

Abstract
Immucillin-H (BCX-1777, forodesine) is a transition state analogue and potent inhibitor of PNP that shows promise as a specific agent against activated human T-cells and T-cell leukemias. The immunosuppressive or antileukemic effects of Immucillin-H (ImmH) require co-administration with deoxyguanosine (dGuo) to attain therapeutic levels of intracellular dGTP. In this study we investigated the requirements for sensitivity and resistance to ImmH and dGuo. 3H-ImmH transport assays demonstrated that the equilibrative nucleoside transporters (ENT1 and ENT2) facilitated the uptake of ImmH in human leukemia CCRF-CEM cells whereas 3H-dGuo uptake was primarily dependent upon concentrative nucleoside transporters (CNTs). Analysis of lysates from an ImmH-resistant CCRF-CEM-AraC-8D cells demonstrated undetectable deoxycytidine kinase (dCK) activity, suggesting that dCK and not deoxyguanosine kinase (dGK) was the rate-limiting enzyme for phosphorylation of dGuo in these cells. Examination of ImmH cytotoxicity in a hypoxanthine-guanine phosphoribosyltransferase (HGPRT)-deficient cell line CCRF-CEM-AraC-8C, demonstrated enhanced sensitivity to low concentrations of ImmH and dGuo. RT-PCR and sequencing of HGPRT from the HGPRT-deficient CCRF-CEM-AraC-8C cells identified an Exon 8 deletion mutation in this enzyme. Thus these studies show that specific nucleoside transporters are required for ImmH cytotoxicity and predict that ImmH may be more cytotoxic to 6-thioguanine (6-TG) or 6-thiopurine-resistant leukemia cells caused by HGPRT deficiency.
Introduction
The immucillins are a novel class of highly potent inhibitors of purine nucleoside phosphorylase (PNP) and are currently in clinical trials for the treatment of T cell specific leukemias 1. Immucillin-H (ImmH, also known as BCX-1777 or forodesine) is a non-cleavable analog of inosine and a transition state inhibitor of PNP that binds to PNP approximately 7×105 fold more tightly than the substrate inosine and inhibits human PNP with a Kd of 56 pM 2. ImmH has a slow rate of dissociation from the enzyme-complexes, causing potent inhibition 3, 4. Inhibition of PNP by immucillins results in an increased conversion of dGuo to intracellular dGTP, inhibition of DNA synthesis, and induction of apoptosis through dGTP-mediated inhibition of ribonucleotide reductase 5–7.
Nucleoside transporters play a critical role in the cellular transport of natural nucleoside substrates and nucleoside analogues used in the treatment of cancer. In mammalian cells, purine nucleoside transport is mediated by members of two families of transporters: the broad substrate specificity-Na+-independent equilibrative transporters (ENTs: ENT1 and ENT2) 8 and Na+-dependent concentrative transporters (CNTs: CNT2 and CNT3) 9. Although multiple nucleoside transporters may contribute to dGuo uptake, the transporters required for the uptake of dGuo, a key component of immucillin cytotoxicity, have not been determined. Moreover, the mechanism of uptake of the immucillins, a family of charged purine analogues also remains to be determined 10. Recent evidence showed that ImmH could potently inhibit the growth and induce apoptosis in human T type leukemia CCRF-CEM cells 1, 5, 6, suggesting that cytotoxic levels of ImmH accumulate in mammalian cells.
ImmH cytotoxicity requires the metabolism of dGuo to dGTP. Under normal conditions, dGuo may be metabolized to dGMP by deoxcytidine kinase (dCK) and/or deoxyguanosine kinase (dGK), or to guanine by purine nucleoside phosphorylase (PNP). When PNP is inhibited, the majority of dGuo is metabolized to dGMP and then to dGTP in two, non-rate limiting kinase reactions. Conversely, dGMP may be catabolized to dGuo by 5’-nucleotidases. T lymphoblasts are particularly sensitive to inhibition of PNP due to their inherently high phosphorylation of dGuo and their slow rate of catabolism of dGMP 11. In the absence of PNP inhibition, dGuo can be converted to guanine by PNP, and further converted to GMP through phosphoribosylation by hypoxanthine-guanine phosphoribosyltransferase (HGPRT) 10. Inhibition of HGPRT has been shown to enable cells to accumulate dGTP 12, 13 and lack of HGPRT activity may facilitate the ability of cancer cells to accumulate intracellular dGTP.
In this study we investigated the requirement for specific nucleoside transporters for the uptake of ImmH and dGuo and the role of the PNP-coupled HGPRT catabolic pathway in determining ImmH sensitivity. The results obtained here demonstrate a requirement for the ENTs in the rapid uptake of ImmH but not the cytotoxic effects of this compound. By contrast, the CNTs are predominantly responsible for the uptake of dGuo and could influence the cytotoxic responses to these agents. Lastly, these studies demonstrate a previously unrecognized but important role for HGPRT in determining the sensitivity to ImmH.
Materials and Methods
Cell culture conditions and reagents used
The nucleoside transport-deficient swine kidney tubular epithelial cell line PK15, and PK15 cell line transfected with human ENT1 (PK15-ENT1), and human ENT2 (PK15-ENT2) were generously provided by Dr. Chung-Ming Tse from the Johns Hopkins University School of Medicine 14. PK15 cells were maintained in Eagle's minimal essential medium/Earles Balanced Salt Solutions (1:1), with 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 5% fetal bovine serum, and 100 U/ml penicillin and 100 µg/ml streptomycin. The CCRF-CEM cell line was obtained from the ATCC (Rockville, Md.). CCRF-CEM and CCRF-CEM-AraC-8D cell line were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum and 100 U/ml penicillin and 100 µ/ml streptomycin. CCRF-CEM-AraC-8C cell line, a nucleoside transport-deficient subline of CCRF-CEM cell line was kindly provided by Dr. Buddy Ullmann (Oregon Health Sciences University) and was grown in Dulbecco’s Modified Eagle Medium (with L-glutamine, without glucose, and without sodium pyruvate). The cells were kept at 37° C in an atmosphere containing 5% CO2. NBMPR (nitrobenzylthioinosine, 6-[(4-nitrobenzyl) thio]-9-(β-D-ribofuranosyl) purine), deoxyguanosine (dGuo), and dipyridamole (DP) were from Sigma (Sigma Chemical Co., USA). Uridine ([5,6-3H], 35–50 Ci/mmol) and [8-14C] hypoxanthine (48 mCi/mmol) were purchased from ICN Biomedicals (Costa Mesa, Calif., USA). 2’-Deoxyguanosine ([8-3H], 10.0 Ci/mmol) was purchased from Moravek Biochemicals (Brea, Calif., USA).
HPRT enzyme activity
Twenty µg of protein extracted from tissues was assayed in a 50 µl reaction volume containing 125 µM [8-14C] hypoxanthine, 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, and 5 mM phosphoribosylpyrophosphate. Reactions were carried out at 37° C for 60 min, and 20 µl of the reaction product was spotted onto thin-layer chromatography plates (cellulose polyethyleneimine-F; J. T. Baker Inc., Phillipsburg, N.J.) and separated in 0.25 M sodium formate (pH 4.3) buffer.
Anion exchange chromatography of CCRF-CEM, CCRF-CEM-AraC-8C cell lysates
Cell extracts of CCRF-CEM, CCRF-CEM-AraC-8C cells were prepared by sonicating cells in buffer A (20 mM histidine (pH 6.0), 1 mM dithiothreitol (DTT). After centrifugation at 10,000 rpm for 15 min, the supernatants (16 mg of protein) were separated on a Mono Q™ HR 5/5 anion exchange column using the Pharmacia FPLC system as described previously with minor modification 15. Briefly, after loading samples to the column, the column was first washed with equilibration buffer A at 1.0 ml/min for 10 min, and the pre-gradient fractions were collected. Bound proteins were then eluted using a linear gradient of 0.1–0.5 M KCl in the equilibration buffer and collecting 1 ml of post-gradient fractions. Fractions were assayed for dCK and dGK activity as described below.
Assay of dCK and dGK
The activity of dCK and dGK was determined by measuring the phosphorylation of [3H]-deoxycytidine or [3H]-dGuo to [3H]-dCMP and [3H]-dGMP, respectively. In brief, 30 µl of each fractions from the anion exchange chromatography was incubated with 1 µCi [3H]-deoxycytidine or [3H]-dGuo, 1 µM cold deoxycytidine or dGuo in a final volume of 50 µl containing 50 mM Tris-HCl buffer (pH 7.4), 5% glycerol, 15 mM β-Me, 10 mM ATP, 1.1 mM magnesium chloride, 10 mM NaF, 25 mM DTT, at 37 °C for 20 min followed by incubation at 95 °C for 3 min to stop the reaction. An aliquot of 35 µL was applied to ion-exchange paper (Whatman DE-81), which was washed three times (10 min each time) with 95% ethanol, and the radioactivity retained on the paper was measured using a liquid scintillation spectrophotometer.
RNA isolation and RT-PCR
Total RNA was isolated from cultured cells using Trizol reagent (GIBCO BRL). Two µgs of total RNA was used for first-strand cDNA synthesis using oligo (dT) as primers and SuperScript II RNase H− Reverse Transcriptase (SuperScript™ First-Strand Synthesis System for RT-PCR, Invitrogen). One tenth of the resulting first-strand cDNA was then used for PCR amplification. Different sets of primers were designed and synthesized for PCR analysis. The primer pairs used for amplifying human HGPRT, GAPDH, PNP, CNT1-3, ENT1 and ENT2 were listed in Table 1. PCR products were analyzed by 1% agarose gel and visualized by staining with ethidium bromide.
Table 1.
| Gene | Primer sequence | Product |
|---|---|---|
| ENT1 | 5’-ctcatccaattcatttggtgcca-3’ (forward) | 278 bp |
| 5’-gcaaggttgagctgcaggtaat-3’ (reverse) | ||
| ENT2 | 5’-tcatcaattccttctgtgccg-3’ (forward) | 316 bp |
| 5’-agctcagctttagtctccagct-3’ (reverse) | ||
| CNT1 | 5’-tggaaggtctgggacatggagaa-3’ (forward) | 612 bp |
| 5’ atgatgctttgagcaggcaa-3’ (reverse) | ||
| CNT2 | 5’-gaggagaacaggagatggagaa-3’ (forward) | 540 bp |
| 5’-ggatcagctgctctggcctt-3’ (reverse) | ||
| CNT3 | 5’-agacaagttcagacttttctggag-3’ (forward) | 456 bp |
| 5’-aggccaaaagagtttagcagcagcc -3’ (reverse) | ||
| PNP | 5’- gtgaaatccc caactttcct -3’ (forward) | 559 bp |
| 5’- gccaaagactcgaagtccac –3 (reverse) | ||
| HGPRT | 5’- ctccgttatggcgacccgcag -3’ (forward) | 667 bp |
| 5’- caaagttgtcatggatgacc -3’ (reverse) | ||
| GAPDH | 5’- ggtgaaggtcggagtcaacg -3’ (forward) | 500 bp |
| 5’- caaagttgtcatggatgacc -3’ (reverse) | ||
PCR and DNA sequencing
For the sequencing of human HGPRT, one tenth of the resulting first-strand cDNA was used for PCR amplification. The blunt end PCR products of human HGPRT were generated by with High Fidelity PFU DNA polymerase (Invitrogen) to amplify the entire coding sequences. The PCR products were first cloned into TOPO blunt cloning vector (Invitrogen) by following the kit manual. The bacterial colonies obtained were randomly picked and examined by BamH1/Xbal digestion to screen the colonies with the potential HGPRT insert, and the resulting plasmids were submitted for automatic sequencing. M13 forward and M13 reverse primer were used when the entire coding region of HGPRT was sequenced.
Uptake assays of [3H]- ImmH, [3H]-uridine and [3H]-dGuo
The uptake assay of [3H]-uridine, [3H]-dGuo and [3H]- ImmH were conducted as described previously in sodium free buffer (20 mM Tris/HCl, 3 mM K2HPO4, 1 mM MgCl2.6H2O, 2 mM CaCl2, 5 mM glucose and 130 mM NMDG (N-methyl-D-glucamine pH 7.4) or sodium- containing buffer (20 mM Tris/HCl, 3 mM K2HPO4, 1 mM MgCl2.6H2O, 2 mM CaCl2, 5 mM glucose and 130 mM NaCl, pH 7.4) 16, 17. Briefly, 5×105 CCRF-CEM cells/sample were washed once with transport assay buffer and then suspended in 400 µl transport assay buffer. After pre-incubation with NBMPR (a selective ENT1 inhibitor), DP (a potent inhibitor of both ENT1 and ENT2), or DMSO for 15 min, uptake assays were started by adding equal volume of transport buffer containing 10µM of [3H]-labelled uridine (4 µCi/ml) or [3H]-labelled dGuo (10 µCi/ml) plus inhibitors or DMSO. Uptake assays were stopped at 5 min or 40 min, respectively, followed by five rapid washes with ice-cold transport buffer containing 1 mM unlabelled uridine or dGuo. The cell pellets were lysed in 10% SDS before quantification of radioactivity. For time course (up to 20 h) of ImmH or dGuo uptake (4 h, 20 h), cells were incubated with [3H]-uridine, [3H]- dGuo and [3H]- ImmH in normal culture medium instead of assay buffer in the absence or presence of 1 µM NBMPR or 10 µM DP.
Measurement of cell proliferation by MTT assay
The effects of ImmH on cell proliferation of CCRF-CEM or CCRF-CEM-AraC-8C, CCRF-CEM-AraC-8D cell lines were determined using the modified MTT assay method 18. Exponentially-growing CCRF-CEM cells were plated in 1 ml aliquots of growth medium into 24-well plates at 4×104 cells per well, respectively, and then incubated with various concentrations of ImmH, dGuo or vehicle alone. After 96 h incubation, cytotoxicity assays were performed by the modified MTT method. Triplicate wells were analyzed for each treatment.
Measurement of [3H]-dGuo incorporation into RNA, DNA and nucleic acid
To measure [3H]-dGuo incorporation into nucleic acid, exponentially-growing CCRF-CEM cells were plated in 1 ml aliquots of growth medium into 24-well plates at 4×104 cells per well, respectively. [3H]-dGuo was added (10 µM, 1 µCi/ml) for the time indicated in the absence or presence of ImmH. At harvest, cells were washed twice with ice-cold phosphate buffered saline (PBS), and precipitated by 5% trichloroacetic acid (TCA) at 4 °C. The pellets were washed with ice-cold 95% ethanol, dried at room temperature, and then dissolved in 200 µl of 1M NaOH for 30 min before quantification of radioactivity. Triplicate wells were analyzed for each treatment. For measuring [3H]-dGuo incorporation into RNA, total RNA was isolated from 1×105 cells/sample by Trizol reagent (GIBCO BRL) as described above. Total DNA was isolated from 1×105 cells/sample by using Wizard Genomic DNA purification kit (Promega) according to the manufacture’s instruction.
Results
dGuo- and ImmH -dependent cytotoxicity on CCRF-CEM cells
The cytotoxic effects of ImmH require the concomitant addition of dGuo to facilitate the formation of dGTP and subsequent inhibition of ribonucleotide reductase 2, 5, 6. To investigate the rate-limiting step in the cytotoxicity of ImmH, human T cell leukemia cells (CCRF-CEM) were incubated with ImmH (0.1–1000 nM) with various concentrations of dGuo (0–10 µM). ImmH induced a dose-dependent inhibition of cell growth in the presence of higher concentrations of dGuo (5–10 uM), whereas lower concentrations (1 µM) were ineffective (Fig. 1). Under these conditions, nanomolar concentrations of ImmH were required for cytotoxic effects in vivo. This contrasted with the reported inhibition of PNP by picomolar concentrations in vitro 2, 6, suggesting that cellular uptake of ImmH could be rate-limiting. In addition, the observation that CCRF-CEM cells were resistant to ImmH in the presence of low concentrations of dGuo (1 µM), indicated that uptake of dGuo could also limit the cytotoxicity of ImmH in vivo.
Fig. 1. Cytotoxic effects of ImmH plus dGuo on the CCRF-CEM cells.
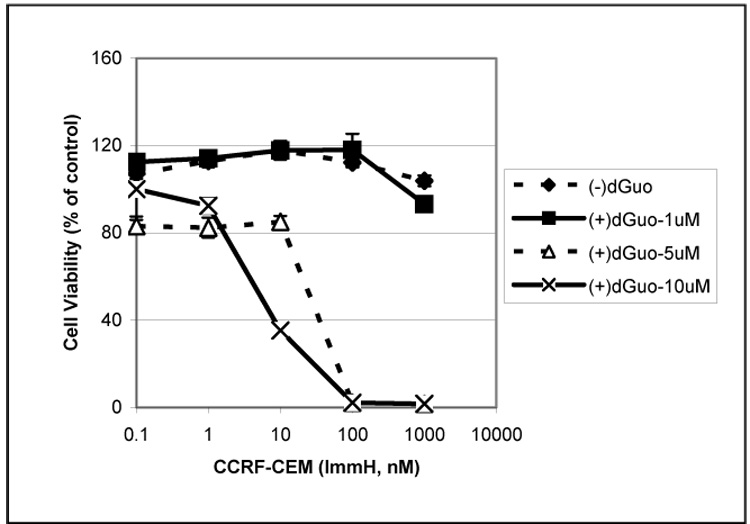
CCRF-CEM cells were seeded in 24-well plates at a density of 2×104 cells/ml and various concentrations of dGuo (0, 1, 5, 10 µM) were added in the presence or absence of various concentrations of ImmH (0, 0.1, 1, 10, 100, 1000 nM). Control values for (−) dGuo were obtained after incubation in the absence of dGuo and ImmH, control values for (+) dGuo were obtained after incubation in 1 uMdGuo and 0 uM ImmH. After 96 h, MTT was added at 0.5 mg/ml for 4 h, and the uptake of MTT was measured as described in Materials and Methods. Each data point represents a mean across 4 identical wells from one representative experiment.
Uptake of dGuo in CCRF-CEM cells is predominantly mediated by a CNT-dependent transport process
To investigate the requirement for dGuo transport and cytotoxicity in CCRF-CEM cells, the uptake of [3H]-dGuo was measured. Cellular accumulation of [3H]-dGuo was diminished in the presence of 1 mM unlabelled dGuo or guanine (Fig. 2A), indicating that passive diffusion did not play a major role in the uptake of dGuo. RT-PCR gene expression analysis of the equilibrative (ENTs) and the concentrative nucleoside transporters (CNTs) showed that both ENT1 and CNT2 were abundantly expressed in CCRF-CEM cells, whereas ENT2, CNT1 and CNT3 were undetectable in this cell line. (Fig. 2B). To test the involvement of ENT1, dGuo uptake was assayed in the presence of 10 µM dipyridamole (DP), a potent inhibitor of both ENT1 and ENT2. The time-dependent uptake of [3H]-dGuo was only slightly reduced by co-incubation with DP (Fig. 2C), whereas the uptake of [3H]-uridine was almost completely abolished (Fig. 2D). These results demonstrated that DP effectively inhibited ENT-dependent uridine uptake under these conditions and suggested that the ENTs are not the major transporters responsible for the uptake of dGuo. To further investigate the relative contribution of CNT versus ENT in the uptake of dGuo, nucleoside transport activity was determined in a sodium-containing buffer (measures both CNT and ENT activity) and sodium free buffer (measures ENT activity only), respectively, as described in the Materials and Methods. As shown in Fig. 2E, uridine uptake occurred under both sodium and sodium-free conditions, by comparison, dGuo uptake was largely mediated by a sodium-dependent transport activity in these cells (~80%). Taken together, these results and the RT-PCR expression data (Fig. 2B), suggests that CNT2 is a potential transporter for dGuo in these cells.
Fig. 2. The effects of unlabeled guanine/dGuo and dipyridamole on the uptake of [3H] dGuo/uridine and profiles of nucleoside transporters expressed in CCRF-CEM cells.
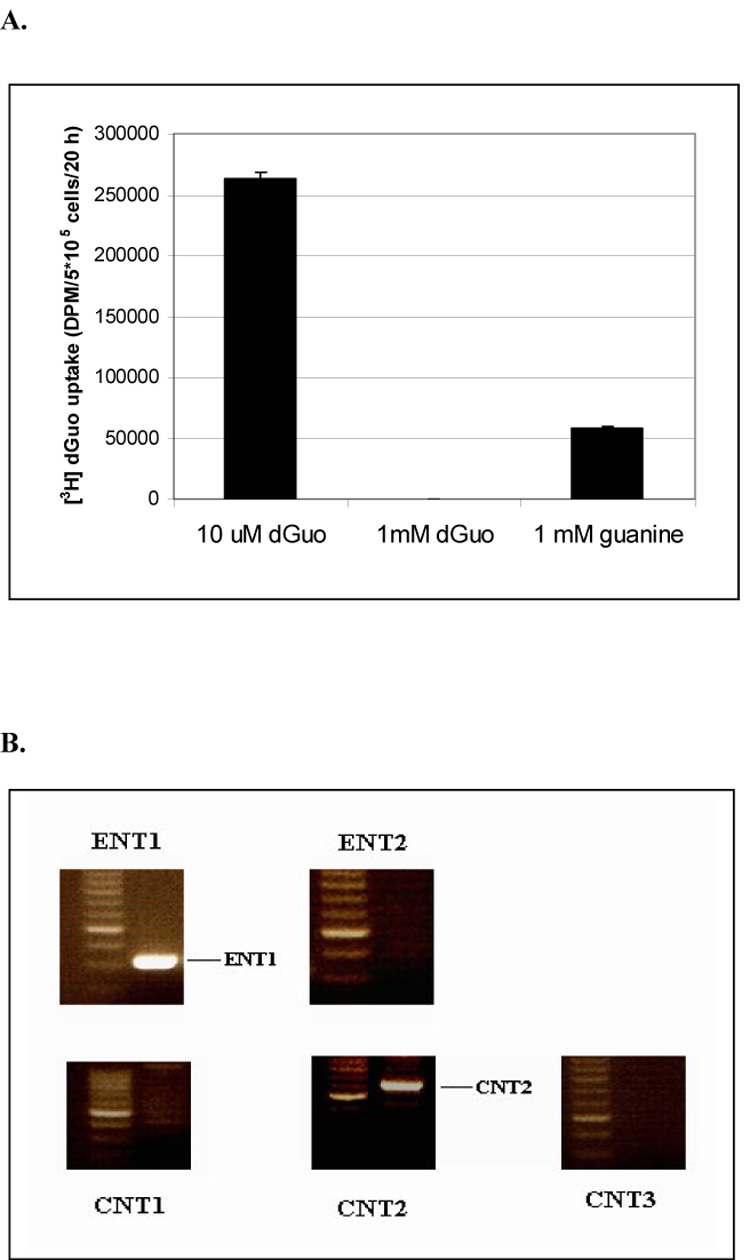

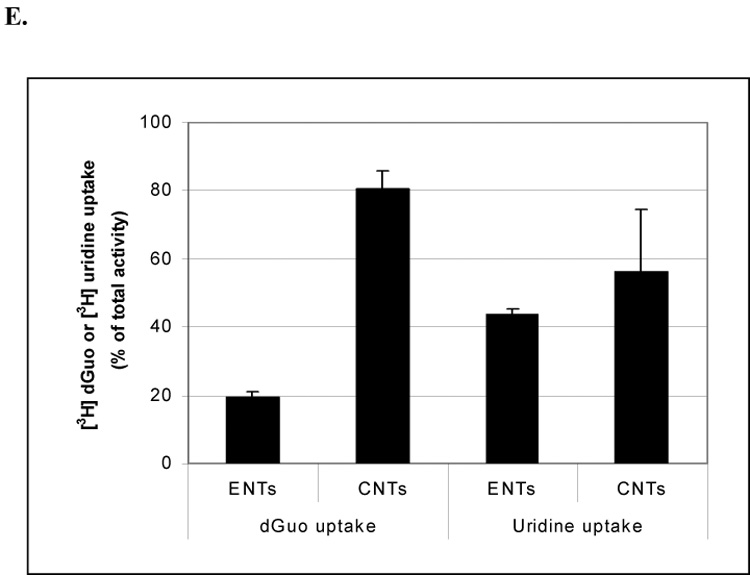
(2A) 3×105 CCRF-CEM cells were incubated with [3H] dGuo (2 µCi/ml) in fresh normal cell culture medium in the presence of 10 µM, 1 mM unlabelled dGuo or guanine, respectively, for 20 h. After five rapid washes with cold PBS buffer, the radioactivity from the lysed cells was measured. (2B) RT-PCR analysis of various types of nucleoside transporters. The mRNA expression of ENT1, ENT2, CNT1, CNT2, CNT3 was determined by RT-PCR as described in Materials and Methods. 3×105 CCRF-CEM cells were incubated with 5 µM [3H] dGuo (2C) or 5 µM [3H] uridine (2D) in fresh normal cell culture medium for time indicated in the presence or absence of 10 µM DP. Alternatively, 3×105 CCRF-CEM cells were incubated with [3H] 5 µM dGuo or [3H] uridine (2E) in sodium free or sodium containing buffer, respectively, for 5 min. After washing with transport assay buffer containing 1 mM unlabelled dGuo or uridine, respectively, for five times the radioactivity in the lysed cells was measured. The results shown are the results are the average of triplicate samples.
Uptake of ImmH is predominantly mediated by ENT1/2 in CCRF-CEM cells
ImmH is a neutral or cationic (pKa = 6.9) inosine analog that is unlikely to enter cells by passive diffusion. To investigate the mechanism of ImmH uptake in CCRF-CEM cells, similar experiments to those described above were performed using [3H]- ImmH as a substrate. As shown in Fig. 3A, uptake of [3H]-ImmH was largely blocked by incubation with the ENT inhibitor DP in CCRF-CEM cell line, indicating that ImmH transport was predominantly facilitated by ENT1. To confirm the role of ENTs, the uptake of [3H]-ImmH was examined in ENTs-deficient cells (PK15), or PK15 cell lines stably expressing human ENT1 and human ENT2. As shown in Fig. 3B, the uptake of [3H]-ImmH was significantly higher in PK15 cells expressing ENT1 and ENT2 compared to PK15 cells. Moreover, the uptake of [3H]-ImmH was inhibited by DP confirming the involvement of ENTs in these cells.
Fig. 3. Effects of DP on the [3H]-ImmH uptake in CCRF-CEM cells and ENT1- and ENT2-dependent transport of [3H]-ImmH.

(3A) CCRF-CEM cells (3×105) were incubated with 50 nM [3H]-ImmH in normal culture medium in the presence or absence of 10 µM DP for 0.5 h, 1.5 h, 20 h. (3B) The nucleoside transporter-deficient PK15 cells, and PK15 cells stably expressing exogenous ENT1 and ENT2 were incubated with 50 nM [3H]-ImmH in the presence or absence of 10 µM DP, or DMSO control for 40 min. in sodium free transport assay buffer as described in Materials and Methods, cells were then washed with transport assay buffer containing 0.5 mM unlabelled ImmH for five times. The radioactivity in the cell lysates was measured and each data point represents a mean from four independent samples.
ENTs are not rate-limiting for ImmH cytotoxicity of CCRF-CEM cells
Although these studies suggested that ENTs were the major transporters responsible for the immediate uptake of ImmH in CCRF-CEM cells, we examined the effects of ImmH on CCRF-CEM-AraC-8C (AraC-8C), a cell line deficient in ENT1- dependent transport 19, 20. Surprisingly, extended exposure (96H) of AraC-8C cells to ImmH showed that ImmH remained cytoxic to these cells, in fact enhanced sensitivity was observed when concentrations of ImmH lower than 10 nM were used (Fig. 4). Neither DP nor NBMPR affected the cytotoxicity of ImmH in this cell line demonstrating that neither ENT1 nor ENT2-dependent uptake was rate-limiting for ImmH cytotoxicity in the AraC-8C cells (data not shown).
Fig. 4. Cytotoxic effects of ImmH on the parental CCRF-CEM and nucleoside transporter-deficient AraC-8C cells.

CCRF-CEM cells were seeded in 24-well plates at a density of 2×104 cells/ml and various concentrations of various concentrations of ImmH (0, 0.25, 0.5, 0.75, 1.0, 2.5, 5.0, 10 nM) were added in the presence or absence of 10 µM of dGuo. After 96 h, MTT was added at 0.5 mg/ml for 4 h, and the uptake of MTT was then measured. Each data point represents a mean across 4 identical wells from one representative experiment.
HGPRT deficiency sensitizes CCRF-CEM cells to ImmH
To investigate why AraC-8C cells were sensitized to low concentrations of ImmH, the incorporation of [3H]-dGuo into nucleic acid or RNA was determined in the absence or presence of ImmH. The incorporation of dGuo into nucleic acid was markedly lower in the mutant AraC-8C cells and ImmH treatment (1 µM) increased the uptake and incorporation of [3H]-dGuo into nucleic acid in these cells (Fig. 5A). Approximately 80% of [3H]-dGuo was incorporated into RNA and 20% of [3H]-dGuo was incorporated into DNA in the parental CCRF-CEM cells, suggesting that the majority of dGuo was metabolized through the PNP-HGPRT-dependent pathway rather than the dCK-dependent pathway (Fig. 5B) (See also Model Fig. 8). In comparison, in the CCRF-CEM cells the incorporation of [3H]-dGuo into RNA was almost abolished in the presence of 1 µM ImmH and was similar to that observed in AraC-8C cells in the absence of ImmH (Fig. 5C). These results suggested that a possible defect in the PNP- HGPRT pathway could sensitize the AraC-8C cells to ImmH.
Fig. 5. Comparison of uptake of [3H]-dGuo, incorporation of [3H]-dGuo into DNA, RNA, total nucleic acids between AraC-8C and the parental CCRF-CEM cells.

(5A) Cells (1×105) were incubated with dGuo (10 µM, 2 µCi/ml) in the presence or absence of 1 µM ImmH for 24 h, the radioactivity in the lysed cells for uptake of dGuo or acid-soluble nucleotides were then determined. (5B) Total RNA or DNA in 1×105 CCRF-CEM or AraC-8C cells was isolated as described in Materials and Methods, and the incorporation of dGuo into RNA or DNA was calculated as radioactivity in RNA or DNA versus total nucleic acids or RNA plus DNA. (5C) CCRF-CEM cells or AraC-8C cells were incubated with dGuo (10 µM, 2 µCi/ml) for 24 h in the presence of 1 µM ImmH, the incorporation of dGuo into RNA was then measured after quantitation of radioactivity in both cell lines.
Fig. 8. A metabolic scheme illustrating the metabolism of dGuo and guanine and inhibition of PNP by ImmH in mammalian cells.
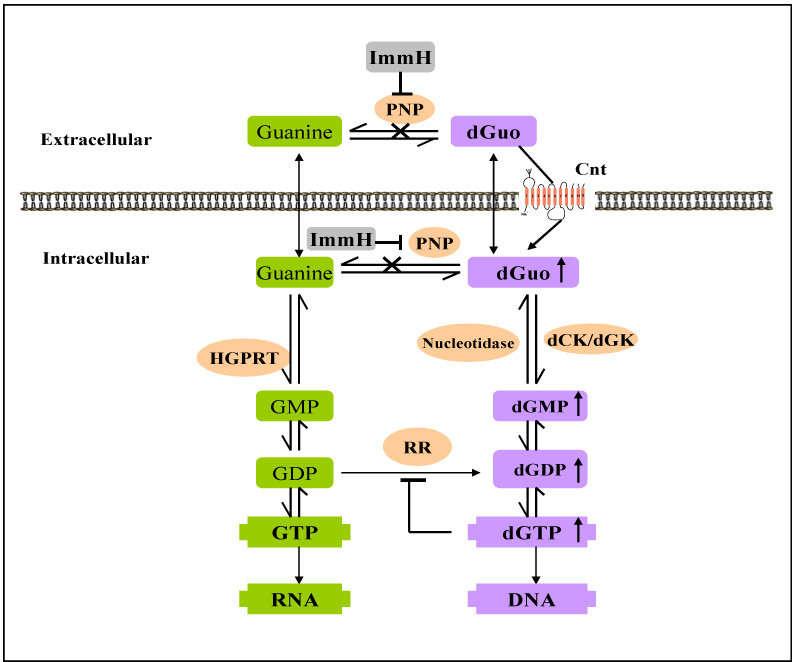
A model showing the inhibition of PNP by ImmH and enzymes involved in the metabolism of dGuo and Guo. Enyzmes shown are purine-nucleoside-phosphorylase (PNP), 5`-nucleotidase (nucleotidase), hypoxanthine-guanine-phosphoribosyltransferase (HGPRT), deoxycytidine kinases (dCK), deoxyguanosine kinase (dGK) and ribonucleotide reductase (RR).
HGPRT activity was previously reported to be deficient in both the AraC-8C and CCRF-CEM-AraC-8D (AraC-8D) cell lines 19, 20. We confirmed that the conversion of hypoxanthine to IMP was absent (Fig. 6A), in addition these cell lines were highly resistant to 6-thioguanine (6-TG) (Fig. 6B). The molecular basis of HGPRT deficiency in these cell lines was investigated. RT-PCR amplification of full-length HGPRT resulted in a smaller HGPRT from both AraC-8C and AraC-8D cells (Fig. 6C), indicating the possibility of a deletion within the HGPRT coding region. To further investigate this, HGPRT was subcloned into Topo-blunt vector and sequenced. Both cell lines contained an identical exon 8 deletion in HGPRT, resulting in a reading frame shift and early termination at exon 9 (Fig. 6D). Thus these findings suggest that increased ImmH sensitivity of AraC-8C cells may result from a deficit in the PNP-HGPRT-dependent metabolism of dGuo, thereby facilitating the cytotoxic accumulation of dGTP.
Fig. 6. Comparison of HGPRT activity, TG cytotoxicity and HGPRT coding sequence in AraC-8C, AraC-8D, and CCRF-CEM cells.


(6A) HGPRT activity was assayed by quantitation of the 3H-labeled hypoxanthine and IMP product in the reaction after separation by Polyethyliminocellulose Thin-layer chromatography (TLC) plates as described in Materials and Methods. (6B) The three leukemia cell lines were seeded in 24-well plates at a density of 2 ×104 cells/ml and were exposed to various concentrations of 6-TG for 96 h and the cytotoxicity determined by MTT assay as described in Materials and Methods. Each data point represents a mean across 4 identical wells from one representative experiment. (6C) Total RNA was isolated from the three leukemia cell lines and full-length HGPRT products from RT-PCR in each cell line were cloned into Topo vector and the sequence verified as described in Materials and Methods. (6D) Shown is a schematic diagram of HGPRT sequence from the parental CCRF-CEM cells and the mutant HGPRTs isolated from the AraC-8C and AraC-8D cells.
dCK, but not dGK is essential for ImmH-dependent cytotoxicity of CCRF-CEM cells
We also examined the requirement for the nucleoside kinases dGK and dCK in metabolism of dGuo in these cells. Both dGK and dCK phosphorylate dGuo in cell free extracts and are believed to be required for the inhibitory effects of ImmH 2, 6. To determine which of these enzymes phosphorylated dGuo in human leukemia cells, the effects of ImmH were examined in AraC-8D, cells previously shown to be deficient in dCK. As shown in Fig. 7A, the growth of the parental CCRF-CEM cells was inhibited by ImmH plus dGuo (10 µM) whereas the AraC-8D cells were resistant up to 1 µM ImmH in the presence of 10 µM dGuo.
Fig. 7. Comparison of the cytotoxic effects of ImmH on the parental CCRF-CEM and dCK-deficient AraC-8D cells.



(7A) CCRF-CEM cells were seeded in 24-well plates at a density of 2×104 cells/ml and were incubated with various concentrations of ImmH (0, 0.1, 1, 10, 100, 1000 nM) in the presence or absence of dGuo (10 µM). After 96 h, MTT was added at 0.5 mg/ml for 4 h, and the uptake of MTT was measured. Each data point represents a mean across 4 identical wells from one representative experiment. Cell lysates (16 mg) were fractionated on Mono Q and fractions (1 ml), were collected and assayed for dCK (7B) and dGK (7C) as described in Materials and Methods.
To confirm the role of dCK in ImmH toxicity, we examined the phosphorylation of dGuo in lysates from CCRF-CEM cells and AraC-8D cells. Reduced dGK activity was also reported in AraC-8D cells 21, however since dCK is capable of phosphorylating both dGuo and deoxycytidine, reduced dGuo phosphorylation may result from a dCK deficiency in these cells. Lysates from CCRF-CEM cells and AraC-8D cells were separated on Mono-Q™ chromatography and the column fractions assayed for deoxycytidine or dGuo phosphorylation. As shown in Fig. 7B, one major peak of deoxycytidine phosphorylating activity was observed in CCRF-CEM cells whereas this activity was absent in the AraC-8D cells. In comparison, two peaks of dGuo phosphorylation were detected in the CCRF-CEM fractions (Fig. 7C). Analysis of fractions from the AraC-8D showed that these cells were devoid of the second peak of dGuo phosphorylation, which correlated with the peak of deoxycytidine phosphorylation in CCRF-CEM cells. These experiments demonstrated that dCK was the major dGuo phosphorylating activity in CCRF-CEM cells and confirmed that this activity was deficient in the AraC-8D cells. In contrast, no difference in dGK activity was observed between the parental CCRF-CEM and AraC-8D cell lines (Fig. 7C).
Discussion
In this study we investigated the factors contributing to the cytotoxicity of ImmH in CCRF-CEM leukemia cells. We observed that the rapid uptake of ImmH was facilitated by one of the equilibrative nucleoside transporters (ENTs). ImmH uptake was significantly enhanced in PK15 cells expressing ENT1 or ENT2 compared to ENT-deficient PK15 cells and the uptake of ImmH into CCRF-CEM cells was blocked by incubation with DP, an ENT1 and ENT2 inhibitor 8, 22. Based on ENT expression analysis, our data suggests that ENT1 is predominantly responsible for ImmH uptake in CCRF-CEM cells. Importantly, although ImmH uptake was decreased in AraC-8C cells lacking ENT activity or after DP treatment, the ImmH-mediated cytotoxicity was not significantly affected, suggesting that therapeutic doses of ImmH were achieved by either diffusion or CNT-mediated uptake of ImmH. This may be important because ENT1 has been observed to be down-regulated in Ara-C-resistant CCRF-CEM and acute myeloid leukemia cells 23, 24. Thus a loss of ENT1 or ENT2 activity is not expected to reduce the therapeutic efficiency of ImmH.
By contrast the uptake of dGuo was primarily dependent on the concentrative nucleoside transporters (CNTs). DP was ineffective in blocking the uptake of dGuo whereas approximately 80% of the total dGuo uptake in CCRF-CEM cells was mediated through a sodium-dependent transport process. RT-PCR analysis showed that both ENT1 and CNT2 are highly expressed in these cells, suggesting that they may be responsible for the uptake of ImmH and dGuo, respectively. The observation that reduced dGuo concentrations affected ImmH cytotoxicity in CCRF-CEM cells indicates that CNT activity or expression may contribute to the sensitivity to ImmH by mediating the uptake of dGuo.
Our study also provides evidence that dCK plays an essential role in the metabolism and cytotoxicity of dGuo during ImmH treatment. Separation of dCK and dGK from crude cytosol extracts of CCRF-CEM cells by Mono-Q™ column chromatography showed that dCK, but not dGK, was responsible for the phosphorylation of dGuo to dGMP in these cells and demonstrated that the activity of dCK was essential for the accumulation of dGTP in cells exposed to dGuo and ImmH. However, in addition to the dCK-dependent pathway, intracellular dGuo can be metabolized through the PNP-HGPRT dependent pathway that converts dGuo to GTP through guanine (See Model Fig. 8). Consistent with the role of HGPRT in this pathway, approximately 43- fold and 15-fold increase in the accumulation of intracellular dATP and dGTP, respectively, were observed upon exposure of CCRF-CEM cells to 50 µM 2'- deoxyguanosine and 20 µM 2'-deoxyadenosine for 6 h 12. This finding suggests that the PNP-HGPRT pathway is important in modulating dGuo metabolism. We hypothesized that a decrease in PNP-HGPRT-dependent metabolism of dGuo, due to reduced HGPRT activity, would increase the sensitivity of leukemia cells to ImmH. In support of this, AraC-8C cells deficient in HGPRT activity showed enhanced sensitivity to ImmH. Although the AraC-8C cells are also deficient in ENT activity, this cell line was more sensitive to low concentrations of ImmH (1 to 10 nM) than the parental cells, confirming that ENT-dependent uptake of ImmH was not rate-limiting as discussed above.
The cytotoxic effects of ImmH may be achieved through inhibiting PNP-mediated degradation of dGuo extracellularly, thus enabling more dGuo to enter AraC-8C cells. Our data suggests that the reduction of PNP-HGPRT-dependent synthesis of GTP (due to the HGPRT deficiency), facilitates the formation of dGTP through the dCK-dependent pathway in the AraC-8C cells. The reduced incorporation of dGuo into RNA in AraC-8C cells was similar to the effects of ImmH in the parental cells, demonstrating that the inhibitory effect of ImmH on the incorporation of dGuo into RNA mimics the deficiency of HGPRT in the AraC-8C cell line. ImmH may allow more dGuo to be metabolized through the dCK-dependent pathway due to the HGPRT deficiency. Thus it is reasonable to propose that accumulation of intracellular dGTP and enhanced ImmH cytotoxicity could be achieved by inhibiting not only PNP but also HGPRT.
CCRF-CEM-AraC-8C and CCRF-CEM Arc-C- 8D cells are clonal derivatives of an hypoxanthine-guanine phosphoribosyltransferase-deficient parental CEM cell line originally isolated in semi-solid agarose containing 8 µM arabinosylcytosine however, the underlining mechanism for HGPRT deficiency was not elucidated 20. The genetic basis for HGPRT deficiency in the AraC-8C and AraC-8D cells was revealed in this study. RT-PCR analysis and sequencing of HGPRT in this cell line demonstrated an exon 8 deletion in the HGPRT coding sequence. Of interest, incubation of CCRF-CEM cells with 50 µM 2'-deoxyguanosine or 20 µM 2'-deoxyadenosine induced approximately a 40 fold increase in mutation frequency at the HGPRT locus, which was attributed to inhibition of DNA repair processes subsequent to the disruption of dNTP pools 12. Interestingly, HGPRT mutations have also been reported in leukemia patients after treatment with chemotherapeutic drugs 25–28. Children treated for acute lymphocyte leukemia (ALL) have significantly increased somatic mutant frequencies (30- to 1300-fold higher) at the HGPRT locus in their non-malignant peripheral T cells compared to control patients 29. Aberrant HGPRT mRNA splicing promoted by splice site mutation or loss is believed to be a common mechanism for loss of HGPRT in human cells 30. The frequencies of spontaneous exon skipping in HGPRT gene has been characterized in T-lymphocytes 31 and human primary fibroblasts 32. Exon 8 is the most prone to skipping with a relative frequency of 0.019 to wild type (approximately one aberrant transcript per 50 wild type transcripts) 32.
We observed that the same exon 8 skipping defect occurred in two different Ara-C selected CCRF-CEM cell lines, raising the possibility that Ara-C selection may favor the survival of HGPRT-deficient cells. Alternatively, a mutational hot spot might exist in proximity to exon 8 of the HGPRT gene. In summary, the occurrence of spontaneous mutations in HGPRT in normal primary cells and the elevated mutational frequencies of HGPRT in drug-treated leukemia patients, suggests the possibility that cells from patients that harbor an HGPRT inactivating mutation are more likely to be sensitive to ImmH.
Acknowledgements
This work was supported by NIH grants RO1-GM069976 to L.M.G., RO1-CA064192 to B.S.M. and R37-GM041916 to V.L.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gandhi V, Kilpatrick JM, Plunkett W, Ayres M, Harman L, Du M, et al. A proof-of-principle pharmacokinetic, pharmacodynamic, and clinical study with purine nucleoside phosphorylase inhibitor immucillin-H (BCX-1777, forodesine) Blood. 2005;98(8):4593–4598. doi: 10.1182/blood-2005-03-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schramm VL. Immucillins as antibiotics for T-cell proliferation and malaria. Nucleosides Nucleotides Nucleic Acids. 2004;23(8–9):1305–1311. doi: 10.1081/NCN-200027564. [DOI] [PubMed] [Google Scholar]
- 3.Miles RW, Tyler PC, Furneaux RH, Bagdassarian CK, Schramm VL. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry. 1998;37(24):8615–8621. doi: 10.1021/bi980658d. [DOI] [PubMed] [Google Scholar]
- 4.Wang F, Miles RW, Kicska G, Nieves E, Schramm VL, Angeletti RH. Immucillin-H binding to purine nucleoside phosphorylase reduces dynamic solvent exchange. Protein Sci. 2000;9(9):1660–1668. doi: 10.1110/ps.9.9.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kicska GA, Long L, Horig H, Fairchild C, Tyler PC, Furneaux RH, et al. Immucillin H, a powerful transition-state analog inhibitor of purine nucleoside phosphorylase, selectively inhibits human T lymphocytes. Proc Natl Acad Sci U S A. 2001;98(8):4593–4598. doi: 10.1073/pnas.071050798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schramm VL. Development of transition state analogues of purine nucleoside phosphorylase as anti-T-cell agents. Biochim Biophys Acta. 2002;1587(2–3):107–117. doi: 10.1016/s0925-4439(02)00073-x. [DOI] [PubMed] [Google Scholar]
- 7.Evans GB, Furneaux RH, Lewandowicz A, Schramm VL, Tyler PC. Synthesis of second-generation transition state analogues of human purine nucleoside phosphorylase. J Med Chem. 2003;46(24):5271–5276. doi: 10.1021/jm030305z. [DOI] [PubMed] [Google Scholar]
- 8.Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004;447(5):735–743. doi: 10.1007/s00424-003-1103-2. [DOI] [PubMed] [Google Scholar]
- 9.Gray JH, Mangravite LM, Owen RP, Urban TJ, Chan W, Carlson EJ, et al. Functional and genetic diversity in the concentrative nucleoside transporter, CNT1, in human populations. Mol Pharmacol. 2004;65(3):512–519. doi: 10.1124/mol.65.3.512. [DOI] [PubMed] [Google Scholar]
- 10.Bzowska A, Kulikowska E, Shugar D. Purine nucleoside phosphorylases: properties, functions, and clinical aspects. Pharmacol Ther. 2000;88(3):349–425. doi: 10.1016/s0163-7258(00)00097-8. [DOI] [PubMed] [Google Scholar]
- 11.Madrid-Marina V, Lestan B, Nowak PJ, Fox IH, Spychala J. Altered properties of human T-lymphoblast soluble low Km 5'-nucleotidase: comparison with B-lymphoblast enzyme. Leuk Res. 1993;17(3):231–240. doi: 10.1016/0145-2126(93)90006-7. [DOI] [PubMed] [Google Scholar]
- 12.Mattano SS, Palella TD, Mitchell BS. Mutations induced at the hypoxanthine-guanine phosphoribosyltransferase locus of human T-lymphoblasts by perturbations of purine deoxyribonucleoside triphosphate pools. Cancer Res. 1990;50(15):4566–4571. [PubMed] [Google Scholar]
- 13.Musk P, Clark JM, Thompson D, Dunn IS, Christopherson RI, Szabados E, et al. Purine deoxynucleoside metabolism in human melanoma cells with a high spontaneous mutation rate. Mutat Res. 1996;350(1):229–238. doi: 10.1016/0027-5107(95)00111-5. [DOI] [PubMed] [Google Scholar]
- 14.Ward JL, Sherali A, Mo ZP, Tse CM. Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J Biol Chem. 2000;275(12):8375–8381. doi: 10.1074/jbc.275.12.8375. [DOI] [PubMed] [Google Scholar]
- 15.Snyder FF, Jenuth JP, Dilay JE, Fung E, Lightfoot T, Mably ER. Secondary loss of deoxyguanosine kinase activity in purine nucleoside phosphorylase deficient mice. Biochim Biophys Acta. 1994;1227(1–2):33–40. doi: 10.1016/0925-4439(94)90103-1. [DOI] [PubMed] [Google Scholar]
- 16.Huang M, Wang Y, Cogut SB, Mitchell BS, Graves LM. Inhibition of nucleoside transport by protein kinase inhibitors. J Pharmacol Exp Ther. 2003;304(2):753–760. doi: 10.1124/jpet.102.044214. [DOI] [PubMed] [Google Scholar]
- 17.Huang M, Wang Y, Collins M, Gu JJ, Mitchell BS, Graves LM. Inhibition of Nucleoside Transport by p38 MAPK Inhibitors. J Biol Chem. 2002;277(32):28364–28367. doi: 10.1074/jbc.C200321200. [DOI] [PubMed] [Google Scholar]
- 18.Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47(4):936–942. [PubMed] [Google Scholar]
- 19.Ullman B. Dideoxycytidine metabolism in wild type and mutant CEM cells deficient in nucleoside transport or deoxycytidine kinase. Adv Exp Med Biol. 1989;253B:415–420. doi: 10.1007/978-1-4684-5676-9_61. [DOI] [PubMed] [Google Scholar]
- 20.Ullman B, Coons T, Rockwell S, McCartan K. Genetic analysis of 2',3' - dideoxycytidine incorporation into cultured human T lymphoblasts. J Biol Chem. 1988;263(25):12391–12396. [PubMed] [Google Scholar]
- 21.Owens JK, Shewach DS, Ullman B, Mitchell BS. Resistance to 1-beta-D-arabinofuranosylcytosine in human T-lymphoblasts mediated by mutations within the deoxycytidine kinase gene. Cancer Res. 1992;52(9):2389–2393. [PubMed] [Google Scholar]
- 22.Hammond JR. Interaction of a series of draflazine analogues with equilibrative nucleoside transporters: species differences and transporter subtype selectivity. Naunyn Schmiedebergs Arch Pharmacol. 2000;361(4):373–382. doi: 10.1007/s002100000214. [DOI] [PubMed] [Google Scholar]
- 23.Takagaki K, Katsuma S, Kaminshi Y, Horio T, Nakagawa S, Tanaka T, et al. Gene-expression profiling reveals down-regulation of equilibrative nucleoside transporter 1 (ENT1) in Ara-C-resistant CCRF-CEM-derived cells. J Biochem. 2004;136:733–740. doi: 10.1093/jb/mvh180. [DOI] [PubMed] [Google Scholar]
- 24.Hubeek I, Stam RW, Peters GJ, Broekhuizen R, Meijerink JPP, van Wering ER, et al. Brit J Cancer. 2005;93:1388–1394. doi: 10.1038/sj.bjc.6602881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang P, Siciliano MJ, Plunkett W. Gene deletion, a mechanism of induced mutation by arabinosyl nucleosides. Mutat Res. 1989;210(2):291–301. doi: 10.1016/0027-5107(89)90090-0. [DOI] [PubMed] [Google Scholar]
- 26.Hanft VN, Fruchtman SR, Pickens CV, Rosse WF, Howard TA, Ware RE. Acquired DNA mutations associated with in vivo hydroxyurea exposure. Blood. 2000;95(11):3589–3593. [PubMed] [Google Scholar]
- 27.Rice SC, Vacek P, Homans AH, Messier T, Rivers J, Kendall H, et al. Genotoxicity of therapeutic intervention in children with acute lymphocytic leukemia. Cancer Res. 2004;64(13):4464–4471. doi: 10.1158/0008-5472.CAN-03-3940. [DOI] [PubMed] [Google Scholar]
- 28.Finette BA, Homans AC, Rivers J, Messier T, Albertini RJ. Accumulation of somatic mutations in proliferating T cell clones from children treated for leukemia. Leukemia. 2001;15(12):1898–1905. doi: 10.1038/sj.leu.2402306. [DOI] [PubMed] [Google Scholar]
- 29.Kendall HE, Vacek PM, Finette BA. Analysis of microsatellite instability in children treated for acute lymphocytic leukemia with elevated HPRT mutant frequencies. Mutagenesis. 2004;19(5):409–412. doi: 10.1093/mutage/geh050. [DOI] [PubMed] [Google Scholar]
- 30.Colgin LM, Hackmann AF, Monnat RJ., Jr Different somatic and germline HPRT1 mutations promote use of a common, cryptic intron 1 splice site. Mutations in brief no. 246. Hum Mutat. 1999;13(6):504–505. doi: 10.1002/(SICI)1098-1004(1999)13:6<504::AID-HUMU15>3.0.CO;2-6. Online. [DOI] [PubMed] [Google Scholar]
- 31.Osterholm AM, Hou SM. Splicing mutations at the HPRT locus in human T-lymphocytes in vivo. Environ Mol Mutagen. 1998;32(1):25–32. [PubMed] [Google Scholar]
- 32.Skandalis A, Ninniss PJ, McCormac D, Newton L. Spontaneous frequency of exon skipping in the human HPRT gene. Mutat Res. 2002;501(1–2):37–44. doi: 10.1016/s0027-5107(02)00013-1. [DOI] [PubMed] [Google Scholar]