

Abstract
p-Nitrobenzenesulfenyl chloride is a stable commercially available sulfenyl chloride that, in conjunction with silver triflate, cleanly activates a wide range of thioglycosides for glycosylation at −78 °C in CH2Cl2.
Thioglycosides1 are some of the most popular glycosyl donors because they are easily prepared and are stable to most functional group modifications. Although many other thiophilic reageants, including NIS–TfOH,2 iodonium dicollidine perchlorate (IDCP),3 methyl trifluoromethanesulfonate (MeOTf)4 are known for the activation of thioglycosides, the sulfenyl/sulfonium class of thiophiles have gained considerable popularity since the discovery of dimethyl(methylthio)sulfonium triflate5 (DMTST) as a promoter. Methanesulfenyl triflate (MeSOTf, MeSBr–AgOTf),6 benzenesulfenyl triflate (PhSOTf, PhSCl–AgOTf)7,8 and p-toluenesulfenyl triflate (p-TolSOTf, p-TolSCl–AgOTf)9 have been investigated as powerful promoters capable of activating thioglycosides rapidly and cleanly at −78 °C with the formation of glycosyl triflates8 as intermediates and stable disulfide byproducts. However, methanesulfenyl bromide (MeSBr), benzenesulfenyl chloride (PhSCl), and p-toluenesulfenyl chloride (p-TolSCl) are not commercially available owing to their limited shelf-life, and must be prepared and distilled prior to use. The 1-benzenesulfinyl piperidine (BSP)/trifluoromethanesulfonic anhydride (Tf2O),10 diphenyl sulfoxide (DPSO)–Tf2O, 11,12 benzenesulfinyl morpholine–Tf2O13 and dimethyl disulfide–Tf2O14 protocols have been developed as shelf-stable substitutes for this sulfenyl halide-based chemistry and have been employed widely in oligosaccharide synthesis. Herein, we report on the use of a stable, commercially available p-nitrobenzenesulfenyl chloride (p-NO2PhSCl) as an activator for glycosylation in conjunction with silver trifluoromethanesulfonate (AgOTf). p-Nitrobenzenesulfenyl chloride is an orange solid that has been previously employed as the precursor to photolabile sulfenate esters and, thus, as a source of alkoxy radicals.15,16
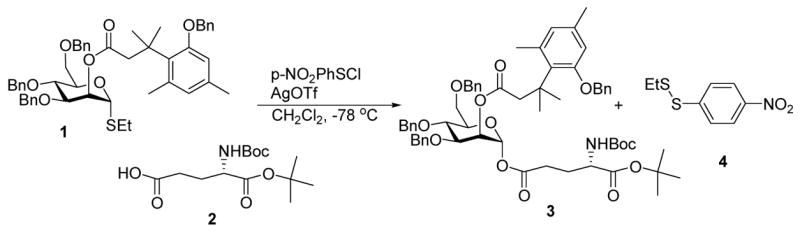
As a first demonstration of the method, the coupling of mannopyranoside donor 1 and an amino acid 2 was conducted using 1 eq p-NO2PhSCl–2.5 eq AgOTf as promoter in CH2Cl2. This reaction was complete in 30 min at −78 °C and gave the α-anomeric product in 80% yield. As expected, the disulfide 4 was also obtained in 83% yield (Scheme 1).
Scheme 1.
A number of other examples were then conducted employing 1.2 eq p-NO2PhSCl, 2.5 eq AgOTf and 1.5 eq acceptor in CH2Cl2, occasionally in admixture with acetonitrile17,18 or 2,4,6-tri-tert-butylpyrimidine (TTBP)19 as additive. The acetate glucose donor 5 and acceptor 6 were pre-mixed and then activated to give disaccharide 7 in 95% yield (Table 1, entry 1). The formation of the β-isomer in this example relies on the neighboring group participation effect. The coupling of 5 with a primary alcohol 8, the acetyl-transfer product 9 predominated (Table 1, entry 2). Preactivation of the 4,6-O-benzylidene mannopyranoside 10 followed by addition of acceptor 6 gave a 13:1 (β:α) anomeric mixture of disaccharides in 72% yield; When 6 and 10 were pre-mixed before activation in the presence of 20% acetonitrile, only a 5:1 (β:α) selectivity was obtained (Table 1, entry 3, 4). The pre-activated 4,6-O-benzylidene glucopyranosyl donor 12 gave a 3:1 (α:β) anomeric mixture of disaccharides after preactivation and then the addition of acceptor 13 as expected on the basis of earlier work (Table 1, entry 5). 6,20 The tetra-O-benzyl glucosyl donor 15 exhibited a β-selective coupling with a primary alcohol (Table 1, entry 6) in keeping with the observation of Hashimoto21 for a related coupling. However, with a less reactive partner 17 the more typical α-selectivity reasserted itself (Table 1, entry 7).
Table 1.
| Entry | Donor | Acceptor | Coupling Product | Yield(α:β)e |
|---|---|---|---|---|
| 1 |
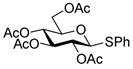
5 |
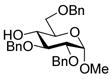
6 |
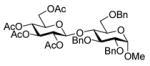
7 |
95%b,d(β only) |
| 2 | 5 |
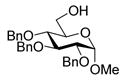
8 |
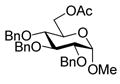
9 |
75%b. |
| 3 |
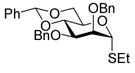
10 |
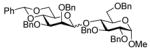
11 |
72%a,c(1:13) | |
| 4 | 6 | 80%b,c,d(1:5) | ||
| 5 |
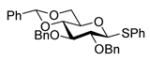
12 |
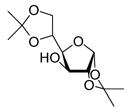
13 |
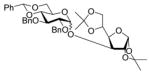
14 |
88%a,c(3:1) |
| 6 |
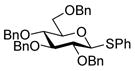
15 |
8 |
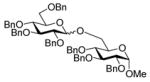
16 |
85%a,c(1:3) |
| 7 | 15 |
17 |
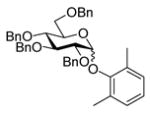
18 |
82%a,c(7:3) |
| 8 |
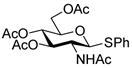
19 |
8 |
20 |
20%b,d(β only) |
| 9 |
21 |
8 |
22 |
60%b,c, d(1.5:1) |
| 10 |
23 |
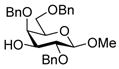
24 |
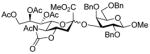
25 |
77%a,d(3:1) |
preactivation;
pre-mixed;
1.2 eq TTBP;
20% (v/v) acetonitrile;
determined by 1H NMR analysis on the crude reaction mixture.
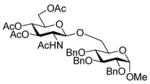
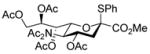
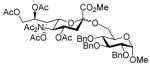
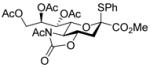
With the N-acetylglucosamine based thioglycoside 19 (Table 1, entry 8), oxazoline ring formation could not be suppressed; nevertheless, the glycosylation product 20 was formed in 20% yield. In the coupling of phenyl thiosialoside donor 21 and acceptor 8, the anomeric ratio of the products was formed to be 1.5:1 (α:β) for a coupling conducted in the presence of acetonitrile, comparable to the result achieved with the DPSO/Tf2O22 promotion system. Encouragingly, the p-NO2PhSOTf promoter could activate even the relatively inert phenyl thiosialoside donor 23 at −78 °C. Thus, premixing of donor 23 and acceptor 24 at −78 °C with acetonitrile as additive before activation afforded the α-sialoside (3:1) in good yield (Table 1, entry 10). This result is comparable to the NIS–TfOH23 activated reaction using the more reactive 1-adamantanyl thiosialoside donor and the same acceptor.
In summary, the shelf stable, commercially available p-NO2PhSCl–AgOTf promoter works well with different kinds of donors. Both 1,2-trans and 1,2-cis products are formed as major products according to the choice of protecting group.
1. Experimental
1.1. General methods
Optical rotations were determined with an Autopol III polarimeter. 1H and 13C NMR spectra were recorded at 500 and 125 MHz, respectively, with chemical shifts reported downfield from tetramethylsilane. All solvents were dried by standard procedures. Commercial reagents were used without purification. Air and/or moisture-sensitive reactions were carried out under an atmosphere of argon or nitrogen using oven-dried glassware.
1.2. Typical procedure for coupling of pre-mixed donors and acceptors: α-tert-Butyl γ-{3,4,6-tri-O-benzyl-2-O-[3′-(2″-benzyloxy-4″,6″-dimethylphenyl) -3′,3′-dimethylpropanoyl]-α-D-mannopyranosyl}-N-tert-butyloxycarbonyl-L-glutamate (3) and 1-ethyl-2-(p-nitrophenyl)disulfane (4)
A suspension of 1 (88 mg, 0.11 mmol), 2 (51 mg, 0.17 mmol), silver triflate (72 mg, 0.28 mmol) and 5 Å molecular sieves in anhydrous CH2Cl2 (1 mL) was stirred, with the exclusion of light, for 10 min at room temperature under N2 before it was cooled to −78 °C. A solution of p-nitrobenzenesulfenyl chloride (22.3 mg, 0.11 mmol, 95% purity) in anhydrous CH2Cl2 (0.5 mL) was dropped into the above suspension at −78 °C. TLC analysis showed the reaction to be finished after stirring for 30 min at −78 °C after which a satd aq NaHCO3 solution (0.2 mL) was added. The reaction mixture was diluted with CH2Cl2 (5 mL) to give a suspension, which was filtered through Celite and the Celite pad was washed with CH2Cl2 (10 mL). The filtrate was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (10:1→3:1, hexane–EtOAc) to give first 4 (20 mg, 0.09 mmol, 83%) as a light yellow color oil, and then 3 (92 mg, 0.09 mmol, 80%) in the form of a viscous oil. Compound 3: [α]18D +22.5 (c, 1.0, CHCl3); lit.24 [α]16D +22.2 (c, 1.0, CHCl3); 1H and 13C NMR spectral data matched that reported.24 Compound 4: 1H NMR (500 MHz, CDCl3): δ: 1.33 (t, J = 7.5 Hz, 3H), 2.80 (q, J = 7.5 Hz, 2H), 7.60 (m, 2H), 8.11 (m, 2H); 13C NMR (125 MHz, CDCl3) δ: 14.5, 33.1, 124.2, 126.0, 146.4, 147.5; HRMS(EI): calcd for C8H9NO2S2[M+]: 215.0075. Found: 215.0079.
1.3. Typical procedure for glycosylation with preactivation of the donor: Methyl 2,3,6-tri-O-benzyl-4-O-(2,3-di-O-benzyl-4,6-O-benzylidene-β-D-mannopyranosyl)-α-D-glucopyranoside (11)
A suspension of 10 (73 mg, 0.15 mmol), TTBP (44.0 mg, 0.18 mmol, 1.2 eq), silver triflate (95 mg, 0.37 mmol, 2.5 eq) and 5Å molecular sieves in anhydrous CH2Cl2 (1 mL) was stirred, with the exclusion of light, for 10 min at room temperature under N2 before it was cooled to −78 °C. A solution of p-nitrobenzenesulfenyl chloride (35.5 mg, 0.18 mmol, 1.2 eq, 95% purity) in anhydrous CH2Cl2 (0.5 mL) was dropped into the above suspension at −78 °C. After 5 min, A solution of 6 (103 mg, 0.22 mmol, 1.5 eq) in anhydrous CH2Cl2 (1 mL) was dropped into this suspension. TLC analysis showed the reaction to be finished after stirring for 1 h at −78 °C after which a satd aq NaHCO3 solution (0.2 mL) was added. The reaction mixture was diluted with CH2Cl2 (5 mL) to give a suspension, which was filtered through Celite and the Celite pad was washed with CH2Cl2 (10 mL). The filtrate was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (9:1, toluene–EtOAc) to give 11 (95 mg, 0.11 mmol, 72%) in the form of a viscous oil. Compound 11: [α]20D −26.3 (c, 1.0, CHCl3); lit.25 [α]28D −26.4 (c, 1.5, CHCl3); 1H and 13C NMR spectral data matched that reported.25
1.4 Methyl 2,3,6-tri-O-benzyl-4-O-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-α-D-glucopyranoside (7)
Colourless syrup; [α]20D −5.1 (c, 1.1, CHCl3); lit.26 [α]20D −5.0 (c, 1.0, CHCl3); 13C NMR spectral data matched that reported.27
1.5 3-O-(2,3-Di-O-benzyl-4,6-O-benzylidene-α-D-glucopyranosyl)-1,2;5,6-di-O-isopropylidene-α-D-glucofuranose (14α)
[α]18D +5.9 (c, 1.0, CHCl3); lit.20 [α]D +5.8 (c, 0.8, CHCl3); 1H and 13C NMR spectral data matched that reported.20
1.6 Methyl 2,3,4-tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-β-D-glucopyranosyl)-α-D-glucopyranoside (16β)
Colourless syrup; [α]20D +19.8° (c, 1.0, CHCl3); lit.28 [α]24D +19.3° (c, 0.2, CHCl3); 1H and 13C NMR spectral data matched that reported.28
1.7 2,6-Dimethylphenyl 2,3,4,6-tetra-O-benzyl-α-D-glucopyranoside (18α)
Colourless syrup; [α]18D +36.5 (c, 1.0, CHCl3); lit.8 [α]20D +36.7 (c, 4.2, CHCl3); 1H and 13C NMR spectral data matched that reported.8 18β was isolated as a mixture together with 18α, the 1H and 13C NMR spectral data matched that reported.8
1.8 Methyl 6-O-(2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-D-glucopyranosyl)-2,3,4-tri-O- benzyl-α-D-glucopyranoside (20)
Colourless syrup; [α]20D −19.9 (c, 1.0, CHCl3); lit.29 [α]25D −20.1 (c, 1.0, CHCl3); 1H and 13C NMR spectral data matched that reported.29
1.9 Methyl [methyl 5-(N-acetylacetamido)-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-α-D-galacto-non-2-ulopyranosylonate]-2,3,4-tri-O-benzyl-α-D-glucopyr anoside (22α) and Methyl [methyl 5-(N-acetylacetamido)-4,7,8,9-tetra-O-acetyl-3,5-dideoxy-D-glycero-β-D-galacto-non-2-ulopyranosylonate]-2,3,4-tri-O-benzyl-α-D-glucopyranoside (22β)
22α and 22β was isolated as an anomeric mixture, the 1H and 13C NMR spectral data matched that reported.22
1.10 Methyl [methyl 5-acetamido-7,8,9-tri-O-acetyl--5-N, 4-O-carbonyl-3,5-dideoxy-D-glycero-α-D-galacto-non-2-ulopyranosylonate]-2,4,6-tri-O-benzyl-β-D-galactopyranoside (25α) and Methyl [methyl 5-acetamido-7,8,9-tri-O-acetyl-5-N, 4-O-carbonyl-3,5-dideoxy-D-glycero-β-D-galacto-non-2-ulopyranosylonate]-2,4,6-tri-O-benzyl-β-D-galactopyranoside (25β)
25α and 25β was isolated as an anomeric mixture, the 1H and 13C NMR spectral data matched that reported.23
Acknowledgments
We thank NIGMS (GM62160) for generous financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ferrier RJ, Hay RW, Vethaviyasar N. Carbohydr Res. 1973;27:55–61. [Google Scholar]
- 2.Konradsson P, Udodong UE, Fraser-Reid B. Tetrahedron Lett. 1990;31:4313–4316. [Google Scholar]
- 3.Veeneman GH, Vuan Boom JH. Tetrahedron Lett. 1990;31:275–278. [Google Scholar]
- 4.Lönn H. Carbohydr Res. 1985;139:105–113. doi: 10.1016/0008-6215(85)90011-4. [DOI] [PubMed] [Google Scholar]
- 5.Fügedi P, Garegg PJ. Carbohydr Res. 1986;149:C9–C12. [Google Scholar]
- 6.Dasgupta F, Garegg PJ. Carbohydr Res. 1988;177:C13–C17. [Google Scholar]
- 7.Martichonok V, Whitesides GM. J Org Chem. 1996;61:1702–1706. doi: 10.1021/jo951711w. [DOI] [PubMed] [Google Scholar]
- 8.Crich D, Sun S. Tetrahedron. 1998;54:8321–8348. [Google Scholar]
- 9.Huang X, Huang L, Wang H, Ye XS. Angew Chem Int Ed. 2004;43:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 10.Crich D, Smith M. J Am Chem Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]
- 11.Garcia BA, Poole JL, Gin DY. J Am Chem Soc. 1997;119:7597–7598. [Google Scholar]
- 12.Codée JDC, Litjens REJN, den Heeten R, Overkleeft HS, van Boom JH, van der Marel GA. Org Lett. 2003;5:1519–1522. doi: 10.1021/ol034312t. [DOI] [PubMed] [Google Scholar]
- 13.Wang C, Wang H, Huang X, Zhang LH, Ye XS. Synlett. 2006:2846–2850. [Google Scholar]
- 14.Tatai J, Füegedi P. Org Lett. 2007;9:4647–4650. doi: 10.1021/ol702139u. [DOI] [PubMed] [Google Scholar]
- 15.Horner JH, Choi SY, Newcomb M. Org Lett. 2000;2:3369–3372. doi: 10.1021/ol006469g. [DOI] [PubMed] [Google Scholar]
- 16.Pasto DJ, Cottard F. Tetrahedron Lett. 1994;35:4303–4306. [Google Scholar]
- 17.Schmidt RR, Ruecker E. Tetrahedron Lett. 1980;21:1421–1424. [Google Scholar]
- 18.Schmidt RR, Behrendt M, Toepfer A. Synlett. 1990:694–696. [Google Scholar]
- 19.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001:323–326. [Google Scholar]
- 20.Crich D, Cai W. J Org Chem. 1999;64:4926–4930. doi: 10.1021/jo990243d. [DOI] [PubMed] [Google Scholar]
- 21.Hashimoto S-i, Umeo K, Sano A, Watanabe N, Nakajima M, Ikegami S. Tetrahedron Lett. 1995;36:2251–2254. [Google Scholar]
- 22.Crich D, Li W. Org Lett. 2006;8:959–962. doi: 10.1021/ol060030s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crich D, Li W. J Org Chem. 2007;72:2387–2391. doi: 10.1021/jo062431r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crich D, Cai F. Org Lett. 2007;9:1613–1615. doi: 10.1021/ol070449y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagai H, Sasaki K, Matsumura S, Toshima K. Carbohydr Res. 2005;340:337–353. doi: 10.1016/j.carres.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 26.Knoben HP, Schlueter U, Redlich H. Carbohydr Res. 2004;339:2821–2833. doi: 10.1016/j.carres.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez EB, Stick RV. Aust J Chem. 1990;43:665–679. [Google Scholar]
- 28.Nguyen HM, Chen Y, Duron SG, Gin DY. J Am Chem Soc. 2001;123:8766–8772. doi: 10.1021/ja015968p. [DOI] [PubMed] [Google Scholar]
- 29.Bongat AFG, Kamat MN, Demchenko AV. J Org Chem. 2007;72:1480–1483. doi: 10.1021/jo062171d. [DOI] [PMC free article] [PubMed] [Google Scholar]