

Abstract
An epidemiological model of tuberculosis has been developed and applied to five regions of the world. Globally, 6.7 million new cases of tuberculosis and 2.4 million deaths from tuberculosis are estimated for 1998. Based on current trends in uptake of the World Health Organization’s strategy of directly observed treatment, short-course, we expect a total of 225 million new cases and 79 million deaths from tuberculosis between 1998 and 2030. Active case-finding by using mass miniature radiography could save 23 million lives over this period. A single contact treatment for tuberculosis could avert 24 million cases and 11 million deaths; combined with active screening, it could reduce mortality by nearly 40%. A new vaccine with 50% efficacy could lower incidence by 36 million cases and mortality by 9 million deaths. Support for major extensions to global tuberculosis control strategies will occur only if the size of the problem and the potential for action are recognized more widely.
Each year, there are more than 6.5 million new cases of tuberculosis and more than 2 million deaths from tuberculosis worldwide (1). Tuberculosis is the seventh most important cause of global premature mortality and disability (2) and is projected to remain among the 10 leading causes of disease burden even in the year 2020 (3). Because of a powerful interaction between tuberculosis and HIV, tuberculosis incidence is rising in sub-Saharan Africa and may rise in Asia. Increasing drug-resistance, which could lead to worse treatment outcomes, also has been reported (4).
By using directly observed treatment, short-course (DOTS), cure rates of 80 to 90% have been achieved for passively diagnosed cases of smear-positive pulmonary tuberculosis (5). Analyses of national programs in Malawi, Tanzania, and Mozambique and in 10 provinces of China have shown that this strategy is both effective and cost-effective (6–8). Based on the success of these programs, the World Health Organization (WHO) adopted DOTS as its strategy for global tuberculosis control, but uptake by national programs has been slow; WHO reports that only 11% of new smear-positive pulmonary tuberculosis cases are enrolled in DOTS programs worldwide (9). Given the pace of uptake of DOTS, the heavy emphasis of WHO on smear-positive pulmonary cases only, and the magnitude of the remaining problem, it is worth examining whether considerable gains against tuberculosis can be achieved through promotion of DOTS alone. In this paper, we evaluate a range of extensions to global control strategies in terms of their potential effects on tuberculosis incidence and mortality by region from 1998 to 2030.
Since the 1960s, simple mathematical models have been used to understand tuberculosis transmission dynamics and to predict the effects of different interventions (10). Recently, there has been renewed interest in using mathematical models to study tuberculosis epidemics (11–14). Blower et al. (11, 12) have used a simple analytical framework to calculate threshold levels of preventive therapy and treatment necessary for tuberculosis eradication. We have elaborated the Blower model to capture some other aspects of the epidemiology of tuberculosis relevant to evaluating control strategies. This model has been applied to real populations, which allows us to study the impact of specific control measures in different regions of the world.
METHODS
The basic structure of the model appears in Fig. 1. The states and parameters in the model are described in the Appendix, with complete technical details available elsewhere†. In the model, individuals enter the uninfected category (U) at birth. An individual may be infected at rate λ and move into one of two categories: fast breakdown (IF), which includes those who progress quickly to clinical tuberculosis over the next 1–5 years; or slow breakdown (IS), which includes those who progress slowly to clinical disease. Only a small fraction of the individuals in the slow breakdown category actually will develop the disease. By modeling the infected state as two categories distinguished by the rate of disease progression, it is possible to capture a composite breakdown rate that declines exponentially from the time of infection‡. Infected individuals who receive isoniazid (INH) preventive therapy and are subject to a reduced rate of breakdown move from IF and IS into the INH slow breakdown category (HS).
Figure 1.

The model of tuberculosis includes nineteen states: uninfected (U); infected subject to fast or slow breakdown (IF and IS, respectively); superinfected subject to fast breakdown (SF); INH recipient subject to slow breakdown (HS); untreated cases (CUi,j), in which the index i takes on values 1, 2, and 3 (smear-positive pulmonary, smear-negative pulmonary, and extra-pulmonary, respectively), and the index j takes on values 1 and 2 (fast and slow rates of diagnosis, respectively); treated cases (CTi,k), in which the index i takes on values 1, 2, and 3 (as above), and the index k takes on values 1 and 2 (good treatment and bad treatment, respectively); recovered cases subject to fast or slow relapse (RF and RS, respectively). A detailed description of the equations and parameters that specify the model appears in the Appendix.
Fig. 1 also shows an alternative path from the IS and HS categories to clinical disease, through the superinfected fast breakdown category (SF). Considerable evidence suggests that superinfection occurs and is epidemiologically significant§. The rate of transfer into SF is the risk of infection, λ, times the proportion developing fast breakdown, (1 − p), times (1 − v), where v is the protection from subsequent fast breakdown that a primary infection affords. Some interventions, such as DOTS, have a much larger epidemiological impact if superinfection is common.
Individuals who progress to clinical disease from the infected, superinfected, or preventive therapy categories enter one of the six states for prediagnosed and otherwise untreated cases, represented collectively as CUi,j. The index i identifies three clinical categories: smear-positive pulmonary, smear-negative pulmonary, and extra-pulmonary tuberculosis. The index j distinguishes individuals subject to a fast rate of diagnosis from those subject to a slow diagnosis rate. Thus, the case-detection rate and diagnostic delay can be controlled separately for each type of clinical disease, which is important to capture the impact of different interventions. Untreated cases may spontaneously remit at rate ɛU or may die at a rate that combines the general mortality risk and the tuberculosis fatality rate (25–27).
Once diagnosed, cases may receive no treatment and remain in the same CU category, or they may begin treatment and move into one of the six states labeled CTi,k. Here, i defines clinical type as above while k distinguishes cases receiving good treatment from those receiving bad treatment. The good and bad treatment categories have different cure rates and death rates (25). The bad treatment category includes a range of circumstances in which an individual fails to complete an adequate course of chemotherapy. Treated cases may be cured at a category-specific rate, ɛTk. Recovered cases, either from spontaneous remission or from treatment cure, may relapse (28–30) or become superinfected. Two categories of recovered cases are included (RF and RS), distinguishing those who are subject to a fast relapse rate from those subject to a slow relapse rate.
To capture the profound effects of HIV on the development of tuberculosis, the model used here actually is composed of two submodels—one for the HIV sero-negative population and one for the HIV sero-positive population. Each submodel follows the structure depicted in Fig. 1. In the HIV-positive submodel, many of the transfer rates are different because of the effects of HIV on the immune system. Individuals move from each category in the HIV-negative submodel to the corresponding category in the HIV-positive submodel at the HIV infection rate, which varies over time. Because the effects of HIV on immune function are not marked with respect to tuberculosis until the CD4 count drops below 500, individuals actually move from the HIV-negative to the HIV-positive submodel after they have been infected with HIV for 3 years. The two submodels also are linked through the infection rate λ because HIV-negative tuberculosis cases can infect HIV-positive individuals, and vice versa. (See Appendix for details on heterogeneous mixing).
The models are solved by using the finite difference method with a time step of 36.5 days. Sensitivity analysis shows that the inaccuracy introduced by this time step is small. Initial conditions have been developed for each state in each region as described below.
Regional Models.
Based on patterns of tuberculosis epidemiology, the world has been divided into five regions, adapted from the World Bank regions used in the Global Burden of Disease Study (8): (i) EME, Established Market Economies; (ii) FSE, Formerly Socialist Economies of Europe; (iii) Asia, composed of India, China, and Other Asia and Islands; (iv) SSA, Sub-Saharan Africa; and (v) LAC/MEC, a combination of Latin America and the Caribbean with the Middle Eastern Crescent.
In order to elaborate regional models, we first defined plausible ranges for each of the model parameters, based on a review of the published and unpublished literature. Details are included in the Appendix. For each of the five regions, we began by modeling a virgin epidemic; that is, we simulated the introduction of a single infectious case into a population of susceptible individuals. Each epidemic was allowed to run to equilibrium. In LAC/MEC, Asia, and SSA, treatment was introduced into the equilibrium models in order to reproduce the declining epidemics of the last half-century. In EME and FSE, where tuberculosis rates have been declining for longer, we have reproduced these declines through a series of incremental changes reflecting reductions in transmission and breakdown, with the addition of treatment starting around the middle of this century.¶
An important determinant of the regional models starting in the 1980s is the HIV epidemic, for which we incorporated estimates developed by the former Global Programme on AIDS (3). In each model, the treatment variables have been adjusted during the 1980s and 1990s to reflect changes in case detection and treatment. The resulting models match closely the trends in the annual risk of infection and levels of incidence and mortality reported by WHO (31–33) and the Global Burden of Disease Study (2). For each region, we have selected only one model; clearly, other combinations of parameter values and shifts in these values over time might also provide adequate fits to known trends in tuberculosis. Uncertainty analysis for a similar model for the United States, however, indicates that evaluations of the impact of different interventions are largely unaffected by the choice of parameter values that fit past trends.‖
Global Control Strategies.
The likely impact of pursuing only the WHO DOTS strategy was evaluated, with a range of assumptions about achievable levels of uptake. Two sets of extensions to the DOTS strategy also were investigated: (i) aggressive applications of existing technologies and (ii) development and application of new technologies. For each scenario, we have estimated incidence and mortality in the HIV-negative and HIV-positive populations, from 1998 to 2030. In order to examine benefits in the longer term, we also have obtained results through the year 2050.
WHO DOTS Strategy.
Three variants of the WHO DOTS strategy were developed based on the extent to which DOTS is adopted, adequately funded, and given sufficient managerial attention. In DOTS-M, recent trends in DOTS uptake are expected to continue, while DOTS-H and DOTS-L refer to a high and a low variant, respectively (Table 1). In all three scenarios, it is assumed that DOTS uptake will continue to increase over the next two decades and will level off by 2020.
Table 1.
Case-detection and cure rates for new smear-positive tuberculosis cases in three DOTS scenarios, 1995 and 2020
| Region | 1995 rate, % | 2020 rate, %
|
||
|---|---|---|---|---|
| DOTS-H | DOTS-M | DOTS-L | ||
| Smear-positive case-detection rate | ||||
| EME | 91 | 96 | 95 | 94 |
| FSE | 70 | 96 | 90 | 81 |
| LAC/MEC | 64 | 83 | 80 | 78 |
| Asia | 50 | 70 | 62 | 56 |
| SSA | 35 | 70 | 50 | 45 |
| Smear-positive cure rate | ||||
| EME | 86 | 98 | 95 | 93 |
| FSE | 70 | 95 | 90 | 85 |
| LAC/MEC | 67 | 88 | 85 | 82 |
| Asia | 50 | 80 | 62 | 56 |
| SSA | 50 | 80 | 75 | 68 |
DOTS-H, high uptake; DOTS-M, medium uptake; DOTS-L, low uptake.
In examining the potential impact of alternative strategies, two important sources of future uncertainty were identified: the course of the HIV epidemic and the possible effects of high coverage of Bacille Calmette-Guerín (BCG) vaccination. Three different Global Programme on AIDS projection scenarios for HIV incidence were used (3). We also considered the possibility of sustained BCG efficacy (beyond 15 years after vaccination). A recent meta-analysis suggested a 50% efficacy, but this conclusion is controversial, as the set of trails shows heterogeneity of efficacy (35). Even in populations where BCG appears to be efficacious, there are few data available on its effects after 15 years (36). Without sustained efficacy, BCG is unlikely to lower tuberculosis infection or incidence significantly because it is given primarily at birth, and children are rarely smear-positive (37). With dramatically increasing BCG coverage since the late 1970s, however, the first birth cohorts with high BCG coverage now are beginning to reach age 15 (38). If BCG has sustained efficacy, it might greatly reduce incidence over the next few decades.
In order to investigate the effects of these two sources of uncertainty, we have defined a baseline DOTS scenario (DOTS-M with baseline HIV projections and no sustained BCG efficacy), a best-case DOTS scenario, which assumes DOTS-H in the most favorable context (optimistic HIV projections and sustained BCG efficacy of 50%), and a worst-case DOTS scenario, which assumes DOTS-L in the least favorable context (pessimistic HIV projections and no sustained BCG efficacy). Each of the alternative strategies described below has been evaluated incrementally to the baseline, best-case, and worst-case DOTS scenarios.
Existing Technologies.
Expanding implementation of DOTS for smear-negative cases (DOTSNeg). It is assumed that, gradually over the next 5 years, an expanded DOTS strategy could achieve the same cure rates in smear-negative patients as in smear-positive patients.
Active case-finding, symptomatic (ACF-Sx). The entire population would be screened for respiratory symptoms every 7 years (≈14% of the population each year). It is assumed that the capacity to implement this strategy would develop gradually over the next 10 years. The 3–4% reporting symptoms (39) would be subject to sputum examination. Using this algorithm, we expect to detect two-thirds of prevalent smear-positive cases (40). Of detected cases, 90% are assumed to start treatment, with cure rates the same as in DOTS-M.
Active case finding by using mass miniature radiography (ACF-MMR). The entire population would be screened every 7 years by using mass miniature radiography (MMR), followed by sputum examination for those with suspicious lesions. It is assumed that capacity to implement this strategy will be built over the next 10 years. This screening approach would detect 95% of prevalent smear-positive cases (40), with 90% of detected cases starting treatment. Smear-negative x-ray suggestives meeting strict clinical criteria also would be treated empirically. We assume that one-third of true smear-negatives would be detected and that 80% of these cases would be enrolled in treatment.
Single-cycle active case-finding (ACF-Sx-1 and ACF-MMR-1). For both the symptomatic and MMR screening algorithms, we also have examined the potential impact of just one complete cycle of screening. Because capacity will develop gradually, this one cycle would require 12 years to achieve full coverage of the population.
Mass preventive therapy for the entire adult population (MassPT). This intervention would include one cycle of active case-finding by using MMR (with assumptions as above), combined with a Mantoux test. Individuals with a positive skin test but without x-ray suggestive findings and clinical symptoms would be offered a 6-month course of INH preventive therapy. It is assumed that two-thirds of the individuals given the Mantoux test would return for the results, 90% of those found positive would start treatment, the efficacy of 6-month INH is 67% (41), and one-third of those starting treatment would complete therapy. This single cycle of MassPT would require 12 years for full implementation.
Preventive therapy for HIV-positive patients (PTHIV5 and PTHIV10). Given the current and likely future access to voluntary HIV testing centers, we have studied two scenarios: in the first, 5% of HIV-positive individuals would be screened and offered PT each year whereas the second scenario would extend to 10% of the HIV-positive population (42–44), with 33% treatment completion assumed in both scenarios.
New Technologies.
Improved sputum examination (Smear75). It is assumed that, within five years, a method could be available that would raise the sensitivity of sputum examination from 50 to 75% (45, 46). In each region, the additional patients diagnosed by using this method over conventional smear are assumed to be smear-negatives in terms of transmission potential; extra cases diagnosed would have the same cure rate as smear-positives.
Nucleic acid amplification test (Smear95). Assumptions are as above, except that the test is assumed to be 95% sensitive (47).
Ultra short course chemotherapy (UltraSCC). It is assumed that, within 10 years, a single contact drug regimen could raise cure rates among treated cases to 95%.
Breakdown vaccines (VacBr20, VacBr50, and VacBr80). These vaccines would require 15 years for development and would prevent breakdown from infection to disease. Vaccine efficacies of 20, 50, and 80% have been studied, with assumed coverages of 80%. It is assumed that the vaccine would be administered to the population gradually over a 10-year period and then subsequently to every child at birth.
Infection vaccines (VacInf20, VacInf50, and VacInf80). Three analogous scenarios have been elaborated for vaccines that would be given to uninfected persons to prevent infection. It is assumed, however, that infected individuals would be identified by using a Mantoux test and that only two-thirds of the individuals tested would return to have their test results read.
RESULTS
Based on the regional models, Table 2 summarizes the regional and global epidemiology of tuberculosis in 1998. Globally, there are 6.7 million new cases and 2.4 million deaths expected in 1998.
Table 2.
Global epidemiology of tuberculosis in 1998
| Region | Annual risk of infection, % | New cases, thousands | Incidence rate per 100,000 | Deaths, thousands | Mortality rate per 100,000 |
|---|---|---|---|---|---|
| EME | 0.03 | 151 | 18 | 13 | 2 |
| FSE | 0.15 | 112 | 32 | 20 | 5 |
| LAC/MEC | 0.32 | 621 | 54 | 191 | 17 |
| Asia | 1.23 | 4,121 | 137 | 1,472 | 49 |
| SSA | 1.95 | 1,734 | 265 | 722 | 111 |
| World | 0.90 | 6,739 | 113 | 2,417 | 40 |
Fig. 2 shows the progression of the global tuberculosis epidemic for the high, medium, and low variants of DOTS uptake, assuming baseline HIV projections and no sustained BCG efficacy. In all three scenarios, rising case numbers and deaths caused by interaction with the HIV epidemic will continue until the middle of the next decade. In DOTS-M, we expect 225 million new cases of tuberculosis and 79 million deaths between 1998 and 2030. With higher DOTS uptake, cases might be reduced by 25.7 million and deaths by 11.4 million whereas lower uptake would lead to 16.4 million extra cases and 8.0 million extra deaths.
Figure 2.

Projections of global deaths from tuberculosis, 1998 to 2030. Three scenarios are shown, reflecting high (DOTS-H), medium (DOTS-M), and low (DOTS-L) assumptions about uptake of the WHO DOTS strategy. All three scenarios assume baseline HIV projections and no sustained efficacy for BCG.
Depending on the course of the HIV epidemic and the possibility of sustained BCG efficacy, there is a range of uncertainty around the potential impact of the DOTS strategy, with the most uncertainty in Asia and SSA. Globally, we expect 171 million new cases and 60 million deaths in the best-case DOTS scenario and 249 million new cases and 90 million deaths in the worst-case DOTS scenario.
Table 3 describes the benefits that might be derived from extensions to the DOTS strategy. Results are reported for each strategy evaluated incrementally to the baseline DOTS scenario. Expanding the coverage of DOTS to include diagnosed smear-negatives would reduce total tuberculosis mortality by 2% between 1998 and 2030. A single cycle of active case-finding with MMR could prevent 11% of the deaths expected in this period in the baseline scenario. Continuous MMR screening could reduce incidence by 22% and mortality by 29% (Fig. 3). The impact of active case-finding by using a symptomatic screen rather than MMR would be ∼40–50% smaller.
Table 3.
Cases and deaths by region in the baseline DOTS strategy and averted cases and deaths by region for alternative strategies, 1998–2030
| Tuberculosis cases, millions
|
Tuberculosis deaths, millions
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EME | FSE | LAC/MEC | Asia | SSA | World | EME | FSE | LAC/MEC | Asia | SSA | World | |
| Total cases, millions | Total deaths, millions | |||||||||||
| Baseline DOTS | 3.3 | 2.2 | 15.4 | 136.7 | 66.9 | 224.5 | 0.2 | 0.3 | 4.2 | 46.6 | 27.5 | 78.8 |
| Cases averted compared to baseline (millions) | Deaths averted compared to baseline (millions) | |||||||||||
| Existing technologies | ||||||||||||
| DOTSNeg | * | * | 0.1 | 1.3 | 0.3 | 1.7 | * | * | 0.1 | 1.3 | 0.4 | 1.9 |
| ACF-Sx-1 | 0.02 | 0.07 | 0.7 | 7.2 | 5.6 | 13.6 | 0.01 | 0.02 | 0.3 | 3.1 | 2.7 | 6.1 |
| ACF-Sx | 0.04 | 0.11 | 1.3 | 15.9 | 12.9 | 30.3 | 0.01 | 0.03 | 0.5 | 6.8 | 5.9 | 13.3 |
| ACF-MMR-1 | 0.04 | 0.13 | 1.3 | 12.9 | 10.1 | 24.4 | 0.01 | 0.04 | 0.5 | 5.8 | 5.0 | 11.4 |
| ACF-MMR | 0.07 | 0.19 | 2.2 | 26.1 | 20.5 | 49.1 | 0.02 | 0.05 | 0.9 | 11.7 | 9.9 | 22.6 |
| MassPT | 0.10 | 0.16 | 2.8 | 24.5 | 15.0 | 42.6 | 0.01 | 0.04 | 0.8 | 9.1 | 6.7 | 16.7 |
| PTHIV5 | 0.00 | 0.00 | 0.1 | 0.5 | 0.6 | 1.2 | 0.00 | 0.00 | 0.0 | 0.2 | 0.3 | 0.5 |
| PTHIV10 | 0.00 | 0.00 | 0.1 | 0.8 | 1.0 | 1.9 | 0.00 | 0.00 | 0.0 | 0.3 | 0.5 | 0.8 |
| New technologies | ||||||||||||
| Smear75 | * | * | 0.1 | 0.8 | 1.0 | 1.9 | * | * | 0.2 | 1.0 | 1.4 | 2.6 |
| Smear95 | * | * | 0.2 | 1.4 | 1.9 | 3.6 | * | * | 0.3 | 1.8 | 2.6 | 4.7 |
| UltraSCC | 0.00 | 0.04 | 0.6 | 18.8 | 4.3 | 23.8 | 0.00 | 0.01 | 0.3 | 8.2 | 2.1 | 10.7 |
| VacBr20 | 0.03 | 0.03 | 0.6 | 10.1 | 5.4 | 16.2 | 0.00 | 0.00 | 0.1 | 2.4 | 1.6 | 4.1 |
| VacBr50 | 0.07 | 0.08 | 1.4 | 22.6 | 12.1 | 36.1 | 0.00 | 0.01 | 0.3 | 5.6 | 3.5 | 9.4 |
| VacBr80 | 0.10 | 0.12 | 2.0 | 32.3 | 17.1 | 51.6 | 0.01 | 0.01 | 0.4 | 8.1 | 5.1 | 13.7 |
| VacInf20 | 0.02 | 0.02 | 0.4 | 6.6 | 3.4 | 10.5 | 0.00 | 0.00 | 0.1 | 1.6 | 1.0 | 2.7 |
| VacInf50 | 0.05 | 0.05 | 0.9 | 15.4 | 8.0 | 24.5 | 0.00 | 0.00 | 0.2 | 3.8 | 2.3 | 6.3 |
| VacInf80 | 0.07 | 0.08 | 1.5 | 23.2 | 12.2 | 37.0 | 0.00 | 0.01 | 0.3 | 5.8 | 3.6 | 9.6 |
| Combination strategies | ||||||||||||
| ACF-MMR + MassPT | 0.13 | 0.23 | 3.7 | 36.4 | 24.4 | 64.8 | 0.02 | 0.06 | 1.2 | 14.4 | 11.1 | 26.7 |
| ACF-MMR + VacBr50 | 0.12 | 0.25 | 3.1 | 39.8 | 25.5 | 68.7 | 0.02 | 0.06 | 1.0 | 14.8 | 11.2 | 27.1 |
| ACF-MMR + UltraSCC | 0.08 | 0.23 | 2.8 | 44.2 | 24.6 | 71.8 | 0.02 | 0.06 | 1.1 | 19.0 | 11.7 | 32.0 |
| Smear95 + UltraSCC | 0.00 | 0.04 | 0.8 | 19.3 | 6.0 | 26.2 | 0.00 | 0.01 | 0.5 | 9.0 | 4.4 | 13.9 |
Indicates no averted cases or deaths over baseline. Cells containing 0.0 indicate >0 but <5,000 cases or deaths averted.
Figure 3.

Projections of global deaths from tuberculosis, 1998 to 2030. Each graph shows the baseline DOTS strategy and alternative strategies incremental to baseline DOTS. The top graph shows expectations of the epidemic given various aggressive applications of existing strategies, including mass preventive therapy (MassPT) and single-cycle or continuous active case-finding using mass miniature radiography (ACF-MMR-1 and ACF-MMR, respectively). The bottom graph shows the results of strategies using new technologies, including a vaccine preventing breakdown with 50% efficacy (VacBr50), a single-dose treatment regimen for active tuberculosis (UltraSCC), and this single-dose regimen combined with active screening (ACF-MMR + UltraSCC).
Preventive therapy for HIV patients diagnosed at voluntary testing centers could save nearly 1 million lives if 10% of the HIV-positive population were screened each year. Of the averted deaths, 36% would be in the HIV-negative population because of reduced transmission. The benefits from a single round of mass preventive therapy would be considerable; even with only 33% treatment completion, this strategy would avert 42.6 million cases and 16.7 million deaths (Fig. 3).
Table 3 also provides estimates of cases and deaths averted if new technologies were developed and were applied widely. Improved sputum smear methods or nucleic acid amplification tests could prevent 1.9 to 3.6 million cases and 2.6 to 4.7 million deaths during the period 1998 to 2030. A single contact treatment for active tuberculosis would prevent 23.8 million cases and 10.7 million deaths (Fig. 3). In combination with active case-finding, single-dose chemotherapy could reduce tuberculosis mortality by 41% from 1998 to 2030 and by 51% over the period 1998 to 2050.
Even with a 15-year time lag for development and clinical trials, vaccines that prevent breakdown or infection would make a major impact between now and 2030. With 50% efficacy, a vaccine that prevents breakdown from infection to disease could prevent 16% of cases and 12% of deaths (Fig. 3). All of the vaccines evaluated prevent a much larger percentage of cases and deaths when the analysis extends farther into the future. Over the period 1998 to 2050, a breakdown vaccine with 50% efficacy would reduce incidence by 29% and mortality by 25%.
We also have evaluated the impact of each strategy incremental to the best-case and worst-case DOTS scenarios. This uncertainty analysis shows that, for many interventions, the expected percentage reduction in tuberculosis mortality is not affected by the range of DOTS uptake, the course of the HIV epidemic, or the efficacy of BCG (although clearly, the absolute numbers of cases and deaths averted are strongly affected). Notable exceptions include new vaccines, which have much smaller benefits if BCG provides sustained protection and a much greater impact in the worst-case scenario. Cases and deaths averted through the introduction of an ultra-short-course regimen also depend strongly on DOTS uptake.
DISCUSSION
Even with high uptake of DOTS in the most favorable context, incidence and mortality from tuberculosis in the next three decades will be enormous. It is therefore imperative to consider means of extending the DOTS strategy. Many of the strategies evaluated in this paper would require substantial resources and an extraordinary mobilization of international attention. Initiating and sustaining such a large-scale effort would be challenging, but clearly the first step must be a realistic assessment of the potential benefits of these ambitious initiatives.
In this study, we have applied epidemic models to real populations to draw conclusions about one of the world’s leading health challenges. We have not attempted to cost different global control strategies. To translate the results in this paper into policy recommendations, it will be important to proceed with a realistic evaluation of the costs of pursuing active case-finding, mass preventive therapy, and the development of new technologies.
WHO, for many years, has recommended passive case-finding as the primary strategy for detecting tuberculosis (48). The results of this analysis, however, indicate that active case-finding could reduce tuberculosis mortality by one-quarter to one-third. Perhaps the time has come for a critical reexamination of the costs, risks, and benefits of active screening.
The concept of INH preventive therapy has been recognized for decades, but in only one case has mass preventive therapy been used to maximum effect (49). In low-incidence communities, the low risk of fatal isoniazid toxicity—0.014% in the U.S. Public Health Service trial (50)—has been a source of great concern. In high incidence regions, however, this small toxicity risk would be far outweighed by the magnitude of the benefits. Even with modest completion rates, a mass preventive therapy campaign could alter significantly the course of the tuberculosis epidemic.
Although aggressive use of existing technologies could produce considerable benefits, tuberculosis likely will kill more than 50 million people over the next three decades, even if these aggressive strategies are pursued. Investing now in new tools could make a substantial difference (51). A single contact treatment for tuberculosis would have an enormous effect and, of all the new technologies, would be the easiest to field. Recent breakthroughs in the treatment of paucibacillary leprosy (52) make consideration of a single contact treatment for tuberculosis worthwhile. By reducing drug and labor costs, such a development probably would prove to be cost-saving. Over the next half-century, the combination of a single contact treatment and active case-finding could reduce mortality from tuberculosis by >50%. Investments in new vaccines also could have a profound impact over this period.
Tuberculosis program managers and control specialists must develop more ambitious objectives for tuberculosis control over the next half-century. Tuberculosis is likely to remain one of the major causes of death in the 21st century, but it is also one of the few for which there are a number of existing and potential new interventions that can make a dramatic difference. A major rethinking of global tuberculosis strategy can occur only if the size of the problem and the potential for action are recognized more widely.
ABBREVIATIONS
- WHO
World Health Organization
- DOTS
directly observed treatment, short-course
- BCG
Bacille Calmette-Guérin
- MMR
mass miniature radiography
- INH
isoniazid
Appendix
The model is determined by the following system of ordinary differential equations:
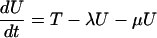 |
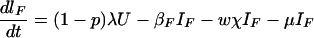 |
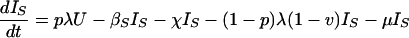 |
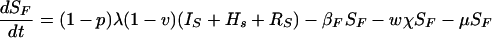 |
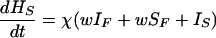 |
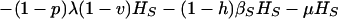 |
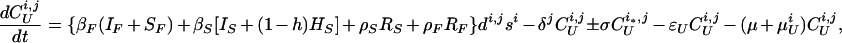 |
 |
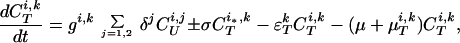 |
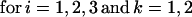 |
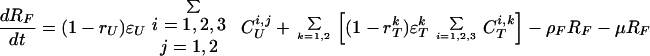 |
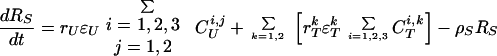 |
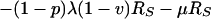 |
where T is the number of new births; λ is the infection rate; μ is the non-tuberculosis death rate; p is the proportion of new infections entering slow breakdown; βF and βS are the fast and slow breakdown rates, respectively; χ is the rate at which infected individuals receive INH preventive therapy; v is the protection from superinfection conferred by primary infection; w is short-term INH effectiveness; h is long-term INH effectiveness; si is the proportion of new cases in clinical category i; di,j is the proportion of prediagnosed cases of clinical type i entering diagnosis category j; δj is the diagnosis rate for category j; σ is the rate of conversion from smear-negative to smear-positive; ɛU is the spontaneous cure rate for untreated cases; μUi is the tuberculosis death rate for untreated cases in clinical category i; gi,k is the proportion of treated cases in clinical category i and treatment category k; ɛTk is the cure rate for treated cases in treatment category k; μTi,k is the tuberculosis death rate for treated cases in category i,k; rU is the proportion of spontaneously recovered cases entering the slow relapse category; rTk is the proportion of recovered cases from treatment category k entering the slow relapse category; and ρF and ρS are the fast and slow relapse rates, respectively. In the equations for CUi,j and CTi,k, the smear rate σ is multiplied by the number of individuals in the respective category i∗, where i∗ = 2 (smear-negative) for i = 1 (smear-positive) and vice versa, and i∗ = ⊘ for i = 3 (extrapulmonary). The term including σ is added for i = 1, subtracted for i = 2, and equal to zero for i = 3. The result of this formulation is that smear-negative patients convert to smear-positive at a rate of σ.
In this model, λ is a function of the prevalence of infectious sources in the population. That is, λ = KL (Y/N), where λ is the instantaneous rate of infection for a susceptible; K is the number of respiratory contacts per person per year; L is the probability that a respiratory contact with an infectious source will lead to tuberculosis infection; Y is the number of infectious cases in the population (in terms of the model states, Y includes all smear-positive cases plus a small proportion of smear-negatives); and N is the population. Thus, it is assumed that the number of secondary infections caused by a source case in a year (KL) does not depend on population size. This formulation of λ often is used in models of sexually transmitted disease (53).
Because HIV-positive individuals may be more likely to come in contact with other HIV-positive individuals, we have elaborated the expression for λ to allow for heterogeneous mixing:
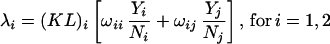 |
where i refers to the population subgroup (HIV sero-positive ≡ 1; HIV sero-negative ≡ 2), j is the complementary subgroup, K, L, Y, and N are as described above, ωii is the proportion of all effective respiratory contacts that are with members of the same subgroup, and ωij is the proportion of all effective contacts that are with members of the complementary subgroup. We note three identities implied by these definitions: (i) ω11 = 1 − ω12; (ii) (KL)2N2ω21 = (KL)1N1ω12; and (iii) ω22 = 1 − ω21. We have formulated ω11 to be proportionate to HIV sero-prevalence; that is, ω11 = c∗ N1/(N1 + N2) for some constant c, which may vary by region. A large value for c implies that HIV prevalence is much higher among the contacts of an HIV-positive individual than among the general population. Thus, proportionate mixing is implied where c = 1 whereas contacts occur exclusively within subgroups when c is the inverse of HIV sero-prevalence; that is, c = (N1 + N2)/N1 ⇒ ω11 = 1.
Following are the ranges considered for the major parameters of interest in the model: transmission parameter (KL), 5–15 infections per infectious case per year; proportion of new infections breaking down to clinical disease within the first 5 years (1 − p), 5–15% in HIV-negatives and 50–70% in HIV-positives; endogenous reactivation rate (βS), 50–150 persons per 100,000 persons per year in HIV-negatives and 5–10% per year in HIV-positives; proportion of new cases of tuberculosis that are smear-positive pulmonary (s1): 40–50%; protection against breakdown from subsequent infection afforded by primary infection (v), 50–100%; death rate for untreated tuberculosis (μU), 0.075–0.20 per person per year in HIV-negatives and 0.3–0.6 per person in HIV-positives; cure rate for untreated tuberculosis (ɛU), 0.05–0.15 per person per year; proportion of recovered cases in fast relapse category (1 − r), 4–15%; slow relapse rate (ρS), 50–150 persons per 100,000 persons per year (11, 12, 16–18, 25–30). Complete details on the parameter values used in each regional model over time are available in ref. 15.
Footnotes
A Commentary on this article begins on page 13352.
The structure and dynamics of the model, as well as technical details on the modeling of alternative control strategies, are described fully in ref. 15. This report is available at http://www.hsph.harvard.edu/organizations/bdu.
Sutherland (16) reported on the time to breakdown in individuals who developed tuberculosis in the Medical Research Council vaccine trial. Nearly 60% developed tuberculosis within 1 year of tuberculin skin test conversion, and the proportion declined exponentially as a function of time since conversion. Krishna Murthy et al. (17) found similar results.
The significance of superinfection has been a highly controversial issue for many years (18). Evidence in favor of superinfection includes anatomical evidence (19), restriction fragment length polymorphism analyses (20, 21), and epidemiological studies (22–24).
See ref. 15 for complete details on parameter values in the regional models.
The model for the United States is part of an ongoing project sponsored by the Centers for Disease Control and Prevention. The methodology for undertaking multivariate uncertainty analysis, while at the same time validating a model by its ability to fit past trends in incidence and mortality, is described in ref. 34. In brief, plausible ranges are defined for each parameter in the model, multiple sets of parameter combinations are generated by randomly sampling each range, and the model is run by using each of these parameter combinations. The subset of combinations that optimize a particular goodness-of-fit criterion comprise the set of valid models, which then are used to evaluate the future impact of alternative policy scenarios.
References
- 1.World Health Organization Global Tuberculosis Programme. WHO Report on the Tuberculosis Epidemic 1997. W. H. O. Geneva; 1997. [Google Scholar]
- 2.Murray C J L, Lopez A D. Science. 1996;274:740–743. doi: 10.1126/science.274.5288.740. [DOI] [PubMed] [Google Scholar]
- 3.Murray C J L, Lopez A D. Lancet. 1997;349:1498–1504. doi: 10.1016/S0140-6736(96)07492-2. [DOI] [PubMed] [Google Scholar]
- 4.The WHO/IUATLD Global Project on Anti-Tuberculosis Drug Resistance Surveillance. Anti-tuberculosis Drug Resistance in the World. W. H. O. Geneva; 1997. [Google Scholar]
- 5.Broekmans J F. In: Tuberculosis: Back to the Future. Porter J D H, McAdam K P W J, editors. New York: Wiley; 1994. pp. 171–192. [Google Scholar]
- 6.Murray C J L, DeJonghe E, Chum H J, Nyangulu D S, Salomao A, Styblo K. Lancet. 1991;338:1305–1308. doi: 10.1016/0140-6736(91)92600-7. [DOI] [PubMed] [Google Scholar]
- 7.China Tuberculosis Control Collaboration. Lancet. 1996;347:358–362. [PubMed] [Google Scholar]
- 8.World Bank. World Development Report. New York: Oxford Univ. Press; 1993. [Google Scholar]
- 9.Kochi A, Nunn P, Dye C, Tayler E. Lancet. 1997;350:142. doi: 10.1016/S0140-6736(05)61845-4. [DOI] [PubMed] [Google Scholar]
- 10.Waaler H, Geser A, Andersen S. Am J Public Health. 1962;52:1002–1013. doi: 10.2105/ajph.52.6.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blower S M, Small P M, Hopewell P C. Science. 1996;273:497–500. doi: 10.1126/science.273.5274.497. [DOI] [PubMed] [Google Scholar]
- 12.Blower S M, McLean A R, Porco T C, Small P M, Hopewell P C, Sanchez M A, Moss A R. Nat Med. 1995;1:815–821. doi: 10.1038/nm0895-815. [DOI] [PubMed] [Google Scholar]
- 13.Brewer T F, Heymann J, Colditz G, Wilson M E, Auerbach K, Kane D, Fineberg H V. JAMA. 1996;276:1898–1903. [PubMed] [Google Scholar]
- 14.Vynnycky E, Fine P E. Epidemiol Infect. 1998;119:183–201. doi: 10.1017/s0950268897007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray C J L, Salomon J A. Using Mathematical Models to Evaluate Global Tuberculosis Control Strategies. Cambridge, MA: Harvard Center for Population and Development Studies; 1998. [Google Scholar]
- 16.Sutherland I. The Ten-Year Incidence of Clinical Tuberculosis Following “Conversion” in 2550 Individuals Aged 14 to 19 Years. The Hague: Tuberculosis Surveillance Research Unit; 1968. [Google Scholar]
- 17.Krishna Murthy V V, Nair S S, Gothi G D, Chakraborty A K. Indian J Tuberc. 1976;33:3–7. [Google Scholar]
- 18.Styblo K. Epidemiology of Tuberculosis. The Hague: Royal Netherlands Tuberculosis Association; 1991. [Google Scholar]
- 19.Canetti G, Sutherland I, Svandova E. Bull Int Union Tuberc. 1972;47:122–143. [PubMed] [Google Scholar]
- 20.Turett G S, Fazal B A, Justman J E, Alland D, Duncalf R M, Telzak E E. Clin Infect Dis. 1997;24:513–514. doi: 10.1093/clinids/24.3.513. [DOI] [PubMed] [Google Scholar]
- 21.Small P M, Shafer R W, Hopewell P C, Singh S P, Murphy M J, Desmond E, Sierra M F, Schoolnik G K. N Engl J Med. 1993;328:1137–1144. doi: 10.1056/NEJM199304223281601. [DOI] [PubMed] [Google Scholar]
- 22.Sutherland I, Svandova E, Radhakrishna S. Tubercle. 1982;63:255–268. doi: 10.1016/s0041-3879(82)80013-5. [DOI] [PubMed] [Google Scholar]
- 23.Krishna Murthy V V, Chaudhuri K. Indian J Tuberc. 1990;37:63–67. [Google Scholar]
- 24.Grzybowski S, Styblo K, Dorken E. Tubercle. 1976;57:s1–s50. doi: 10.1016/0041-3879(76)90059-3. [DOI] [PubMed] [Google Scholar]
- 25.National Tuberculosis Institute Bangalore. Bull W H O. 1974;51:473–488. [PMC free article] [PubMed] [Google Scholar]
- 26.Drolet G J. Am Rev Tuberc. 1938;37:125–151. [Google Scholar]
- 27.Berg G. Acta Tuberc Scand, Suppl. 1939;IV:1–207. [Google Scholar]
- 28.Horwitz O. Am Rev Respir Dis. 1969;99:183–193. doi: 10.1164/arrd.1969.99.2.183. [DOI] [PubMed] [Google Scholar]
- 29.Chan-Yeung M, Galbraith J D, Schulson N, Brown A, Grzybowski S. Am Rev Respir Dis. 1971;104:861–865. doi: 10.1164/arrd.1971.104.6.861. [DOI] [PubMed] [Google Scholar]
- 30.Chakraborty A K, Gothi G D. Indian J Tuberc. 1976;33:9–13. [Google Scholar]
- 31.Raviglione M C, Snider D E, Kochi A. JAMA. 1995;273:220–226. [PubMed] [Google Scholar]
- 32.World Health Organization. Global Tuberculosis Programme. Tuberculosis—A Global Emergency: Case Notification Update, February 1996. W. H. O. Geneva; 1996. [Google Scholar]
- 33.Cauthen G M, Pio A, ten Dam H G. Annual Risk of Tuberculosis Infection. W. H. O. Geneva; 1988. [PMC free article] [PubMed] [Google Scholar]
- 34.Murray C, Sawert H. Tuberculosis in the United States: Strategies for Developing and Validating Models. Cambridge, MA: Harvard Center for Population and Development Studies; 1994. [Google Scholar]
- 35.Colditz G A, Brewer T F, Berkey C S, Wilson M E, Burdick E, Fineberg H V. JAMA. 1994;271:698–702. [PubMed] [Google Scholar]
- 36.Rodrigues L C, Smith P G. Trans R Soc Trop Med Hyg. 1990;84:739–744. doi: 10.1016/0035-9203(90)90172-b. [DOI] [PubMed] [Google Scholar]
- 37.Styblo K, Meijer J. Tubercle. 1976;57:17–43. doi: 10.1016/0041-3879(76)90015-5. [DOI] [PubMed] [Google Scholar]
- 38.United Nations Children’s Fund. The State of the World’s Children. New York: Oxford Univ. Press; 1991. [Google Scholar]
- 39.Aluoch J A, Swai O B, Edwards E A, Stott H, Darbyshire J H, Fox W, Sutherland I. Am Rev Respir Dis. 1984;129:915–920. doi: 10.1164/arrd.1984.129.6.915. [DOI] [PubMed] [Google Scholar]
- 40.Gothi G D, Narayan R, Nair S S, Chakraborty A K, Srikantaramu N. Indian J Med Res. 1976;64:1150–1159. [PubMed] [Google Scholar]
- 41.International Union Against Tuberculosis Committee on Prophylaxis. Bull W H O. 1982;60:555–564. [PMC free article] [PubMed] [Google Scholar]
- 42.Pape J W, Jean S S, Ho J L, Hafner A, Johnson W D. Lancet. 1993;342:268–272. doi: 10.1016/0140-6736(93)91817-6. [DOI] [PubMed] [Google Scholar]
- 43.Aisu T, Raviglione M C, van Praag E, Eriki P, Narain J P, Barugahare L. AIDS. 1995;9:267–273. [PubMed] [Google Scholar]
- 44.Ngamvithayapong J, Uthaivoravit W, Yanai H, Akarasewi P, Sawanpanyalert P. AIDS. 1997;11:107–112. doi: 10.1097/00002030-199701000-00016. [DOI] [PubMed] [Google Scholar]
- 45.Gebre N, Karlsson U, Jonsson G, Macaden R, Wolde A, Assefa A, Miorner H. Trans R Soc Trop Med Hyg. 1995;89:191–193. doi: 10.1016/0035-9203(95)90491-3. [DOI] [PubMed] [Google Scholar]
- 46.Saceanu C A, Pfeiffer N C, McLean T. J Clin Microbiol. 1993;31:2371–2374. doi: 10.1128/jcm.31.9.2371-2374.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forbes B A. Immunol Invest. 1997;26:106–116. doi: 10.3109/08820139709048919. [DOI] [PubMed] [Google Scholar]
- 48.World Health Organization. Ninth Report of the WHO Expert Committee on Tuberculosis. W. H. O. Geneva; 1974. [PubMed] [Google Scholar]
- 49.Chakraborty A K, Channabasavaiah R, Krishnamurthy M S, Shashidhara A N, Motiram G. Indian J Tuberc. 1991;38:201–211. [Google Scholar]
- 50.Kopanoff D E, Snider D E, Caras G J. Am Rev Respir Dis. 1978;117:991–1001. doi: 10.1164/arrd.1978.117.6.991. [DOI] [PubMed] [Google Scholar]
- 51.Ad Hoc Committee on Health Research Relating to Future Intervention Options. Investing in Health Research and Development. W. H. O. Geneva; 1996. [Google Scholar]
- 52.Single-lesion Multicentre Trial Group. Indian J Leprosy. 1997;69:121–129. [PubMed] [Google Scholar]
- 53.Hethcote H W, Van Ark J W. Math Biosci. 1987;84:85–118. [Google Scholar]