

Abstract
Recently, we identified a thiocarbazate that exhibits potent inhibitory activity against human cathepsin L. Since this structure represents a novel chemotype with potential for activity against the entire cysteine protease family, we designed, synthesized and assayed a series of analogs to probe the mechanism of action, as well as the structural requirements for cathepsin L activity. Molecular docking studies using coordinates of a papain-inhibitor complex as a model for cathepsin L provided useful insights.
Human cathepsin L is an endosomal cysteine protease that has been implicated in a variety of physiological and pathophysiological processes.1-3 Cathepsin L is widely distributed, and plays key roles in bone remodeling and the immune response, as well as in disease states such as cancer,4 rheumatoid arthritis5 and osteo-arthritis.6,7 Furthermore, a number of infectious agents (e.g., Ebola, SARS, and Leishmania) have been reported to require cathepsin L or cathepsin L-like activity for viral processing and infectivity.8-12 As such, the identification of inhibitors of cathepsin L would provide valuable tools to probe the role of this enzyme in biological systems, as well as to provide potential starting points for drug discovery efforts.
The Penn Center for Molecular Discovery (PCMD), a member of the Molecular Libraries Screening Center Network (MLSCN), has conducted a series of high throughput screening (HTS) campaigns of the Molecular Libraries Small Molecular Repository (MLSMR) to identify inhibitors of cysteine (cathepsins B,13 L, and S) and serine (cathepsin G, Factor XIa, and XIIa) proteases.14 This effort recently led to the identification15 and characterization16 of (-)-1, a novel and potent inhibitor of human cathepsin L (Figure 1).17
Figure 1.

Thiocarbazate cathepsin L inhibitor (−)-1.
Most cysteine protease inhibitors require the presence of an electrophilic warhead that provides a site of reaction (either reversible or irreversible) for the active site thiolate. Selectivity and potency are often dictated by the reactivity of the warhead in conjunction with additional binding interactions of the molecule across the enzyme active site. Classic warheads include epoxides, nitriles, activated carbonyls, vinyl sulfones, oxocarbazates, and aza-peptides.2,18-20 Indeed, incorporation of such warheads has led to cathepsin K and cathepsin S inhibitors currently in clinical trials.3 Potent inhibitors of cathepsin L that incorporate azepanones and cyanamides have also been described recently.21,22 To the best of our knowledge, thiocarbazates and their corresponding biological activity have not been described prior to our original report.15 Since the thiocarbazate core embodies the potential for broad utility as a cysteine protease inhibitor scaffold, we sought to understand further the requirements for activity within this substructure.
In an effort to evaluate the potential binding mode of (−)-1 with cathepsin L, we performed docking studies using the publicly available X-ray coordinates for papain complexed to a succinyl epoxide inhibitor (1cvz.pdb).23 The papain model was a relevant model for Cathepsin L due to the high degree of sequence homology between the binding sites of these two cysteine proteases. In these studies we observed the simultaneous occupation of the S2, S3 and S1′ subsites by hydrophobic and aromatic functionalities of thiocarbazate (−)-1 as shown in Figure 2; the indole side-chain occupies the S2 subsite, the -NHBoc group group the S3 subsite, and the 2-ethylphenyl aniline the S1′ subsite. A key hydrogen bond is observed between the Gly66 backbone NH and the amino acid derived carbonyl of the diacyl hydrazine. In other inhibitor systems, the absence of a hydrogen bonding interaction between Gly66 and inhibitor has been reported to lead to a loss of inhibition in numerous cathepsins, including cathepsin L.22 Details of the molecular docking studies are reported elsewhere,24 however they suggest that (a) the thiocarbazate carbonyl is in sufficient proximity to the active site Cys25 to permit nucleophilic thiolate addition; and (b) significant binding interactions (both hydrogen bonding and van der Waals) are present between the inhibitor and protease subsites. These observations supported our hypothesis that specific binding interactions as well as appropriate reactivity of (−)-1 are essential for the observed inhibitory properties.
Figure 2.
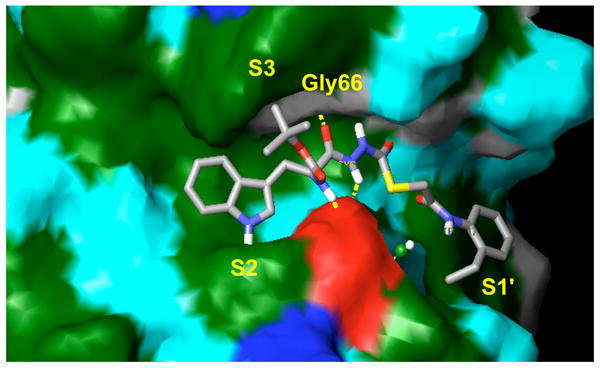
Thiocarbazate (−)-1 (IC50 = 56 nM) in the binding subsite of papain. The indole forms hydrophobic contacts within the S2 subsite, the -NHBoc group forms hydrophobic contacts within the S3 subsite, and the 2-ethylphenyl anilide occupies the S1′ subsite.
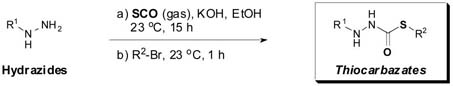
The docking studies were validated by the synthesis of analogs in which key residues occupying the S2, S3 and S1′ subsites were modified. Specifically, replacement of the indole side-chain, the NHBoc, and the 2-ethylphenyl anilide of (−)-1 were targeted. Thiocarbazates for this study were prepared from the requisite hydrazides exploiting our previously developed chemistry.15 In a one-pot reaction, hydrazides were treated with carbonyl sulfide gas followed by an appropriate electrophile (i.e., R2-Br). Preparative reverse phase HPLC was employed to purify the final products,25 which were assayed for their ability to inhibit cathepsin L.26
As illustrated by the results listed in Table 1, occupation of the S2 subsite is essential for cathepsin L inhibition. Partial occupation, as in (−)-2 where the indole side chain is replaced with the smaller phenyl group, results in less potent activity (IC50 = 115 nM vs. 56 nM). Thiocarbazate 3, in which the entire indole side chain has been eliminated, exhibits no inhibition. Also pronounced are the -NHBoc group's contributions to potency, as illustrated by thiocarbazate 4's significantly reduced activity (IC50 = 22 μM). In this case, we reason that the loss of a key hydrogen bond between the NHBoc group and the Asp158 residue, leads to diminished activity. These results support the importance of maintaining hydrophobic and hydrogen bonding interactions in the active site, consistent with the mode of docking proposed.
Table 1.
Synthesis and cathepsin L inhibitory activity of thiocarbazates.
 | |||
|---|---|---|---|
| thiocarbazate | R1 | R2 | IC50 (? M) |
| (-)-1 | Boc-l-Trp |
|
0.056 ± 0.001 |
| (-)-2 | Boc-l-Phe | 0.115 ± 0.006 | |
| 3 | Boc-Gly | inactive | |
| 4 |
|
21.797 ± 1.836 | |
| (-)-5 | Boc-l-Trp |
|
0.041 ± 0.002 |
| 6 | Boc-l-Trp |
|
0.110 ± 0.003 |
| (-)-7 | Boc-l-Trp |
|
0.201 ± 0.012 |
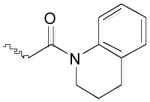
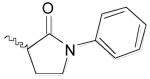
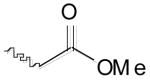
From the docking studies of thiocarbazate (−)-1,16,24 we hypothesized that additional room for structural modifications and ring constraints was available in the anilide portion of this thiocarbazate (S1′ subsite). Based on this observation, a tetrahydroquinoline anilide (−)-5 was substituted for the 2-ethylphenyl anilide moiety (Table 1).27 An improvement in potency was observed (IC50 = 41 nM), further supporting our hypothesis. To explore this area further, two additional analogs were prepared: thiocarbazate 6, in which constraints were imposed by incorporation of an N-phenyl pyrrolidinone group, and a methyl ester thiocarbazate (−)-7.28,29 Both analogs 6 and (−)-7 exhibited reduced activity against cathepsin L with IC50 values of 110 nM and 201 nM, respectively.
Our thiocarbazates are structurally related to oxocarbazates (e.g., A) and aza-peptides (e.g., B), known protease inhibitors (Figure 3)30-32 that are active by the virtue of their activated carbonyl groups. Depending on the nature of the leaving group present, these inhibitors often bind and react with the active site serine or cysteine, resulting in the formation of a stable acyl-enzyme complex, which then undergoes slow hydrolysis.30,31 Alternatively, oxocarbazate and aza-peptide inhibitors, with poor leaving groups are believed to form stable tetrahedral intermediates without acyl enzyme adduct formation.18,33,34 To further understand the cathepsin L inhibitory activity of thiocarbazates, (−)-1 was incubated in the presence of stoichiometric amounts of cysteine or cathepsin L over prolonged time periods in the presence of assay buffer. The reaction was monitored by LC-MS for the disappearance of (−)-1 as well as the appearance of reaction products such as cysteine adducts and products of hydrolysis.35 In both experiments, thiocarbazate (−)-1 was found to remain unchanged after greater than 24 hours, and no evidence of newly formed reaction products were detected. As the formation of a stable acyl-enzyme complex would have resulted in the generation of a new product based on the leaving group embedded in (-)-1, we hypothesize that a stable acyl-enzyme adduct is not formed. These data are consistent with detailed kinetic analysis16 indicating (-)-1 is a slowly reversible inhibitor of Cathepsin L.
Figure 3.

Examples of known oxocarbazate A and aza-peptide B inhibitors.
To probe the necessity of the thiocarbazate core in inhibitors such as (−)-1 and (-)-5, a series of analogs were prepared in which a carbon, oxygen, or nitrogen atom replaced of the sulfur atom. This series was designed to incorporate the preferred S1′ subsite substituent, the tetrahydroquinoline anilide. Toward this end, the corresponding diacyl hydrazine (−)-9, aza-peptide (−)-11, and oxocarbazate (−)-13 were prepared as illustrated in Scheme 1.
Scheme 1.

Synthesis of tetrahydroquinoline derived diacyl hydrazine (−)-9, aza-peptide (−)-11, and oxocarbazate (−)-13 analogs starting from Boc-l-Trp-NHNH2.
Diacyl hydrazine (−)-9 was prepared from succinanilic acid368 via an EDC mediated coupling reaction with Boc-l-Trp-NHNH2. Aza-peptide (−)-11 was generated via the reaction of preformed isocyanate 10 with Boc-l-Trp-NHNH2.37 Oxocarbazate (−)-13 was synthesized using a three-component protocol using Boc-l-Trp-NHNH2, α-bromoanilide 12, and carbon dioxide.38,39 While yields for these analogs were modest 33-65%, 1 mmol scale reactions yielded material of >99% purity for biological assay. When tested for inhibition of cathepsin L, diacyl hydrazine (−)-9 was found to be inactive and aza-peptide analog (−)-11 displayed only modest potency. In these cases, the presence of the preferred tetrahydroquinoline anilide could not compensate for the lack of an activated carbonyl [e.g., (-)-9] or unoptimized carbonyl reactivity (e.g., (-)-11]. In contrast, oxocarbazate (−)-13, in which an oxygen atom was substituted for the sulfur atom of (−)-5, was the most potent cathepsin L inhibitor identified within this study (IC50 = 7 nM).40,41
As in the case of (−)-1, oxocarbazate (−)-13 and thiocarbazate (−)-5 were found to be unreactive to transesterification by cysteine and DTT nucleophiles. Furthermore, (-)-13 remained intact for greater than 24 hours when incubated with Cathepsin L under stoichiometric conditions, and in the presence of assay buffer.26 Electrostatic potential calculations were also conducted to determine the relative electrophilicities of the carbonyls within the tetrahydroquinoline substituted inhibitors; however the results from these studies gave no clear correlation between electrophilicity and activity, further supporting our hypothesis that both reactivity as well as binding interactions dictate potency.
In summary, through the design, synthesis and assay of a series of thiocarbazates we have characterized the activity of a novel family of cathepsin L inhibitors. Based on these studies we conclude that full occupancy of the S2, S3 and S1′ subsites is required for potent inhibition. With these requirements met, the activated carbonyl group is positioned in close proximity to the Cys25 active site residue. Although we observed no evidence of reaction between the protein and the inhibitors, changes in the functionality adjacent to the putative reactive carbonyl (i.e., sulfur, carbon, oxygen, and nitrogen analogs) strongly influenced potency. In the course of these studies, we designed and synthesized a highly potent cathepsin L inhibitor, oxocarbazate (−)-13 that contains a preferred tetrahydroquinoline anilide group. Future efforts in our laboratory will focus on the thiocarbazate chemotype and its potential to exhibit broad cysteine protease inhibitory activity.
Acknowledgments
Financial support for this work was provided by the NIH (5U54HG003915-02 and 5U54HG003915-03). We thank Dr. Carlo E. Ballatore and Mr. Onur Atasoylu for electrostatic potential calculations and Professor Barry S. Cooperman for helpful discussions. Finally we thank Dr. G. T. Furst and Dr. R. Kohli at the University of Pennsylvania for assistance in obtaining NMR and high-resolution mass spectra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.McGrath ME. Annu Rev Biophys Biomol Struct. 1999;28:181. doi: 10.1146/annurev.biophys.28.1.181. [DOI] [PubMed] [Google Scholar]
- 2.Lecaille F, Kaleta J, Brömme D. Chem Rev. 2002;102:4459. doi: 10.1021/cr0101656. [DOI] [PubMed] [Google Scholar]
- 3.Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B. Curr Pharm Des. 2007;13:385. doi: 10.2174/138161207780162962. [DOI] [PubMed] [Google Scholar]
- 4.Felbor U, Dreier L, Bryant RAR, Ploegh HL, Olsen BR, Mothes W. EMBO J. 2000;19:1187. doi: 10.1093/emboj/19.6.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schedel J, Seemayer CA, Pap T, Neidhart M, Kuchen S, Michel BA, Gay RE, Müller-Ladner U, Gay S, Zacharias W. Gene Therapy. 2004;11:1040. doi: 10.1038/sj.gt.3302265. [DOI] [PubMed] [Google Scholar]
- 6.Esser RE, Angelo RA, Murphey MD, Watts LM, Thornburg LP, Palmer JT, Talhouk JW, Smith RE. Arthritis Rheum. 1994;37:236. doi: 10.1002/art.1780370213. [DOI] [PubMed] [Google Scholar]
- 7.Hill PA, Buttle DJ, Jones SJ, Boyde A, Murata M, Reynolds JJ, Meikle MC. J Cell Biochem. 1994;56:118. doi: 10.1002/jcb.240560116. [DOI] [PubMed] [Google Scholar]
- 8.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Science. 2005;308:1643. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. J Virol. 2006;80:4174. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaletsky RL, Simmons G, Bates P. J Virol. 2007;81:13378. doi: 10.1128/JVI.01170-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Proc Natl Acad Sci USA. 2005;102:11876. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rafati S, Salmanian AH, Hashemi K, Schaff C, Belli S, Fasel N. Mol Biochem Parasitol. 2001;113:35. doi: 10.1016/s0166-6851(00)00377-7. [DOI] [PubMed] [Google Scholar]
- 13.Myers MC, Napper AD, Motlekar N, Shah PP, Chiu CH, Beavers MP, Diamond SL, Huryn DM, Smith AB., III Bioorg Med Chem Lett. 2007;17:4761. doi: 10.1016/j.bmcl.2007.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penn Center for Molecular Discovery (PCMD): http://www.seas.upenn.edu/∼pcmd/Molecular Library Screening Center Network (MLSCN): http://nihroadmap.nih.gov/molecularlibraries/Molecular Libraries Small Molecular Repository (MLSMR): http://mlsmr.glpg.com/MLSMR_HomePage/index.htmlPubChem: http://pubchem.ncbi.nlm.nih.gov/
- 15.Myers MC, Shah PP, Diamond SL, Huryn DM, Smith AB., III Bioorg Med Chem Lett. 2008;18:210. doi: 10.1016/j.bmcl.2007.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah PP, Myers MC, Beavers MP, Purvis JE, Jing H, Grieser HJ, Sharlow ER, Napper AD, Huryn DM, Cooperman BS, Smith AB, III, Diamond SL. Mol, Pharm. doi: 10.1124/mol.108.046219. Accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.PubChem Substance number for (−)-1 is SID 26681509
- 18.Otto HH, Schirmeister T. Chem Rev. 1997;97:133. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- 19.Schirmeister T, Kaeppler U. Mini Rev Med Chem. 2003;3:361. doi: 10.2174/1389557033488079. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez AA, Roush WR. Curr Opin Chem Bio. 2002;6:459. doi: 10.1016/s1367-5931(02)00345-9. [DOI] [PubMed] [Google Scholar]
- 21.Falgueyret JP, Oballa RM, Okamoto O, Wesolowski G, Aubin Y, Rydzewski RM, Prasit P, Riendeau D, Rodan SB, Percival MD. J Med Chem. 2001;44:94. doi: 10.1021/jm0003440. [DOI] [PubMed] [Google Scholar]
- 22.Marquis RW, James I, Zeng J, Trout REL, Thompson S, Rahman A, Yamashita DS, Xie R, Gress CJ, Blake S, Lark MA, Hwang SM, Tomaszek T, Offen P, Head MS, Cummings MD, Veber DF. J Med Chem. 2005;48:6870. doi: 10.1021/jm0502079. [DOI] [PubMed] [Google Scholar]
- 23.Tsuge H, Nishimura T, Tada Y, Asao T, Turk D, Turk V, Katunuma N. Biochem Biophys Res Commun. 1999;266:411. doi: 10.1006/bbrc.1999.1830. [DOI] [PubMed] [Google Scholar]
- 24.Beavers, M. P.; Myers, M. C.; Shah, P. P.; Purvis, J. E.; Diamond, S. L.; Cooperman, B. S.; Huryn, D. M.; Smith, A. B., III. (Manuscript submitted for publication).
- 25.General procedure to form amino acid-substituted thiocarbazates: Boc-protected amino acid hydrazide (1.0 mmol, 1.0 equiv.) was added to a 25 mL round bottom flask followed by a solution of KOH in 95% EtOH (0.25 M, 4.4 mL, 1.1 equiv.). After stirring for 5 min at 23 °C, a balloon of carbonyl sulfide gas was attached to the flask. The flask was purged with the gas (5 s) and a full balloon was reattached. The reaction was stirred for 15 h at 23 °C. After stirring overnight, the α-bromo anilide (1.1 mmol, 1.1 equiv.) was added in one portion and the reaction was monitored by LC-MS. The α-bromo anilides were typically consumed within 20 to 60 min, and the reaction mixture was filtered, using a Büchner funnel. The filtrate was concentrated on a rotatory evaporator and purified by preparative reverse phase HPLC.
- 26.High throughput screening (HTS), followup (cherry picks) studies, and analog activity analysis were conducted with the following assay buffer: 20 mM sodium acetate, 1 mM EDTA, and 5 mM DTT, pH 5.5. Confirmatory results were obtained utilizing the following assay conditions, replacing DTT with cysteine in the assay buffer: Compounds were serially diluted in DMSO and transferred into a 96-well Corning 3686 assay microplate to give 16 dilutions ranging from 50 μM to 1.5 nM. Human liver cathepsin L (Calbiochem 219402) was activated by incubating with assay buffer for 30 min. Assay buffer consisted of 20 mM sodium acetate, 1 mM EDTA, and 5 mM cysteine, pH 5.5. Upon activation, cathepsin L (300 pM) was incubated with 1 μM Z-Phe-Arg-AMC substrate and test compound in 100 μL of assay buffer for 1 h at room temperature. Fluorescence of AMC released by enzyme-catalyzed hydrolysis of Z-Phe-Arg-AMC was read on a PerkinElmer Envision microplate reader (excitation 355 nm, emission 460 nm). Data was scaled using internal controls and fit to a four parameter logistic model (IDBS XLfit equation 205) to obtain IC50 values in triplicate.
- 27.2-bromo-1-(3,4-dihydro-2H-quinolin-1-yl)-ethanone was used as the α-bromo anilide electrophile. For a general procedure for the preparation of α-bromo anilides, see: von Geldern TW, Tasker AS, Sorensen BK, Winn M, Szczepankiewicz BG, Dixon DB, Chiou WJ, Wang L, Wessale JL, Adler A, Marsh KC, Nguyen B, Opgenorth TJ. J Med Chem. 1999;42:3668. doi: 10.1021/jm990170q.
- 28.(±)-3-bromo-1-phenyl-2-pyrrolidinone was used as the α-bromo anilide electrophile. The resulting thiocarbazate 6 was assayed as a mixture of diastereomers.
- 29.Methyl bromoacetate was used as the α-bromo electrophile.
- 30.Magrath J, Abeles RH. J Med Chem. 1992;35:4279. doi: 10.1021/jm00101a004. [DOI] [PubMed] [Google Scholar]
- 31.Baggio R, Shi YQ, Wu Yq, Abeles RH. Biochemistry. 1996;35:3351. doi: 10.1021/bi952879e. [DOI] [PubMed] [Google Scholar]
- 32.Xing R, Hanzlik RP. J Med Chem. 1998;41:1344. doi: 10.1021/jm970802d. [DOI] [PubMed] [Google Scholar]
- 33.Rich DH, Brown MA, Barrett AJ. Biochem J. 1986;235:731. doi: 10.1042/bj2350731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bondebjerg J, Fuglsang H, Valeur KR, Kaznelson DW, Hanse JA, Pedersen RO, Krogh BO, Jensen BS, Lauritzen C, Petersen G, Pedersen J, Naerum L. Bioorg Med Chem. 2005;13:4408. doi: 10.1016/j.bmc.2005.04.048. [DOI] [PubMed] [Google Scholar]
- 35.The following standards were utilized as potential reaction products during the stoichiometric incubation of cathepsin L with (−)-1: L-Boc-Trp-NHNH2, N-(2-ethyl-phenyl)-2-mercapto-acetamide, and 2-ethyl-phenylamine.
- 36.For a general procedure for the preparation of succinanilic acids, see: Kar A, Argade NP. Synthesis. 2002:221.
- 37.Aza-peptides are commonly prepared from phosgene equivalents, resulting in a reactive isocyanate intermediate. For a representative example, see: Boeglin D, Lubell WD. J Comb Chem. 2005;7:864. doi: 10.1021/cc050043h.
- 38.For a general procedure used to prepare oxocarbazates such as (−)-13, see: Fox DL, Ruxer JT, Olive JM, Alford KL, Salvatore RN. Tetrahedron Lett. 2004;45:401.
- 39.Dyker H, Scherkenbeck J, Gondol D, Goehrt A, Harder A. J Org Chem. 2001;66:3760. doi: 10.1021/jo001749v. [DOI] [PubMed] [Google Scholar]
- 40.PubChem Substance number for (−)-13 is SID 46493575
- 41.Characterization data for (−)-13 mp = 157 °C; [α]D24 = -36.4 (c 0.1, AcOH); IR (KBr) 3413, 2926, 1685, 1654, 1162, 1121, 1061 cm-1; 1H NMR (500 MHz, DMSO-d6, VT-350K) δ 10.62 (br s, 1H), 9.76, (br s, 1H), 9.10 (br s, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.20 (t, J = 7.0 Hz, 2H), 7.20-7.17 (m, 1H), 7.12 (t, J = 7.5 Hz, 1H), 7.06 (t, J = 7.0 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.32 (br s, 1H), 4.82 (br s, 2H), 4.31 (br m, 1H), 3.69 (t, J = 6.5 Hz, 2H) 3.16-3.12 (m, 1H) 2.97-2.92 (m, 1H), 2,74 (t, J = 6.5 Hz), 1.92 (pentet J = 6.5 Hz), 1.30 (br s, 9H); 13C NMR (125 MHz, DMSO-d6, major rotamer) δ 171.9, 166.5, 155.6, 155.1, 137.7, 136.0, 132.0, 128.7, 127.3, 125.9, 125.0, 124.9, 123.8, 120.8, 118.5, 118.1, 111.2, 109.9, 77.9, 62.1, 53.4, 42.8, 28.1, 27.6, 26.1, 23.2; high resolution mass spectrum (ES+) m/z 558.2325 [(M+Na)+; calcd for C28H33N5O6Na: 558.2329].