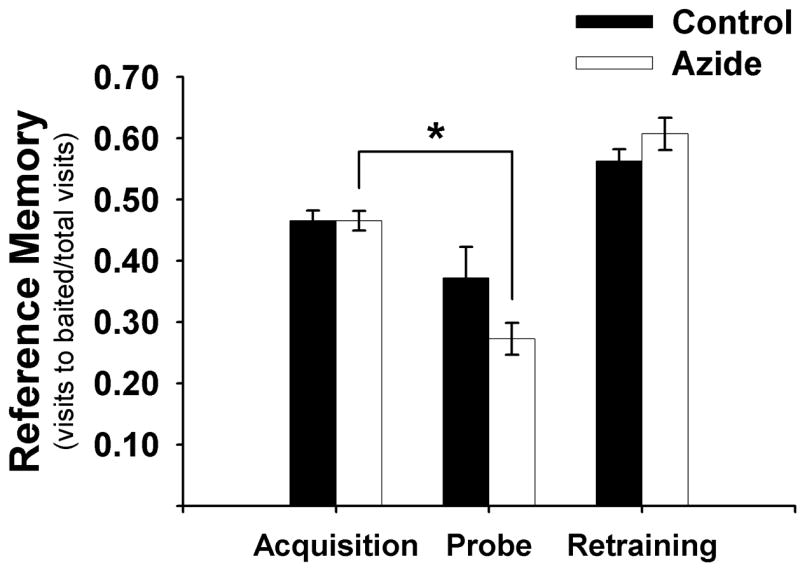
Mean ± S.E.M. of reference memory before (acquisition) and after (probe and retraining) sodium azide injection into the PCC. * significantly different from acquisition, p < .001.
Retraining
Analysis of retraining scores using simple contrasts did not reveal a significant group difference or interaction (group X day) when daily retraining averages were compared to probe scores (p > .05). In addition, there was not a significant group difference or interaction when comparing daily retraining averages to one another using repeated contrasts (p > .05). Therefore, sodium azide-treated rats only showed a memory retention deficit in the probe and were able to learn the task again to control levels with continued retraining over days 13–16 (Figure 4).
General performance
The sodium azide-treated rats spent a longer amount of time in between the initial pokes into previously-baited holes (t(21) = 2.51, p = .02) although the time to complete the probe trial was not different than control animals (t(21) = 2.027, p > .05). In addition, the total number of nosepokes in sodium azide-treated rats was not different than control rats (26 versus 23, respectively) (t(21) = 0.565, p > .05). There were no group differences in the probe trial in the number of novel or repeated nosepokes in baited or unbaited holes (p > .05).
Regional Cytochrome Oxidase Activity
Quantitative histochemistry revealed a specific local decrease in C.O. activity in the PCC in sodium azide-treated rats as compared to control animals. Other brain regions with purported anatomical connections to the PCC did not show group differences in average C.O. activity.
Optical density of the selected regions of interest was converted to enzyme activity units (Table 2). Out of 26 regions examined, the only area to show a group difference was the PCC (t(21) = 4.165, p < .001). Further examination of this area revealed that hypometabolism due to sodium azide in the PCC was localized to the dorsal aspect of the PCC (Figure 5; t(21) = 5.601, p < .001). The rats injected with 3M sodium azide showed a 23% decrease in C.O. activity in the dorsal PCC five days after administration. Significant decreases in activity were found from Bregma −2.3 to −4.2 mm, confirming that our injection was within the boundary of the PCC (p < .05).
Table 2.
Regional differences in cytochrome oxidase activity (Mean ± S.E.M. μmol/min/g tissue) in brains of rats injected with sodium azide in the PCC or sham control. AP refers to the antero-posterior coordinates from Bregma. The number of brains analyzed for each region is given as (n). Some samples had a smaller n to exclude brains with section artifacts in the region.
| REGION OF INTEREST | CONTROL | AZIDE | ||
|---|---|---|---|---|
| abbreviation | Bregma level (mm) | Mean ± SEM (n) | Mean ± SEM (n) | |
| CINGULATE REGIONS | ||||
| posterior cingulate | PCC | −1.80 to −5.30 | 270 ± 6 (12) | 233 ± 7 (11) * |
| anterior cingulate | ACC | 1.60 to −1.40 | 251 ± 3 (10) | 260 ± 5 (9) |
| cingulum bundle | cb | −1.80 to −5.30 | 33 ± 3 (12) | 29 ± 3 (11) |
| THALAMIC REGIONS | ||||
| anterior dorsal | AD | −1.80 | 453 ± 16 (11) | 451 ± 19 (11) |
| anterior ventral | AV | −1.80 | 349 ± 5 (11) | 338 ± 11 (11) |
| anterior medial | AM | −1.80 | 275 ± 5 (11) | 262 ± 9 (11) |
| reticular | Rt | −1.80 | 299 ± 5 (11) | 303 ± 8 (11) |
| rhomboid/reuniens | Rh/Re | −1.80 | 250 ± 10 (11) | 246 ± 6 (11) |
| lateral dorsal | LD | −2.30 | 298 ± 12 (12) | 292 ± 12 (11) |
| HIPPOCAMPAL FORMATION | ||||
| anterior/dorsal | ||||
| CA1 | CA1d | −3.80 | 236 ± 7 (12) | 227 ± 7 (11) |
| CA3 | CA3d | −3.80 | 221 ± 6 (12) | 216 ± 8 (11) |
| dentate gyrus | DGd | −3.80 | 306 ± 7 (12) | 292 ± 11 (11) |
| posterior/ventral | ||||
| CA1 | CA1v | −5.30 | 253 ± 8 (12) | 252 ± 8 (11) |
| CA3 | CA3v | −5.30 | 277 ± 10 (12) | 273 ± 11 (10) |
| dentate gyrus | DGv | −5.30 | 226 ± 8 (12) | 230 ± 7 (11) |
| subiculum | SUB | −5.30 | 226 ± 13 (11) | 241 ± 10 (11) |
| mammillary bodies | MB | −4.80 | 356 ± 15 (11) | 350 ± 13 (9) |
| entorhinal | Ent | −4.80 | 148 ± 12 (12) | 142 ± 11 (11) |
| perirhinal | PRh | −4.80 | 175 ± 12 (12) | 165 ± 12 (11) |
| OTHER | ||||
| secondary motor cortex | M2 | −0.26 | 276 ± 8 (10) | 279 ± 8 (9) |
| secondary visual cortex | ||||
| medial area | V2M | −5.30 | 253 ± 9 (12) | 251 ± 8 (11) |
| lateral area | V2L | −5.30 | 260 ± 8 (12) | 254 ± 5 (11) |
| caudate putamen | CPu | −1.80 | 295 ± 6 (12) | 280 ± 7 (11) |
| zona incerta | ZI | −3.80 | 219 ± 12 (12) | 207 ± 14 (11) |
| periaqueductal gray | PAG | −5.30 | 269 ± 12 (10) | 271 ± 10 (10) |
| raphe nuclei | RLi | −5.30 | 292 ± 11 (10) | 303 ± 8 (10) |
signficantly different from control (p < 0.001 )
Figure 5.
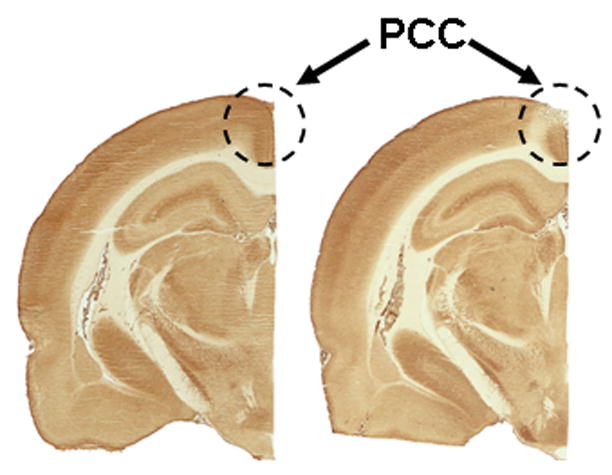
Micrographs of cytochrome oxidase-stained sections around Bregma −2.80 mm. Hemisections from two different animals are shown side by side to compare metabolic effects after vehicle (left section) or sodium azide (right section) PCC injections.
Interregional PCC Correlations and Network Changes
Interregional pair-wise correlations suggested that regions were functionally dissociated from the PCC in the azide-treated group. Covariance analysis of C.O. activity revealed significant correlations between the PCC and several other brain regions in the control group that were absent in the sodium azide-treated group. Correlations between C.O. activity in the PCC and other brain regions were differently observed in the two groups (Table 3). In the control group, there were 9 significant correlations (p < .05) in C.O. activity between the PCC and other regions of interest. An interesting finding were the large number of positive correlations (6) between the PCC and hippocampal areas (DGv, CA1v, CA3v, SUB, PRh and Ent). Significant positive correlations were also found with the CPu and secondary visual areas (V2M, V2L).
Table 3.
Significant Pearson correlation coefficients (r) between C.O. activity in the PCC and other brain regions for each group.
| PCC–Interregional Correlations
| ||
|---|---|---|
| CONTROL | AZIDE | |
| CA3d | 0.19 | −0.66 * |
| CA1v | 0.60 * | −0.16 |
| CA3v | 0.71 * | 0.02 |
| SUB | 0.67 * | 0.28 |
| PRh | 0.63 * | 0.01 |
| Ent | 0.64 * | 0.09 |
| DGv | 0.86 * | 0.49 |
| CPu | 0.64 * | −0.05 |
| V2M | 0.77 * | 0.39 |
| V2L | 0.73 * | 0.04 |
significantly different from zero (p < 0.05)
In the sodium azide-treated group, there was only one significant correlation—one negative correlation between the PCC and CA3d. Positive correlations between C.O. activity in the PCC and other regions suggested functional network coupling in the control group that was absent in the azide-treated group.
Multivariate network changes between the control and azide groups were demonstrated using discriminant analysis. Using the stepwise method, one function comprising a network of four regions, namely RSA, CA3d, SUB, and AV, accounted for 100% of the variance between groups (Eigenvalue = 6.28, Chi-square = 23.828, p < .0001). The stepwise statistics and discrimination coefficients for each region in this network are shown in Table 4.
Table 4.
Discriminant analysis stepwise statistics.
| Step | Region | Wilks’ Lambda | Standardized canonical coefficient |
|---|---|---|---|
| 1 | RSA | 0.459 | 1.921 |
| 2 | CA3d | 0.308 | 1.217 |
| 3 | SUB | 0.209 | −1.150 |
| 4 | AV | 0.137 | 0.875 |
PCC Cell Counts
There were no overall differences in neuronal loss or gliosis in the RSG or RSA between the sham and vehicle-treated control subjects and no differences were detected when changes were compared by layer of the PCC regions (Table 5).Therefore, since there were no differences in reference memory between vehicle and sham groups, nor were there differences in C.O. activity or cell counts, these rats were treated as a single control group for all analyses.
Table 5.
Cell counts in vehicle-treated rats and sham controls.
| RSG | Superficial RSA | Intermediate RSA | Deep RSA | |||||
|---|---|---|---|---|---|---|---|---|
| Neuron | Glia | Neuron | Glia | Neuron | Glia | Neuron | Glia | |
| Vehicle | 15.29 | 8.79 | 25.78 | 4.37 | 15.87 | 7.79 | 15.75 | 7.89 |
| Sham | 14.80 | 11.05 | 25.95 | 6.41 | 15.19 | 12.31 | 16.67 | 9.46 |
The neurotoxic effect of sodium azide in PCC was evidenced by dramatic changes in both neuronal and glial cell density. Sodium azide induced significant decreases in neuronal density and increases in glial cell density in all the analyzed PCC samples as compared to control animals (Figure 6, p < .05). Since the superficial layers of the dorsal PCC (RSA superficial) had relatively higher ratios of neuronal to glial cells, different PCC subregions were analyzed separately, but they all showed the same general group effect. Furthermore, the majority of the remaining PCC neurons in sodium azide-treated subjects showed morphological signs of necrotic degeneration such as cell edema, vacuolization, and karyorrhexis (Figure 7).
Figure 6.
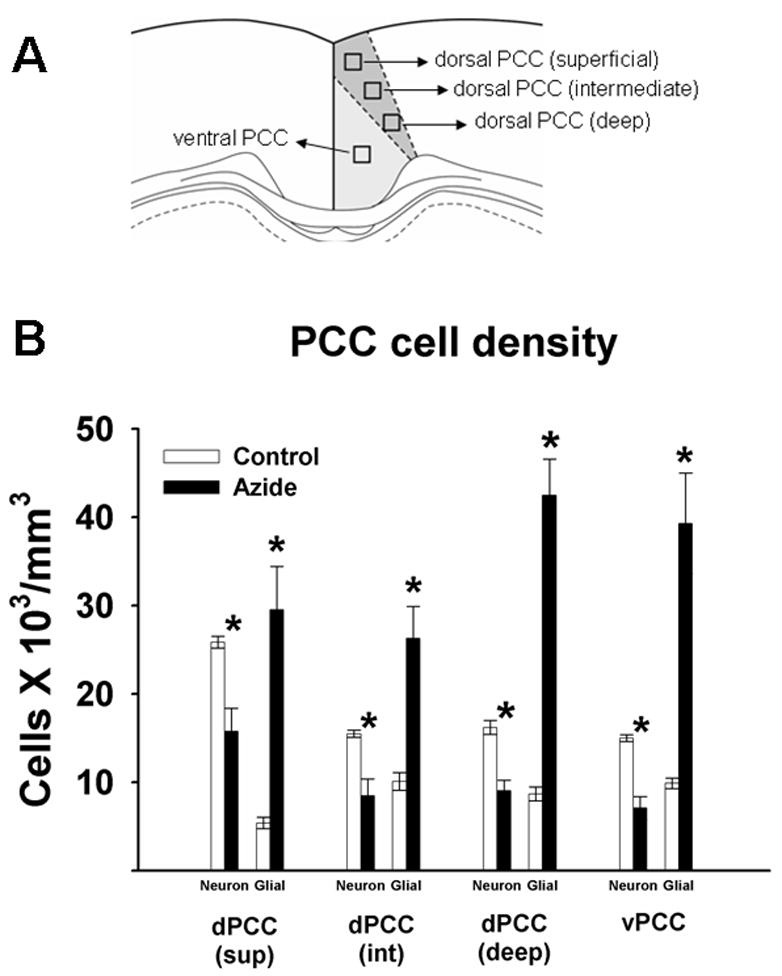
A. PCC regional and laminar breakdown corresponding to the cell counts samples (Bregma −2.80 mm). B. Neuronal and glial cell densities in the PCC in sodium azide-injected rats and control rats. *significantly different from control values, p < .05.
Figure 7.
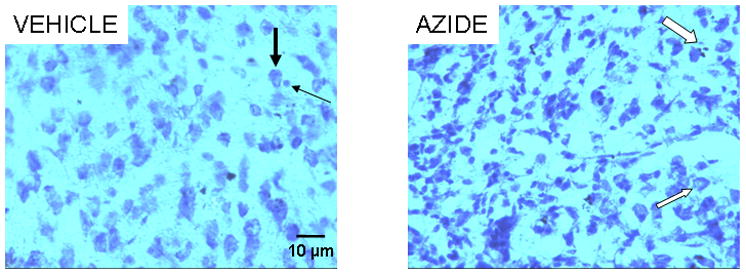
Cresyl violet-stained cells in the dorsal PCC from rats with vehicle (left panel) or sodium azide injections (right panel). Neurons with ovoid-shaped, large, homogeneous and lightly-stained cell bodies (thick black arrow) predominate over the small round-shaped glia (thin black arrow) in vehicle-treated subjects (left). Sodium azide (right) significantly increased gliosis and neuronal loss. Signs of degeneration such as karyorrhexis (thick white arrow) and vacuolization (thin white arrow) are observed in the remaining neurons. Scale bar = 10 μm.
Although the percent decrease in the mean neuronal density in the RSA was similar in the three layers (39–44%) (Figure 6), the extent of neuronal loss in different layers varied between subjects, a phenomenon that has been previously observed in patients with AD. Vogt et al described a neuropathological subtyping based on the laminar distribution of neuronal losses in the PCC of patients with AD (Vogt et al., 1990). Using a similar approach, it can be noted that in the present study (Table 6), neuronal loss was less than 40 per cent in any of the three layers (Vogt’s class 1) in three subjects; it predominated in the intermediate layers (Vogt’s class 3) in two subjects, and one subject presented generalized neuronal loss that affected the overall layered architecture (Vogt’s class 5). Thus, this model emulates the heterogeneous distribution of neurodegeneration in different layers of the PCC observed in patients with AD, which could have its onset during the stage of MCI.
Table 6.
Percent decrease in neuronal density in the RSA after sodium azide treatment.
| Subject | RSA layer | Vogt’s class | ||
|---|---|---|---|---|
| Superficial | Intermediate | Deep | ||
| 1 | 20.4 | 42.4 | 43.9 | 1 |
| 2 | 1.1 | 13.4 | 9 | 1 |
| 3 | 53.3 | 75 | 43 | 3 |
| 4 | 38.8 | 9.8 | 52.5 | 1 |
| 5 | 51.1 | 79.6 | 53.7 | 3 |
| 6 | 69.9 | 50 | 63.5 | 5 |
Lipid Peroxidation
Sodium azide potently increased brain lipid peroxidation in a concentration-dependent fashion (Figure 8, F(4, 23) = 12.8, p < .001). Compared to control, the lowest sodium azide concentration tested (5 mM), produced a two-fold increase in lipid peroxidation. A seven-fold increase in lipid peroxidation was produced by the sodium azide concentration estimated in the PCC injection in the in vivo experiment (100 mM).
Figure 8.
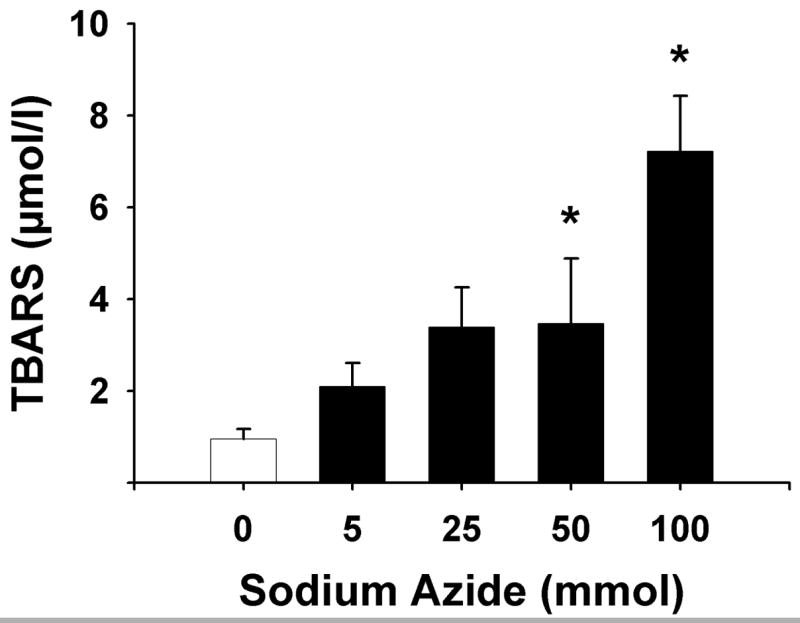
Sodium azide treatment increases lipid peroxidation. Data represent the Mean ± S.E.M. of TBARS reaction product in rat brain homogenate. *significantly different from untreated control, p < .001, Dunnett’s test.
DISCUSSION
We addressed the hypothesis that isolated PCC hypometabolism in rats may produce a mild impairment in a spatial memory task. The behavioral findings support this hypothesis since isolated hypometabolism in the PCC due to local sodium azide treatment produced memory impairment in a holeboard task that required using spatial cues. The present study supports previous human and animal studies showing that the PCC is involved in spatial memory, and it served to verify that a local injection of sodium azide in this region induces local inhibition of C.O. activity and neurotoxicity.
The behavioral deficits in the probe trial are unlikely due to a change in response to sucrose reward. Rats with excitotoxic lesions in the PCC were not different than control animals in a sucrose consumption test (Bussey et al., 1996). Also, previous studies have shown that rats treated systemically with sodium azide have no change in sensory or motor function, or in exploratory activity in an open field chamber (Bennett and Rose, 1992; Szabados et al., 2004). However, they did have impaired memory for spatial tasks such as the eight-arm radial maze (Bennett and Rose, 1992), Morris water maze (Bennett et al., 1992), and holeboard task (Callaway et al., 2002). The present study did not find a difference in the total number of nosepokes between sodium azide-treated and control rats, or in the ability of the rats to relearn the task with subsequent trials, which suggests that the impaired behavior was due to a deficit in memory retrieval for the baited pattern and not due to differences in motor performance or general learning ability.
The fact that sodium azide-treated rats were not different from control rats during the retraining phase in the holeboard maze may be due to the availability of a competing learning process not dependent on the PCC. The baited pattern could be learned either by forming and accessing a cognitive map, or by response/procedural learning (Whishaw et al., 2001). In the present study, subjects were placed in the same corner of the holeboard for every trial and thus, response/procedural learning might also yield successful results. Brain systems consisting of different structures are involved in processes for similar types of learning and memory (“parallel processing”) (Lee and Kesner, 2003; McIntyre et al., 2003; Mizumori et al., 2004). Which neural system has more relative influence on behavioral expression may vary depending upon a number of factors such as age, experience, or incentive. If the PCC is damaged, the tendency to use cognitive maps may be impaired and the competing response/procedural learning may proceed. Furthermore, C.O. inhibition in the PCC was not complete. Similar performance in the two groups during retraining may have resulted from the spared ventral portion of the PCC. Finally, the most anterior and posterior aspects of the PCC were also spared from the inhibitory metabolic effects of sodium azide, and some components of the stored information may have remained undisturbed. It is possible that the metabolically normal tissue was sufficient for retraining in the maze.
Quantitative C.O. histochemistry verified that the hypometabolism was localized within the PCC and that significant hypometabolism of connecting structures did not occur by 5 days after sodium azide administration. The observation of hypometabolism in the dorsal, but not ventral aspect of the PCC can be explained by the depth of injection during surgery. We wanted the injection to be specific to the PCC and avoid damage to the underlying cingulum fiber bundle. There is debate in the literature that previous findings of behavioral impairment from PCC damage can be, in part, due to inadvertent damage to this fiber tract (Neave et al., 1994; Warburton et al., 1998). Having C.O. inhibition limited to the more superficial PCC is also consistent with our previous findings in AD brains where the most pronounced C.O. inhibition is localized to the superficial layers of the PCC (Valla et al., 2001). Furthermore, the dorsal PCC showed a mean 23% inhibition of C.O. activity in the rat model that is similar to the mean 28% inhibition of C.O. activity in PCC found in AD patients (Valla et al., 2001).
Memory of the baited holeboard may be associated with network functional changes in neuronal metabolism, as suggested by the significant positive inter-correlations of metabolic activity among the PCC and other limbic areas. Interestingly, most of the PCC correlations in the control animals were with hippocampal and limbic cortex components of the Papez circuit. This circuit was originally hypothesized to underlie emotion (Papez, 1937). It has been modified to include memory because lesions of these structures in humans and rodents often lead to disruption in learning and memory (Harker and Whishaw, 2002; Holdstock et al., 2000; Valenstein et al., 1987; van Groen et al., 2004). The inter-regional correlations in control rats support the idea that impairment of the functional relationships in the Papez circuit can impair spatial memory. PCC hypometabolism by sodium azide eliminated all the significant correlations between the PCC and other brain regions that were found in the control group, especially the strong positive correlations between the PCC and hippocampal regions. This network dissociation found in the rat model is remarkably similar to the disruption in the functional correlations of a network of brain regions involving the PCC and interconnected medial temporal regions in MCI patients at risk for AD (Sorg et al., 2007). Recent ideas about the progression from MCI to AD suggest that an early PCC hypometabolism may lead to a functional disconnection of a resting-state network in individuals at risk for AD (Sorg et al., 2007).
A potential caveat of the model is whether sodium azide leads to a metabolic lesion as opposed to simply cell loss. The action of local azide administration as used in our model appears to be mediated by an initial metabolic lesion, not just simply a nonspecific cell loss. At the 3M concentration used, sodium azide has been similarly injected locally into the striatum in rats (Brouillet et al., 1994). The effect of this injection was monitored using MRI signal intensities to evaluate focal lactate increase and neuronal loss in vivo. Increased lactate concentration could be detected in vivo by 3 hours post-injection, a time at which no neuronal loss is detectable. This ruled out the possibility that the metabolic alteration produced by local injection of this concentration of sodium azide is merely secondary to cell loss. The azide metabolic lesion leads to neuron loss and glial cell increases, at least after 5 days post-injection, the time when we counted the cells. This metabolic causation would not be the same in another model that uses a nonspecific chemical or surgical lesion to just kill the cells locally.
While it is possible to damage the PCC by many other means, such as aspiration or non-specific toxins, such other lesions would not help to model the sort of metabolic lesion found in AD linked to C.O. inhibition. The C.O. inhibitor sodium azide at the dose used in this study (3M) was sufficient to produce a mild memory impairment that was accompanied by cellular changes such as partial decreases in C.O. activity and neuron numbers in PCC. These metabolic alterations may have contributed to the neurodegenerative effects we observed in the Nissl-stained tissue; specifically, there were significant decreases in neurons and increases in glial cells. Loss of somata of neurons in the PCC would affect their axons in the cingulum bundle that project to the hippocampal regions in Papez circuit. This neurodegenerative alteration in the rat model is consistent with a disruption of the integrity of PCC axons traveling in the cingulum bundle between PCC and hippocampal regions. This feature of the rat model is also compatible with recent findings in patients with amnestic MCI and AD, as determined by diffusion tensor imaging of the cingulum fibers (Zhang et al., 2007).
In the present azide model the PCC metabolic lesion is not secondary to hippocampal atrophy. However, it is possible that in AD a cingulum disruption affecting the PCC may result from hippocampal atrophy. Although the cingulum carries reciprocal fibers between the PCC and parahippocampal areas (Schmahmann & Pandya, 2006), two facts argue against the possibility that hippocampal atrophy causes PCC hypometabolism in AD. First, PCC hypometabolism precedes hippocampal atrophy and is not the result of tissue atrophy (Chetelat et al., 2008; Minoshima et al., 1997; Mosconi et al., 2006; Samuraki et al., 2007). Second, the degree of hypometabolism is significantly greater than the degree of atrophy in the PCC (Chetelat et al., 2008). This is not the case regarding the hippocampus. This discrepancy in AD indicates a metabolic alteration in the PCC that is not explained by tissue loss, which may explain the hypometabolism in the hippocampus.
We also showed in vitro that lipid peroxidation, a marker of oxidative stress, was increased in a dose-dependent manner in sodium azide-treated brain homogenates. Compared to control samples, a seven-fold increase in lipid peroxidation was produced by the concentration of sodium azide used in the in vivo experiment. Therefore, inhibition in C.O. activity caused increased lipid peroxidation, in agreement with increasing evidence implicating C.O. inhibition and oxidative damage as neurodegenerative mechanisms in MCI and AD (Bennett et al.,1992; Benzi and Moretti, 1995; Blass et al., 2002; Mielke and Lyketsos, 2006; Reddy and Beal, 2005; Valla et al., 2001; Valla et al., 2006).
The PCC may be a metabolically vulnerable brain region. The PCC has a high baseline metabolic activity in rats (Gonzalez-Lima and Cada, 1998) as well as healthy human subjects (Minoshima et al., 1997) and it contains numerous glutaminergic excitatory synapses. To fulfill a high energy demand, C.O. expression is upregulated (Wong-Riley et al., 1998). A high energy demand will render the PCC vulnerable to C.O. inhibition as well as other metabolic insults such as vascular problems (de la Torre, 2006) or neurotoxins (Li et al., 2002). For example, an intraperitoneal injection of an NMDA antagonist produces region-specific neurotoxicity, and the PCC is the most affected region (Li et al., 2002). Likewise, the PCC would be one of the first areas affected by C.O. inhibition that compromises energy generation and eventually neuron survival. While in vivo the PCC shows high metabolic activity in normal subjects, early-stage AD patients display a 21–22% decrease in glucose metabolism that is significantly greater than that found in other cortical regions (Minoshima et al., 1997). One possibility may be that the underlying cause for this early PCC hypometabolism may be the amyloid plaques or neurofibrillary tangles seen in AD brains postmortem. However, recent in vivo neuroimaging studies have found high amyloid load detected by [11C]PIB-PET in AD in the face of spared glucose metabolism, suggesting that amyloid plaque formation may not be directly responsible for neuronal dysfunction in this disease (Edison et al., 2007). The metabolic lesion appears to be happening earlier than post-mortem AD histopathological signs such as amyloid deposition and neurofibrillary tangles (Valla et al., 2001; Blass et al., 2002). However, histopathology and other insults affecting metabolic energy and producing oxidative stress in any vulnerable region are likely to secondarily aggravate a primary C.O. decrement in that region (Valla et al., 2001).
In summary, C.O. inhibition in the PCC resulted in neurotoxicity, reduced inter-regional correlations in brain activity, and memory impairment in a spatial holeboard task. This model of hypometabolism in the rat PCC resembles behavioral, physiological, histochemical, morphological, and biochemical changes similar to those seen in amnestic MCI and AD patients. The PCC displays the largest decrement in glucose utilization in early-stage AD (Minoshima et al., 1997), and histochemical analysis of C.O. activity in the PCC of AD patients reveals a greater decrease in the energy-demanding superficial layers that contain synaptic neuropil (Valla et al., 2001). PCC hypometabolism appears before the onset of memory deficits in persons at genetic risk for AD, but who are not yet cognitively impaired (Reiman et al., 1996), as well as in subjects with MCI who later develop AD (Mosconi, 2005). Metabolic reductions in the PCC and medial temporal cortices (Mosconi, 2005) and reduced integrity in cingulum fibers connecting these regions (Zhang et al., 2007) are currently the most reliable neuroimaging indicators of early-stage AD. In human fMRI, there is strong functional connectivity between the PCC and hippocampal regions in healthy controls that is absent in patients with amnestic MCI, at high risk for AD (Sorg et al., 2007). C.O. inhibition in the rat PCC changed inter-regional correlations with hippocampal regions and increased brain lipid peroxidation, which resulted in memory impairment in a spatial holeboard task. AD patients show disrupted correlations in metabolism between cortical regions (Horwitz et al., 1987) and increased brain lipid peroxidation (Mielke and Lyketsos, 2006). Therefore, sodium azide-treated PCC may be a useful animal model for characterizing behavior and brain changes associated with amnestic mild cognitive impairment, and for testing neuroprotective and metabolic enhancing strategies to improve memory (Messier, 2004; Riha et al., 2005; Gold, 2006; Wrubel et al., 2007). This novel animal model presenting a specific PCC metabolic lesion, similar to that in patients with MCI who later convert to early-stage AD, may provide opportunities to characterize neurobehavioral alterations and to test therapeutics to reverse memory impairment.
Acknowledgments
This research was submitted by Dr. Penny Riha in partial fulfillment of the requirements for the Ph.D. degree at the University of Texas at Austin. We gratefully acknowledge the helpful comments and review of this work by the members of the dissertation committee: Drs. Theresa Jones, Michelle Lane, Timothy Schallert, and David Tucker. We also thank Alison Crane for her skillful assistance in the surgical procedures and Christian Balderrama for his help with tissue processing and quantitative imaging. This research was funded by NIH grants R01 MH076847 and T32 MH65728 to FGL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alsop DC, Detre JA, Grossman M. Assessment of cerebral blood flow in Alzheimer’s disease by spin-labeled magnetic resonance imaging. Annals of Neurology. 2000;47:93–100. [PubMed] [Google Scholar]
- Bennett MC, Diamond DM, Stryker SL, Parks JK, Parker WD., Jr Cytochrome oxidase inhibition: a novel animal model of Alzheimer’s disease. Journal of Geriatric Psychiatry and Neurology. 1992;5:93–101. doi: 10.1177/002383099200500206. [DOI] [PubMed] [Google Scholar]
- Bennett MC, Rose GM. Chronic sodium azide treatment impairs learning of the Morris water maze task. Behavioral and Neural Biology. 1992;58:72–75. doi: 10.1016/0163-1047(92)90967-9. [DOI] [PubMed] [Google Scholar]
- Benzi G, Moretti A. Age- and peroxidative stress-related modifications of the cerebral enzymatic activities linked to mitochondria and the glutathione system. Free Radical Biology and Medicine. 1995;19:77–101. doi: 10.1016/0891-5849(94)00244-e. [DOI] [PubMed] [Google Scholar]
- Berthoz A. Parietal and hippocampal contribution to topokinetic and topographic memory. Philosophical transactions of the Royal Society of London. 1997;352:1437–1448. doi: 10.1098/rstb.1997.0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass JP, Gibson GE, Hoyer S. The role of the metabolic lesion in Alzheimer’s disease. Journal of Alzheimer’s Disease. 2002;4:225–232. doi: 10.3233/jad-2002-4312. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Hyman BT, Jenkins BG, Henshaw DR, Schulz JB, Sodhi P, Rosen BR, Beal MF. Systemic or local administration of azide produces striatal lesions by an energy impairment-induced excitotoxic mechanism. Experimental Neurology. 1994;129:175–182. doi: 10.1006/exnr.1994.1159. [DOI] [PubMed] [Google Scholar]
- Bussey TJ, Muir JL, Everitt BJ, Robbins TW. Dissociable effects of anterior and posterior cingulate cortex lesions on the acquisition of a conditional visual discrimination: Facilitation of early learning vs. impairment of late learning. Behavioural Brain Research. 1996;82:45–56. doi: 10.1016/s0166-4328(97)81107-2. [DOI] [PubMed] [Google Scholar]
- Bussey TJ, Muir JL, Everitt BJ, Robbins TW. Triple dissociation of anterior cingulate, posterior cingulate, and medial frontal cortices on visual discrimination tasks using a touchscreen testing procedure for the rat. Behavioral Neuroscience. 1997;111:920–936. doi: 10.1037//0735-7044.111.5.920. [DOI] [PubMed] [Google Scholar]
- Cada A, Gonzalez-Lima F, Rose GM, Bennett MC. Regional brain effects of sodium azide treatment on cytochrome oxidase activity: a quantitative histochemical study. Metabolic Brain Disease. 1995;10:303–320. doi: 10.1007/BF02109361. [DOI] [PubMed] [Google Scholar]
- Callaway NL, Riha PD, Wrubel KM, McCollum D, Gonzalez-Lima F. Methylene blue restores spatial memory retention impaired by an inhibitor of cytochrome oxidase in rats. Neuroscience Letters. 2002;332:83–86. doi: 10.1016/s0304-3940(02)00827-3. [DOI] [PubMed] [Google Scholar]
- Cammalleri R, Gangitano M, D’Amelio M, Raieli V, Raimondo D, Camarda R. Transient topographical amnesia and cingulate cortex damage: a case report. Neuropsychologia. 1996;34:321–326. doi: 10.1016/0028-3932(95)00108-5. [DOI] [PubMed] [Google Scholar]
- Chen LL, Lin LH, Green EJ, Barnes CA, McNaughton BL. Head-direction cells in the rat posterior cortex. I. Anatomical distribution and behavioral modulation. Experimental Brain Research. 1994;101:8–23. doi: 10.1007/BF00243212. [DOI] [PubMed] [Google Scholar]
- Chetelat G, Desgranges B, Landeau B, Mezenge F, Poline JB, de la Sayette V, Viader F, Eustache F, Baron JC. Direct voxel-based comparison between grey matter hypometabolism and atrophy in Alzheimer’s disease. Brain. 2008;131:60–71. doi: 10.1093/brain/awm288. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. How do heart disease and stroke become risk factors for Alzheimer’s disease? Neurological Research. 2006;28:637–644. doi: 10.1179/016164106X130362. [DOI] [PubMed] [Google Scholar]
- Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Finch DM, Derian EL, Babb TL. Afferent fibers to rat cingulate cortex. Experimental Neurology. 1984;83:468–485. doi: 10.1016/0014-4886(84)90116-x. [DOI] [PubMed] [Google Scholar]
- Gabriel M. Discriminative avoidance learning: A model system. In: Vogt BA, Gabriel M, editors. Neurobiology of cingulate cortex and limbic thalamus. Boston: Birkhauser; 1993. pp. 478–523. [Google Scholar]
- Gold PE. Glucose and age-related changes in memory. Neurobiology of Aging. 2005;26:60–64. doi: 10.1016/j.neurobiolaging.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Lima F, Cada A. Cytochrome oxidase atlas of rat brain. In: Gonzalez-Lima F, editor. Cytochrome oxidase in neuronal metabolism and Alzheimer’s disease. New York: Plenum Press; 1998. pp. 263–280. [Google Scholar]
- Gonzalez-Lima F, Cada A. Quantitative histochemistry of cytochrome oxidase activity. In: Gonzalez-Lima F, editor. Cytochrome oxidase in neuronal metabolism and Alzheimer’s disease. New York: Plenum Press; 1998. pp. 55–90. [Google Scholar]
- Gonzalez-Lima F, Jones D. Quantitative mapping of cytochrome oxidase activity in the central auditory system of the gerbil: a study with calibrated activity standards and metal-intensified histochemistry. Brain Research. 1994;660:34–49. doi: 10.1016/0006-8993(94)90836-2. [DOI] [PubMed] [Google Scholar]
- Harding AJ, Halliday GM, Cullen K. Practical considerations for the use of the optical disector in estimating neuronal number. Journal of Neuroscience Methods. 1994;51:83–89. doi: 10.1016/0165-0270(94)90028-0. [DOI] [PubMed] [Google Scholar]
- Harker KT, Whishaw IQ. Impaired spatial performance in rats with retrosplenial Lesions: Importance of the spatial problem and the rat strain in identifying lesion effects in a swimming pool. Journal of Neuroscience. 2002;22:1155–1164. doi: 10.1523/JNEUROSCI.22-03-01155.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirono N, Mori E, Ishii K, Ikejiri Y, Imamura T, Shimomura T, Hashimoto M, Yamashita H, Sasaki M. Hypofunction in the posterior cingulate gyrus correlates with disorientation for time and place in Alzheimer’s disease. Journal Of Neurology, Neurosurgery, and Psychiatry. 1998;64:552–554. doi: 10.1136/jnnp.64.4.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdstock JS, Mayes AR, Cezayirli E, Isaac CL, Aggleton JP, Roberts N. A comparison of egocentric and allocentric spatial memory in a patient with selective hippocampal damage. Neuropsychologia. 2000;38:410–425. doi: 10.1016/s0028-3932(99)00099-8. [DOI] [PubMed] [Google Scholar]
- Horwitz B, Grady CL, Schlageter NL, Duara R, Rapoport SI. Intercorrelations of regional cerebral glucose metabolic rates in Alzheimer’s disease. Brain Research. 1987;407:294–306. doi: 10.1016/0006-8993(87)91107-3. [DOI] [PubMed] [Google Scholar]
- Ishii K, Sasaki M, Yamaji S, Sakamoto S, Kitagaki H, Mori E. Demonstration of decreased posterior cingulate perfusion in mild Alzheimer’s disease by means of H215O positron emission tomography. European Journal of Nuclear Medicine. 1997;24:670–673. doi: 10.1007/BF00841407. [DOI] [PubMed] [Google Scholar]
- Lannert H, Hoyer S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behavioral Neuroscience. 1998;112:1199–1208. doi: 10.1037//0735-7044.112.5.1199. [DOI] [PubMed] [Google Scholar]
- Lee I, Kesner RP. Time-dependent relationship between the dorsal hippocampus and the prefrontal cortex in spatial memory. Journal of Neuroscience. 2003;23:1517–1523. doi: 10.1523/JNEUROSCI.23-04-01517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Clark S, Lewis DV, Wilson WA. NMDA receptor antagonists disinhibit rat posterior cingulate and retrosplenial cortices: A potential mechanism of neurotoxicity. Journal of Neuroscience. 2002;22:3070–3080. doi: 10.1523/JNEUROSCI.22-08-03070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire EA. Hippocampal involvement in human topographical memory: evidence from functional imaging. Philosophical transactions of the Royal Society of London. 1997;352:1475–1480. doi: 10.1098/rstb.1997.0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh AR, Gonzalez-Lima F. Network interactions among limbic cortices, basal forebrain, and cerebellum differentiate a tone conditioned as a Pavlovian excitor or inhibitor: fluorodeoxyglucose mapping and covariance structural modeling. Journal of Neurophysiology. 1994;72:1717–1733. doi: 10.1152/jn.1994.72.4.1717. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Marriott LK, Gold PE. Patterns of brain acetylcholine release predict individual differences in preferred learning strategies in rats. Neurobiology of Learning and Memory. 2003;79:177–183. doi: 10.1016/s1074-7427(02)00014-x. [DOI] [PubMed] [Google Scholar]
- Mello e Souza T, Roesler R, Madruga M, de-Paris F, Quevedo J, Rodrigues C, Sant’Anna MK, Medina JH, Izquierdo I. Differential effects of post-training muscimol and AP5 infusions into different regions of the cingulate cortex on retention for inhibitory avoidance in rats. Neurobiology of Learning and Memory. 1999;72:118–127. doi: 10.1006/nlme.1998.3892. [DOI] [PubMed] [Google Scholar]
- Messier C. Glucose improvement of memory: a review. European Journal of Pharmacology. 2004;490:33–57. doi: 10.1016/j.ejphar.2004.02.043. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Nobre AC, Kim YH, Parrish TB, Gitelman DR. Heterogeneity of cingulate contributions to spatial attention. NeuroImage. 2001;13:1065–1072. doi: 10.1006/nimg.2001.0768. [DOI] [PubMed] [Google Scholar]
- Mielke MM, Lyketsos CG. Lipids and the pathogenesis of Alzheimer’s disease: Is there a link? International Review of Psychiatry. 2006;18:173–186. doi: 10.1080/09540260600583007. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Foster NL, Kuhl DE. Posterior cingulate cortex in Alzheimer’s disease. Lancet. 1994;344:895–895. doi: 10.1016/s0140-6736(94)92871-1. [DOI] [PubMed] [Google Scholar]
- Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Annals of Neurology. 1997;42:85–94. doi: 10.1002/ana.410420114. [DOI] [PubMed] [Google Scholar]
- Mizumori SJY, Yeshenko O, Gill KM, Davis DM. Parallel processing across neural systems: implications for a multiple memory system hypothesis. Neurobiology of Learning and Memory. 2004;82:278–298. doi: 10.1016/j.nlm.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. European Journal of Nuclear Medicine and Molecular Imaging. 2005;32:486–510. doi: 10.1007/s00259-005-1762-7. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Sorbi S, de Leon MJ, Li Y, Nacmias B, Myoung PS, Tsui W, Ginestroni A, Bessi V, Fayyazz M, Caffarra P, Pupi A. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. Journal of Nuclear Medicine. 2006;47:1778–1786. [PubMed] [Google Scholar]
- Nair HP, Gonzalez-Lima F. Extinction of behavior in infant rats: Development of functional coupling between septal, hippocampal, and ventral tegmental regions. Journal of Neuroscience. 1999;19:8646–8655. doi: 10.1523/JNEUROSCI.19-19-08646.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neave N, Lloyd S, Sahgal A, Aggleton JP. Lack of effect of lesions in the anterior cingulate cortex and retrosplenial cortex on certain tests of spatial memory in the rat. Behavioural Brain Research. 1994;65:89–101. doi: 10.1016/0166-4328(94)90077-9. [DOI] [PubMed] [Google Scholar]
- Ohnishi T, Matsuda H, Hirakata M, Ugawa Y. Navigation ability dependent neural activation in the human brain: an fMRI study. Neuroscience Research. 2006;55:361–369. doi: 10.1016/j.neures.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Papez JW. A proposed mechanism of emotion. Archives of Neurology & Psychiatry. 1937;38:725–743. [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates San Diego. 3. San Diego: Academic Press; 1997. [Google Scholar]
- Reddy PH, Beal MF. Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Research Reviews. 2005;49:618–632. doi: 10.1016/j.brainresrev.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. New England Journal of Medicine. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Riha PD, Bruchey AK, Echevarria DJ, Gonzalez-Lima F. Memory facilitation by methylene blue: dose-dependent effect on behavior and brain oxygen consumption. Eur J Pharmacol. 2005;511:151–158. doi: 10.1016/j.ejphar.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Samuraki M, Matsunari I, Chen WP, Yajima K, Yanase D, Fujikawa A, Takeda N, Nishimura S, Matsuda H, Yamada M. Partial volume effect-corrected FDG PET and grey matter volume loss in patients with mild Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging. 2007;34:1658–1669. doi: 10.1007/s00259-007-0454-x. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, Pandya DN. Fiber pathways of the brain. New York: Oxford University Press; 2006. [Google Scholar]
- Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, Lavretsky H, Miller K, Siddarth P, Rasgon NL, Mazziotta JC, Saxena S, Wu HM, Mega MS, Cummings JL, Saunders AM, Pericak-Vance MA, Roses AD, Barrio JR, Phelps ME. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:6037–6042. doi: 10.1073/pnas.090106797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg C, Riedl V, Muhlau M, Calhoun VD, Eichele T, Laer L, Drzezga A, Forstl H, Kurz A, Zimmer C, Wohlschlager AM. Selective changes of resting-state networks in individuals at risk for Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:18760–18765. doi: 10.1073/pnas.0708803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland RJ, Whishaw IQ, Kolb B. Contributions of cingulate cortex to two forms of spatial learning and memory. The Journal of Neuroscience. 1988;8:1863–1872. doi: 10.1523/JNEUROSCI.08-06-01863.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabados T, Dul C, Majtenyi K, Hargitai J, Penzes Z, Urbanics R. A chronic Alzheimer’s model evoked by mitochondrial poison sodium azide for pharmacological investigations. Behavioural Brain Research. 2004;154:31–40. doi: 10.1016/j.bbr.2004.01.016. [DOI] [PubMed] [Google Scholar]
- Valenstein E, Bowers D, Verfaellie M, Heilman KM, Day A, Watson RT. Retrosplenial amnesia. Brain. 1987;110:1631–1646. doi: 10.1093/brain/110.6.1631. [DOI] [PubMed] [Google Scholar]
- Valla J, Berndt JD, Gonzalez-Lima F. Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: Superficial laminar cytochrome oxidase associated with disease duration. Journal of Neuroscience. 2001;21:4923–4930. doi: 10.1523/JNEUROSCI.21-13-04923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valla J, Schneider L, Niedzielko T, Coon KD, Caselli R, Sabbagh MN, Ahern GL, Baxter L, Alexander G, Walker DG, Reiman EM. Impaired platelet mitochondrial activity in Alzheimer’s disease and mild cognitive impairment. Mitochondrion. 2006;6:323–330. doi: 10.1016/j.mito.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Groen T, Kadish I, Wyss JM. Retrosplenial cortex lesions of area Rgb (but not of area Rga) impair spatial learning and memory in the rat. Behavioural Brain Research. 2004;154:483–491. doi: 10.1016/j.bbr.2004.03.016. [DOI] [PubMed] [Google Scholar]
- van Groen T, Wyss JM. Connections of the retrosplenial dysgranular cortex in the rat. The Journal of Comparative Neurology. 1992;315:200–216. doi: 10.1002/cne.903150207. [DOI] [PubMed] [Google Scholar]
- Vann SD, Aggleton JP. Extensive cytotoxic lesions of the rat retrosplenial cortex reveal consistent deficits on tasks that tax allocentric spatial memory. Behavioral Neuroscience. 2002;116:85–94. [PubMed] [Google Scholar]
- Vogt BA, Van Hoesen GW, Vogt LJ. Laminar distribution of neuron degeneration in posterior cingulate cortex in Alzheimer’s disease. Acta Neuropathologica. 1990;80:581–589. doi: 10.1007/BF00307624. [DOI] [PubMed] [Google Scholar]
- Warburton EC, Aggleton JP, Muir JL. Comparing the effects of selective cingulate cortex lesions and cingulum bundle lesions on water maze performance by rats. European Journal of Neuroscience. 1998;10:622–634. doi: 10.1046/j.1460-9568.1998.00074.x. [DOI] [PubMed] [Google Scholar]
- Whishaw IQ, Maaswinkel H, Gonzalez CLR, Kolb B. Deficits in allothetic and idiothetic spatial behavior in rats with posterior cingulate cortex lesions. Behavioural Brain Research. 2001;118:67–76. doi: 10.1016/s0166-4328(00)00312-0. [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, Jack CR., Jr 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer’s disease. Brain. 2007;130:1777–1786. doi: 10.1093/brain/awm112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley MT, Nie F, Hevner RF, Liu S, Gonzalez-Lima F. Cytochrome oxidase in neuronal metabolism and Alzheimer’s disease. New York: Plenum Press; 1998. Brain cytochrome oxidase: Functional significance and bigenomic regulation in the CNS; pp. 1–53. [Google Scholar]
- Wrubel KM, Riha PD, Maldonado MA, McCollum D, Gonzalez-Lima F. The brain metabolic enhancer methylene blue improves discrimination learning in rats. Pharmacology, Biochemistry, and Behavior. 2007;86:712–717. doi: 10.1016/j.pbb.2007.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Rojas JC, Gonzalez-Lima F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotoxicity Research. 2006;9:47–57. doi: 10.1007/BF03033307. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Schuff N, Jahng GH, Bayne W, Mori S, Schad L, Mueller S, Du AT, Kramer JH, Yaffe K, Chui H, Jagust WJ, Miller BL, Weiner MW. Diffusion tensor imaging of cingulum fibers in mild cognitive impairment and Alzheimer disease. Neurology. 2007;68:13–19. doi: 10.1212/01.wnl.0000250326.77323.01. [DOI] [PMC free article] [PubMed] [Google Scholar]