

Abstract
Fast transverse relaxation of 1H, 15N, and 13C by dipole-dipole coupling (DD) and chemical shift anisotropy (CSA) modulated by rotational molecular motions has a dominant impact on the size limit for biomacromolecular structures that can be studied by NMR spectroscopy in solution. Transverse relaxation-optimized spectroscopy (TROSY) is an approach for suppression of transverse relaxation in multidimensional NMR experiments, which is based on constructive use of interference between DD coupling and CSA. For example, a TROSY-type two-dimensional 1H,15N-correlation experiment with a uniformly 15N-labeled protein in a DNA complex of molecular mass 17 kDa at a 1H frequency of 750 MHz showed that 15N relaxation during 15N chemical shift evolution and 1HN relaxation during signal acquisition both are significantly reduced by mutual compensation of the DD and CSA interactions. The reduction of the linewidths when compared with a conventional two-dimensional 1H,15N-correlation experiment was 60% and 40%, respectively, and the residual linewidths were 5 Hz for 15N and 15 Hz for 1HN at 4°C. Because the ratio of the DD and CSA relaxation rates is nearly independent of the molecular size, a similar percentagewise reduction of the overall transverse relaxation rates is expected for larger proteins. For a 15N-labeled protein of 150 kDa at 750 MHz and 20°C one predicts residual linewidths of 10 Hz for 15N and 45 Hz for 1HN, and for the corresponding uniformly 15N,2H-labeled protein the residual linewidths are predicted to be smaller than 5 Hz and 15 Hz, respectively. The TROSY principle should benefit a variety of multidimensional solution NMR experiments, especially with future use of yet somewhat higher polarizing magnetic fields than are presently available, and thus largely eliminate one of the key factors that limit work with larger molecules.
NMR spectroscopy with proteins based on observation of a small number of spins with outstanding spectral properties, which either may be present naturally or introduced by techniques such as site-specific isotope labeling, yielded biologically relevant information on human hemoglobin (M = 65,000) as early as 1969 (1), and subsequently also for significantly larger systems such as Igs (2). In contrast, the use of NMR for de novo structure determination (3, 4) so far has been limited to relatively small molecular sizes, with the largest NMR structure below molecular weight 30,000. Although NMR in structural biology may, for practical reasons of coordinated use with x-ray crystallography (5), focus on smaller molecular sizes also in the future, considerable effort goes into attempts to extend the size limit to bigger molecules (for example, see refs. 6–8). Here we introduce transverse relaxation-optimized spectroscopy (TROSY) and present experimental data and theoretical considerations showing that this approach is capable of significantly reducing transverse relaxation rates and thus overcomes a key obstacle opposing solution NMR of larger molecules (7).
At the high magnetic fields typically used for studies of proteins and nucleic acids, chemical shift anisotropy interaction (CSA) of 1H, 15N, and 13C nuclei forms a significant source of relaxation in proteins and nucleic acids, in addition to dipole–dipole (DD) relaxation. This leads to increase of the overall transverse relaxation rates with increasing polarizing magnetic field, B0. Nonetheless, transverse relaxation of amide protons in larger proteins at high fields has been reduced successfully by complete or partial replacement of the nonlabile hydrogen atoms with deuterons and, for example, more than 90% of the 15N, 13Cα, and 1HN chemical shifts thus were assigned in the polypeptide chains of a protein-DNA complex of size 64,000 (6). TROSY uses spectroscopic means to further reduce T2 relaxation based on the fact that cross-correlated relaxation caused by DD and CSA interference gives rise to different relaxation rates of the individual multiplet components in a system of two coupled spins ½, I and S, such as the 15N–1H fragment of a peptide bond (9, 10). Theory shows that at 1H frequencies near 1 GHz nearly complete cancellation of all transverse relaxation effects within a 15N–1H moiety can be achieved for one of the four multiplet components. TROSY observes exclusively this narrow component, for which the residual linewidth is then almost entirely because of DD interactions with remote hydrogen atoms in the protein. These can be efficiently suppressed by 2H-labeling, so that in TROSY-type experiments the accessible molecular size for solution NMR studies no longer is primarily limited by T2 relaxation.
Theory
We consider a system of two scalar coupled spins ½, I and S, with a scalar coupling constant JIS, which is located in a protein molecule. T2 relaxation of this spin system is dominated by the DD coupling of I and S and by CSA of each individual spin, because the stereochemistry of the polypeptide chain restricts additional interactions of I and S to weak scalar and DD couplings with a small number of remote protons, Ik. The relaxation rates of the individual multiplet components of spin S in a single quantum spectrum then may be widely different (9, 11, 12). They can be described by using the single-transition basis operators S34± and S12± (13), which refer to the transitions 1→2 and 3→4 in the standard energy-level diagram for a system of two spins ½, and are associated with the corresponding resonance frequencies, ωS12 = ωS + πJIS and ωS34 = ωS − πJIS (13–16):
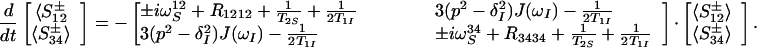 |
1 |
ωS and ωI are the Larmor frequencies of the spins S and I, T2S and T1I account for the transverse relaxation of spin S and the longitudinal relaxation time of spin I, respectively, by all mechanisms of relaxation except DD coupling between the spins S and I and CSA of the spins S and I.
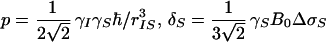 |
and
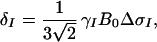 |
where γI and γS are the gyromagnetic ratios of I and S, ℏ is the Planck constant divided by 2π, rIS the distance between S and I, B0 the polarizing magnetic field, and ΔσS and ΔσI are the differences between the axial and the perpendicular principal components of the axially symmetric chemical shift tensors of spins S and I, respectively. R1212 and R3434 are the transverse relaxation rates of the individual components of the S doublet (11) given by Eqs. 2 and 3,
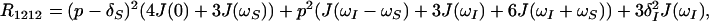 |
2 |
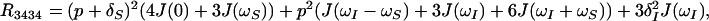 |
3 |
where J(ω) represents the spectral density functions at the frequencies indicated:
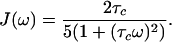 |
4 |
In deriving Eqs. 2 and 3, parallel orientation of the principal symmetry axis of the chemical shift tensor and the vector rIS was assumed. These equations show that whenever CSA and DD coupling are comparable, i.e. p ≈ δS, the resonance at frequency ωS12 may exhibit slow transverse relaxation even for very large molecules.
For a treatment of the relaxation of spin I by Eq. 1 the symbols I and S simply can be interchanged. The single-transition operators I13± and I24± then refer to the transitions between the energy levels 1→3 and 2→4, respectively, which are associated with the frequencies ωI13 = ωI + πJIS and ωI24 = ωI − πJIS, and the relaxation rates R1313 and R2424 are determined by equations obtained by permutation of the S and I indices in Eqs. 2 and 3, respectively.
To evaluate the contributions from other mechanisms of relaxation we identify I and S as the 1HN and 15N spins in a 15N–1H moiety. The relaxation of 15N then is mainly determined by the CSA of 15N and the DD interactions with the directly attached proton (17), so that the contributions from other interactions, 1/T1S and 1/T2S, to a good approximation can be neglected. For 1HN, 1/T1I and 1/T2I are dominated by DD interactions with other protons Ik at distance rk. These can be accounted for by spectral density functions Jk(ω), which describe the motions of the vectors joining the individual 1HN–1Hk spin pairs (17):
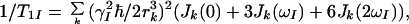 |
5 |
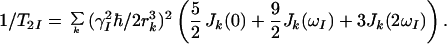 |
6 |
Here, Eqs. 1-6 were used to calculate theoretical lineshapes of spin multiplets for given sets of the relaxation parameters, which subsequently were compared with the experimental NMR data. In particular, the in-phase absorptive spectrum was calculated using Eq. 7 (14),
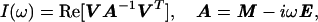 |
7 |
where V = (1,1), and the relaxation matrix A is the (2×2) matrix on the right side of Eq. 1, and E is the unity matrix.
Experimental Procedures
NMR spectra were recorded on Bruker DRX 750 and Varian Unityplus 400 spectrometers with a 2 mM solution of the specific 1:1 complex formed between a uniformly 15N-labeled 70-residue fushi tarazu (ftz) homeodomain polypeptide and an unlabeled 14-bp DNA duplex (18, 19) in 95% 1H2O/5% 2H2O at pH 6.0 and 4°C.
The isotropic rotational correlation time, τc, of the complex was estimated from the T1/T2 ratio of the relaxation times of the backbone 15N nuclei (20). The experimental schemes of Farrow et al. (21) were used for measurements of T1 (15N) and T2 (15N) for backbone nitrogen atoms.
The TROSY approach (Fig. 1) and conventional [15N,1H]correlation spectroscopy (COSY) (22, 23) experiments were used to correlate 1H and 15N resonances. For all spectra t1max = 90 ms and t2max = 171 ms were used. In TROSY the evolution of the I,S spin system due to the 1JIS scalar coupling was not refocused during t1 and t2, thus avoiding suppression of cross-correlated relaxation during these periods. To obtain the pure absorptive spectrum containing only the most slowly relaxing component of the two-dimensional multiplets, the scheme of Fig. 1 was used (see also Appendix: Quantitative Analysis of TROSY).
Figure 1.

Experimental scheme for TROSY-type two-dimensional 1H,15N correlation spectroscopy. In the rows marked 1H and 15N, narrow and wide bars stand for nonselective 90° and 180° rf-pulses, respectively. Water suppression is achieved by watergate (34), using the two off-resonance rf-pulses indicated by curved shapes. The 1H and 15N carrier frequencies are placed at 9 and 127 ppm, respectively. The delay τ1 corresponds to 1/(41J(1H,15N)) = 2.7 ms. Phases used are ψ1 = {y,−y,−x,x,y,−y,−x,x}; ψ2 = {4(x),4(-x)}; φ1 = {4(y),4(-y)}; φ2 (receiver) = {x,−x,−y,y,x,−x,y,−y}; x on all other pulses. The row marked PFG (pulsed field gradient) indicates the applied magnetic field gradients along the z-axis: G1, amplitude = 30 G/cm, duration = 0.4 ms; G2, −60 G/cm, 1 ms; G3, 50 G/cm, 0.4 ms; G4, 48 G/cm, 0.6 ms. Two free induction decays are recorded per t1 delay, with ψ1 incremented by 90° in between and stored as the real and imaginary parts of the interferogram in t1. The Fourier transformation results in a two-dimensional 1H,15N correlation spectrum that contains only the component of the four-line 15N–1H multiplet that has the slowest T2 relaxation rates for both nuclei. With this scheme, DD/CSA relaxation interference, which has been known for many years (35, 36), can be used to extend the limits of protein NMR.
Results
The NMR experiments with the uniformly 15N-labeled ftz homeodomain complexed with a 14-bp DNA duplex were performed at 4°C. The T1/T2 ratio of 15N was used to estimate the effective global correlation time, τc, of the complex (20). For the backbone amide groups, average T1 (15N) and T2 (15N) values of 0.720 ± 0.03 and 0.042 ± 0.005 s, respectively, were measured at 400 MHz, resulting in a global rotational correlation time of τc = 20 ± 2 ns. This τc value corresponds to that expected for a spherical protein of size 40 kDa in H2O solution at 35°C.
Fig. 2 shows a small region from 15N–1H correlation spectra of the ftz homeodomain-DNA complex that contains the resonance of the indole 15N–1H moiety of Trp-48, which is buried in the core of the protein (19). In the conventional [15N,1H]COSY experiment (22, 23), decoupling of 1H and 15N during the time periods t1 and t2, respectively, leads to detection of a single correlation peak per 15N–1H moiety (Fig. 2a). If the same [15N,1H]COSY spectrum is recorded without decoupling, four cross-peaks are observed per 15N–1H moiety, which show largely different linewidths (Fig. 2b). The cross-peak at (ω1 = 130.7 ppm, ω2 = 10.78 ppm) exhibits the broadest linewidths in both dimensions, which shows that it originates from the rapidly relaxing components of both 1HN and 15N. One-dimensional cross-sections taken along ω2 and ω1 at the positions indicated by arrows in the spectra presented in Fig. 2 show that the two cross-peaks at (ω1 = 132.1 ppm, ω2 = 10.78 ppm) and (ω1 = 130.7 ppm, ω2 = 10.65 ppm) are broadened either along ω1 or along ω2 (Fig. 3). The cross-peak at (ω1 = 132.1 ppm, ω2 = 10.65 ppm) displays narrow linewidths in both dimensions, showing that it originates from the two slowly relaxing components of the 15N–1H doublets. The TROSY-type correlation experiment, which does not use decoupling either during t1 or t2, contains only this narrowest correlation peak (Fig. 2c), which shows about 60% and 40% decrease in the linewidths of the 15N and 1H resonances, respectively, when compared with the collapsed cross-peak in the conventional, broadband-decoupled spectrum (Fig. 2).
Figure 2.

Contour plots of 15N,1H correlation spectra showing the indole 15N–1H spin system of Trp-48 recorded in a 2 mM solution of uniformly 15N-labeled ftz homeodomain complexed with an unlabeled 14-bp DNA duplex in 95% H2O/5% 2H2O at 4°C, pH = 6.0, measured at the 1H frequency of 750 MHz. (a) Conventional broad-band decoupled [15N,1H]COSY spectrum (22, 23). The evolution caused by the 1J(1H,15N) scalar coupling was refocused in the ω1 and ω2 dimensions by a 180° proton pulse in the middle of the 15N evolution time t1, and by waltz composite pulse decoupling of 15N during data acquisition, respectively. (b) Conventional [15N,1H]COSY spectrum recorded without decoupling during t1 and t2. (c) TROSY-type 15N,1H correlation spectrum recorded with the pulse scheme of Fig. 1. Chemical shifts relative to DSS in ppm and shifts in Hz relative to the center of the multiplet are indicated in both dimensions. The arrows identify the locations of the cross-sections shown in Fig. 3.
Figure 3.

Cross-sections through the spectra of Fig. 2 (solid lines). To facilitate a comparison of the linewidths in the different spectra the cross-sections were normalized to the same maximal signal amplitude. (a1), (a2), etc. refer to the arrows in Fig. 2. Simulated line shapes (dashed lines in a and b) were calculated using 1J(1H,15N) = −105 Hz, a rotational correlation time of τc = 20 ns, and chemical shift anisotropies of ΔσH = −16 ppm and ΔσN = −160 ppm. A long-range scalar coupling 2J(1Hδ1,15Nɛ1) = −5 Hz was included in the simulation of the 15N lineshapes (24), but possible effects of the small scalar couplings 3J(1Hδ1,1Hɛ1) and 3J(1Hζ2,15Nɛ1) were neglected. For 1HN the relaxation due to DD coupling with other protons in the nondeuterated complex was approximated by three protons placed at a distance of 0.24 nm from 1HN.
The fits of the experimental line shapes shown in Fig. 3 were obtained with line-shape calculations using the parameters τc = 20 ns and 1J(1H,15N) = 105 Hz, where the chemical shift anisotropies, ΔσH and ΔσN, were adjusted for the best fit. Because there was an otherwise unaccountable deviation from the Lorentzian lineshape we included a long-range scalar coupling 2J(1Hδ1,15Nɛ1) = −5 Hz (24) in the calculations, and T1 and T2 relaxation of 1HN because of DD coupling with other protons was modeled by placing three protons at a distance of 0.24 nm from 1HN. Application of one or a series of 180° pulses on spin I during the evolution of spin S interchanges the slowly and rapidly relaxing components of the S multiplet, which results in averaging of the slow and fast relaxation rates and elimination of the CSA/DD interference (25, 26). Indeed, the line shapes of the 1HN and 15N resonances measured with conventional [15N,1H]COSY are well reproduced if the average of the two relaxation rates is used in the simulation (Fig. 3 a1 and b1). The best-fit values of ΔσH = −16 ppm and ΔσN = −160 ppm correspond closely to the experimentally measured chemical shift anisotropies of 1H and 15N in 15N–1H moieties. With solid-state NMR studies of 15N–2D moieties, values for ΔσD near −14 ppm (27) and for ΔσN of −160 ppm (28) were determined. Independently, solution NMR experiments yielded values for ΔσH of backbone amide protons in the range 3 to 15 ppm (37) and ΔσN near −170 ppm (10).
Discussion
In the experiments with the ftz homeodomain-DNA complex the overall transverse relaxation rates of 15N and 1HN in the indole 15N–1H moiety of a buried tryptophan were reduced by 60% and 40%, respectively, when using a TROSY-type [15N,1H]correlation experiment instead of the conventional [15N,1H]COSY scheme. At a first glance this may appear to be a modest improvement, but a closer look reveals that DD coupling with remote protons, which could be nearly completely suppressed by replacement of the nonlabile hydrogen atoms with 2H (e.g., refs. 6 and 8), accounts for 95% of the residual T2(1HN) relaxation and 75% of the residual T2(15N) relaxation. In a corresponding DNA complex with the perdeuterated ftz homeodomain the reduction of the T2 relaxation rates of the 15N–1H moieties by the use of TROSY at 750 MHz would be about 40-fold for 1HN and about 10-fold for 15N.
Using Eqs. 1–6 with ΔσH = −16 ppm, ΔσN = −160 ppm, rHN = 0.101 nm and parallel orientation of the principal axis of the CSA tensor with the vector rHN, we evaluated the dependence of the residual T2 relaxation rates of 15N and 1H in TROSY-type experiments on the polarizing magnetic field B0 and the molecular size. These calculations showed that nearly complete compensation of T2 relaxation because of DD and CSA within the 15N–1H moieties is obtained at a B0 strength corresponding to a 1H frequency near 1,100 MHz, i.e., at this field strength (p − δS) ≅ 0 and (p − δI) ≅ 0 in Eq. 2. Theory further predicts that the residual TROSY T2 relaxation rates because of DD and CSA interactions within the 15N–1H fragment are practically independent of the molecular size. For perdeuterated proteins the size limit for TROSY-type [15N,1H]correlation experiments thus is not critically determined by T2 relaxation, but one needs nonetheless to consider that the effect of deuteration of the nonlabile proton sites in the protein is dependent on conformation. For the 15N–1H moieties in β-sheet secondary structure, DD and CSA interactions within the 15N–1HN fragment are the only sources of transverse relaxation that need to be considered, whereas in α-helices the two sequentially adjacent 1HN protons (3) contribute significantly to the transverse relaxation of the 15N and 1HN spins.
To provide a tangible illustration (Fig. 4) we calculated the 1HN and 15N line shapes for two perdeuterated spherical proteins in 1H2O solution with rotational correlation times τc of 60 and 320 ns, which corresponds to molecular masses of 150 and 800 kDa, respectively. A magnetic field B0 corresponding to a resonance frequency 750 MHz for protons was assumed. To account for the worst possible situation for DD interaction with remote protons, two protons were placed at 0.29 nm from 1HN. The linewidth of the narrow component of the 15N doublet increases only slightly with molecular mass and is about 5 Hz at 150 kDa and 15 Hz for a 800-kDa protein (Fig. 4). The 1HN linewidth depends more strongly on the residual DD interactions with remote protons and is about 15 Hz at 150 kDa and 50 Hz for a 800-kDa protein. For the 150-kDa protein these numbers correspond to 10- and 4-fold reduction of the 15N and 1HN TROSY linewidths, respectively, when compared with a conventional [15N,1H]COSY experiment with broad-band decoupling of 15N and 1H. For large molecular sizes the experimental scheme of Fig. 1 may, in principle, be further improved by elimination of the 180° refocusing radio frequency (rf)-pulses during the insensitive nuclei enhanced by polarization transfers (INEPTs), because during the INEPT mixing times these pulses mix the multiplet components with slow and fast T2 relaxation in a similar way as during the entire experiment in conventional [15N,1H]COSY. The elimination of decoupling sequences and 180° pulses from TROSY-type NMR pulse sequences also may have implications for future probe designs, because the constraints by the requirements for minimal radio frequency heating and maximal B1 homogeneity then may be relaxed, permitting a better optimization of other parameters such as sensitivity or sample diameter.
Figure 4.

1H and 15N lineshapes predicted for the broad and narrow multiplet components of 1HN and 15N of the 15N–1H moiety in a [15N,1H]COSY experiment of the type of Fig. 3 b1 and b2 for large proteins in H2O solution at 20°C and a 1H frequency of 750 MHz. (a1 and a2) Spherical protein of size 150 kDa. For the calculation a rotational correlation time of 60 ns, ΔσH = −16 ppm and ΔσN = −160 ppm were used, and all nonlabile protons were replaced with deuterons. Relaxation due to DD coupling with other labile protons was modeled by placing two protons at a distance of 0.29 nm from 1HN. The full linewidths at half height are indicated. (b1 and b2) Spherical protein of size 800 kDa. The calculation used τc = 320 ns and otherwise the same parameters as in a.
The TROSY principle drastically reduces all major sources of relaxation throughout the entire NMR experiment, including signal acquisition, and is clearly distinct from the use of heteronuclear multiple-quantum coherence to reduce dipolar relaxation between heteronuclei (29), which previously was used for measurements of 3JHαHβ scalar coupling constants in proteins (30). Heteronuclear multiple-quantum coherences are subject to dipolar relaxation with remote spins as well as to CSA relaxation, which limits the use of these coherences at high polarizing magnetic fields. Moreover, the slow relaxation of the multiple-quantum coherences cannot be used during signal acquisition (14), which is critical for large molecules.
The following are some initial considerations on practical applications of the TROSY principle: (i) Because only one of the four multiplet components of 15N–1H moiety is retained in TROSY-type experiments, the conventional [15N,1H]COSY is intrinsically more sensitive. However, for measurements with proteins at 1H frequencies higher than 500 MHz, TROSY will provide a much better ratio of signal height to noise. (ii) TROSY-type [13C,1H]correlation experiments with the 13C–1H moieties of the aromatic rings of Tyr, Phe, and Trp yield comparable results to those for 15N–1H moieties (unpublished results). (iii) Two-dimensional nuclear Overhauser effect spectroscopy (NOESY) experiments correlating amide protons and aromatic protons can be relayed by TROSY-type heteronuclear correlation experiments. In favorable cases this might result in low resolution structures for several-fold larger proteins than have been accessible so far. (iv) We anticipate that a wide variety of NMR experiments currently used for resonance assignments and collection of conformational constraints can be optimized for larger molecular sizes by use of the TROSY approach in one or several dimensions.
Acknowledgments
We thank M. Wahl for the preparation of the ftz homeodomain-DNA complex and Dr. R. Brüschweiler for critical reading of the manuscript. Financial support was obtained from the Schweizerischer Nationalfonds (Project 31.49047.96).
ABBREVIATIONS
- rf
radio frequency
- DD
dipole–dipole
- CSA
chemical shift anisotropy
- COSY
correlation spectroscopy
- TROSY
transverse relaxation-optimized spectroscopy
- ftz homeodomain
fushi tarazu homeodomain polypeptide of 70 amino acid residues, with the homeodomain in positions 3–62
Quantitative Analysis of TROSY
The coherence transfer during the pulse sequence of Fig. 1 was evaluated using the product operator formalism (31) as implemented in the program poma (32), and the resulting phases of the rf-pulses and the receiver were transferred into the experimental pulse program according to (33). The transverse proton magnetization after the first 90° pulse on protons (a in Fig. 1) then is given by Eq. 8:
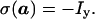 |
8 |
During the delay 2τ1 the scalar coupling 1J(1H,15N) evolves, so that the first 90°(15N) pulse generates two-spin coherence. With τ1 = 1/(41J(1H,15N)) we have at time b for the first step of the phase cycle (Fig. 1):
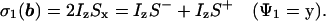 |
9 |
The evolution of these terms during t1, including relaxation, was evaluated using the single-transition basis operators S12± and S34±:
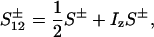 |
10 |
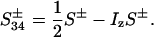 |
11 |
The time evolution of the expectation values of these operators can be obtained by integration of Eq. 1 with initial conditions derived from Eq. 9 and the assumption that 1/T1I ≪ 1J(1H,15N), which results in the following density matrix at time c:
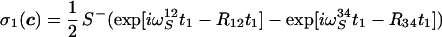 |
12 |
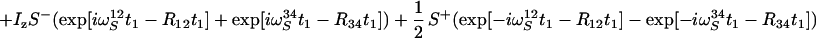 |
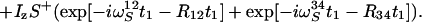 |
The relaxation factors Rij are related to the individual relaxation rates of the multiplet components by Eqs. 13 and 14:
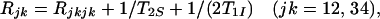 |
13 |
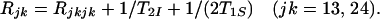 |
14 |
The subsequent polarization transfer step (time period c to d in Fig. 1) links the evolution period t1 with the acquisition period t2. The density matrix at time point c is represented by Eq. 15, where only those I− and I−Sz coherences are retained that result in detectable signals during data acquisition:
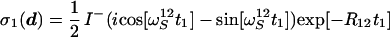 |
15 |
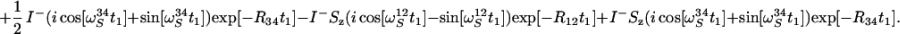 |
The other steps in the phase cycle of Fig. 1 can be analyzed in an analogous fashion. Accumulation of the eight transients of the pulse sequence results in the following density matrix for the real part of the interferogram:
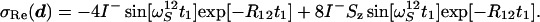 |
16 |
Incrementation of the phase ψ1 by 90° at each discrete value of t1 leads to the corresponding imaginary part:
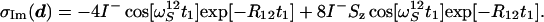 |
17 |
Eqs. 16 and 17 are combined to the hypercomplex interferogram that represents pure phase correlation in the ω1 dimension.
The treatment of the relaxation of the 1HN coherences during the acquisition period t2 is similar to the treatment of 15N during t1 (see Theory). The signals generated by I− and I−Sz coherences that are received during t2 are described by Eqs. 18 and 19, respectively:
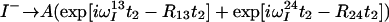 |
18 |
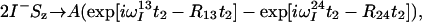 |
19 |
where A is a proportionality coefficient. Substitution of Eqs. 18 and 19 into Eqs. 16 and 17 results in the hypercomplex interferogram corresponding to the 1→2 and 2→4 transitions of the 1H,15N spin system:
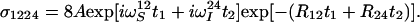 |
20 |
Finally, the Fourier transformation of the hypercomplex interferogram represented by Eq. 20 results in the pure absorptive correlation spectrum, with resonance frequencies in ω1 and ω2 corresponding to the desired individual component of the 15N–1H multiplet.
References
- 1.Shulman R G, Ogawa S, Wüthrich K, Yamane T, Peisach J, Blumberg W E. Science. 1969;165:251–257. doi: 10.1126/science.165.3890.251. [DOI] [PubMed] [Google Scholar]
- 2.Arata Y, Kato K, Takahashi H, Shimada I. Methods Enzymol. 1994;239:440–464. doi: 10.1016/s0076-6879(94)39017-7. [DOI] [PubMed] [Google Scholar]
- 3.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 4.Wüthrich K. NMR in Structural Biology. Singapore: World Scientific; 1995. [Google Scholar]
- 5.Wüthrich K. Acta Cryst D. 1995;51:249–270. doi: 10.1107/S0907444994010188. [DOI] [PubMed] [Google Scholar]
- 6.Shan X, Gardner K H, Muhandiram D R, Rao N S, Arrowsmith C H, Kay L E. J Am Chem Soc. 1996;118:6570–6579. [Google Scholar]
- 7.Wagner G. J Biomol NMR. 1993;3:375–385. doi: 10.1007/BF00176005. [DOI] [PubMed] [Google Scholar]
- 8.Nietlispach D, Clowes R T, Broadhurst R W, Ito Y, Keeler J, Kelly M, Ashurst J, Oschkinat H, Domaille P J, Laue E D. J Am Chem Soc. 1996;118:407–415. [Google Scholar]
- 9.Guéron M, Leroy J L, Griffey R H. J Am Chem Soc. 1983;105:7262–7266. [Google Scholar]
- 10.Tjandra N, Szabo A, Bax A. J Am Chem Soc. 1996;118:6986–6991. [Google Scholar]
- 11.Farrar T C, Stringfellow T C. In: Encyclopedia of NMR. Grant D M, Harris R K, editors. Vol. 6. New York: Wiley; 1996. pp. 4101–4107. [Google Scholar]
- 12.Vold R R, Vold R L. Prog NMR Spectrosc. 1978;12:79–133. [Google Scholar]
- 13.Ernst R R, Bodenhausen G, Wokaun A. The Principles of Nuclear Magnetic Resonance in One and Two Dimensions. Oxford: Clarendon; 1987. [Google Scholar]
- 14.Abragam A. The Principles of Nuclear Magnetism. Oxford: Clarendon; 1961. [Google Scholar]
- 15.Goldman M. J Magn Reson. 1984;60:437–452. [Google Scholar]
- 16.London R E. J Magn Reson. 1990;86:410–415. [Google Scholar]
- 17.Peng J W, Wagner G. J Magn Reson. 1992;98:308–332. [Google Scholar]
- 18.Percival-Smith A, Müller M, Affolter M, Gehring W J. EMBO J. 1990;9:3967–3974. doi: 10.1002/j.1460-2075.1990.tb07617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian Y Q, Furukubo-Tokunaga K, Resendez-Perez D, Müller M, Gehring W J, Wüthrich K. J Mol Biol. 1994;238:333–345. doi: 10.1006/jmbi.1994.1296. [DOI] [PubMed] [Google Scholar]
- 20.Kay L E, Torchia D A, Bax A. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 21.Farrow N A, Muhandiram R, Singer A U, Pascal S M, Kay C M, Gish G, Shoelson S E, Pawson T, Forman-Kay J D, Kay L E. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 22.Müller L. J Am Chem Soc. 1979;101:4481–4484. [Google Scholar]
- 23.Bodenhausen G, Ruben D J. Chem Phys Lett. 1980;69:185–189. [Google Scholar]
- 24.Bystrov V F. Prog NMR Spectrosc. 1976;10:41–81. [Google Scholar]
- 25.Palmer A G, Skelton N J, Chazin W J, Wright P E, Rance M. Mol Phys. 1992;75:699–711. [Google Scholar]
- 26.Kay L E, Nicholson L K, Delaglio F, Bax A, Torchia D A. J Magn Reson. 1992;97:359–375. [Google Scholar]
- 27.Michal C A, Wehman J C, Jelinski L W. J Magn Reson Ser B. 1996;111:31–39. doi: 10.1006/jmrb.1996.0057. [DOI] [PubMed] [Google Scholar]
- 28.Hiyama Y, Niu C, Silverton J V, Bavoso A, Torchia D A. J Am Chem Soc. 1988;110:2378–2383. [Google Scholar]
- 29.Griffey R H, Redfield A G. Quart Rev Biophys. 1987;19:51–82. doi: 10.1017/s0033583500004029. [DOI] [PubMed] [Google Scholar]
- 30.Grzesiek S, Kuboniwa H, Hinck A P, Bax A. J Am Chem Soc. 1995;117:5312–5315. [Google Scholar]
- 31.Sørensen O W, Eich G W, Levitt M H, Bodenhausen G, Ernst R R. Prog NMR Spectrosc. 1983;16:163–192. [Google Scholar]
- 32.Güntert P, Schaefer N, Otting G, Wüthrich K. J Magn Reson. 1993;101:103–105. [Google Scholar]
- 33.Levitt M H. J Magn Reson. 1997;126:164–182. [Google Scholar]
- 34.Piotto M, Saudek V, Sklenar V J. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 35.McConnell H M. J Chem Phys. 1956;25:709–715. [Google Scholar]
- 36.Shimizu H. J Chem Phys. 1964;40:3357–3364. [Google Scholar]
- 37.Tjandra N, Bax A. J Am Chem Soc. 1997;119:8076–8082. [Google Scholar]