

Abstract
The resting membrane potential of the human erythrocyte is largely determined by a constitutive Cl- conductance ∼100-fold greater than the resting cation conductance. The 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS)-sensitive electroneutral Cl- transport mediated by the human erythroid Cl-/HCO3- exchanger, AE1 (SLC4A1, band 3) is ≥10,000-fold greater than can be accounted for by the Cl- conductance of the red cell. The molecular identities of conductive anion pathways across the red cell membrane remain poorly defined. We have examined red cell Cl- conductance in the Ae1-/- mouse as a genetic test of the hypothesis that Ae1 mediates DIDS-sensitive Cl- conductance in mouse red cells. We report here that wildtype mouse red cell membrane potential resembles that of human red cells in the predominance of its Cl- conductance. We show with four technical approaches that the DIDS-sensitive component of erythroid Cl- conductance is reduced or absent from Ae1-/- red cells. These results are consistent with the hypothesis that the Ae1 anion exchanger polypeptide can operate infrequently in a conductive mode. However, the fragile red cell membrane of the Ae1-/- mouse red cell exhibits reduced abundance or loss of multiple polypeptides. Thus, loss of one or more distinct, DIDS-sensitive anion channel polypeptide(s) from the Ae1-/- red cell membrane cannot be ruled out as an explanation for the reduced DIDS-sensitive anion conductance.
Keywords: Cl-/HCO3- exchange, patch clamp, isotopic flux, light scattering, ionophore
Introduction
The red cell Cl-/HCO3- exchanger, AE1 (SLC4A1, Band 3) is present in 1.2 × 106 copies per red cell. The main anion function of AE1 is to allow erythroid cytosolic carbonic anhydrase to increase the total CO2 carrying capacity of the blood. This process maximizes the transfer of CO2 from the respiring tissues to the lungs, while the hemoglobin of the red cell maximizes transfer of O2 from the lungs to the respiring tissues [1]. The high anion permeability of the red cell membrane exceeds by 104 -fold its anion conductance [2; 3], which itself exceeds by 10- to 100-fold the membrane's resting cation conductance [4; 5; 6].
It has long been assumed that red cell anion exchange and anion conductance are both mediated by AE1, since both anion exchange and conductance exhibit parallel sensitivities to the irreversible stilbene disulfonate inhibitor 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS) [2] and the reversible inhibitor 4,4′-diintrostilbene-2,2′-disulfonic acid (DNDS) [7]. The complications incurred by attribution of a conductive pathway to a normally electroneutral anion exchanger with an apparently obligatory sequential (“ping-pong”) mechanism gave rise to postulates of “slippage” (a stoichiometrically uncoupled or loosely coupled transporter-like translocation across the lipid bilayer permeability barrier of the empty anion binding site) or “tunneling” (a channel-like conductive anion transfer following an unusual gating event, or in the absence of any conformational change in the polypeptide). Several lines of evidence have been advanced against the “slippage” mechanism [8; 9; 10], and the data have been considered compatible with “tunneling” [7].
Numerous solute transporters and exchangers expressed in Xenopus oocytes or reconstituted in proteoliposomes or lipid bilayer exhibit uncoupled conductive modes of transport [11; 12]. Among these is trout AE1, [13; 14], unique in this property among AE1 polypeptides studied to date. The maximal oocyte surface expression of AE1 (estimated ≤5 ×108 per oocyte based on unidirectional influx values at 20°C; Alper et al., unpublished) and its estimated conductive turnover number (3-30 sec-1 in red cells) are likely too low to detect the postulated AE1-mediated current over the considerable background anion conductance of the oocyte. We thus addressed AE1's role in red cell anion conductance through comparison of DIDS-sensitive Cl- conductance in red cells from wild-type and Ae1-/- mice [15]. We reasoned that persistence of this conductance in Ae1-/- red cells would prove its independence from the Ae1 polypeptide. In contrast, absence of the conductance from these cells would be consistent with DIDS-sensitive anion exchange and DIDS-sensitive anion conductance representing two transport modes of the Ae1 polypeptide.
We assessed red cell Cl- conductance indirectly in three ways. The first approach monitored change in membrane potential measured as the change in extracellular pH (pHo) in a suspension of CCCP-treated red cells in the presence and absence of valinomycin [4]. The second approach measured the rate of change of corpuscular hemoblobin concentration (mean) (CHCM) and mean corpuscular volume (MCV) in valinomycin-treated red cells [16]. The third approach measured 86Rb efflux from valinomycin-treated red cells [2; 3; 9; 17]. We then directly measured whole cell Cl- currents by patch-clamp recording of nystatin-permeabilized mouse red cells. Each of these methods revealed the DIDS-sensitive component of Cl- conductance in Ae1-/- red cells to be reduced or undetectable, consistent with a role for mouse Ae1 polypeptide in the DIDS-sensitive component of erythroid Cl- conductance.
Materials and Methods
Drugs and chemicals
Dimethylsulfoxide (DMSO) was from Fisher (Pittsburgh, PA). DIDS and choline chloride were from Calbiochem (LaJolla, CA). Tris(hydroxymethyl)aminomethane (TRIS) was from Mallinckrodt (St. Louis, MO), 3-(N-morpholino)propanesulfonic acid (MOPS) was from Research Organics (Cleveland, OH). Sucrose was from Serva Biochemicals (Paramus, NJ). NS-3623 was the kind gift of P. Christophersen, (Neurosearch, Ballerup, Denmark). 86RbCl was from NEN-Dupont (Boston, MA). Carbonyl cyanide m-chlorophenylhydrazone (CCCP), valinomycin, ruthenium red, Triton X-100, and all other salts and chemicals were from Sigma (St. Louis, MO).
Erythrocyte preparation
Human blood was collected from normal donors in accordance with protocols approved by the Clinical Investigations Committee of Children's Hospital Boston. Blood collected in a heparinized Vacutainer tube was centrifuged at 3000 × g for 10 min in an SS34 rotor. After removal of the buffy coat, the cells were washed five times with human wash solution containing (in mM) 152 choline chloride, 1 MgCl2, 10 Tris-MOPS, pH 7.40 at 4°.
Mouse blood was collected from ether-anesthetized or Inactin-anesthetized mice by cardiac puncture into heparinized tubes, in accordance with protocols approved by the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center and of The Jackson Laboratories. All mouse red cell experiments except patch clamp were performed with mouse blood pooled from three to six mice of identical genotype. After removal of the buffy coat, the cells were washed three times in mouse wash solution containing (in mM) 172 choline chloride, 1 MgCl2, 10 Tris-MOPS, pH 7.40 at 4°. Care was used in handling and centrifuging the fragile Ae1-/- red cells to minimize hemolysis.
Red cell indices were determined by H*3 RTX blood analyzer (Siemens Medical Solutions Diagnostics, Tarrytown, NY) calibrated as specified by the manufacturer for human or for mouse red cells [18]. Red cell cation content was measured by atomic absorption spectrometry as previously described [19].
Measurement of red cell membrane potential as reflected by the equilibrium proton distribution
Indirect measurement of membrane potential (Vm) by pH equilibration was as described [4] with modifications [20]. Erythrocytes washed twice at room temperature with unbuffered medium of desired composition were added to 0.5 ml of the same medium under stirring to a final 5% hematocrit. All media contained the protonophore CCCP (50 μM) with final DMSO ≤0.5%. In the presence of CCCP, changes in Vm are reflected by change in pHo since H+ is kept in equilibrium across the membrane, such that Vm = RT/F (pHi - pHo). The medium was maintained at 37°, and pHo was recorded for 1 min prior and 5 min following addition of red cells. The high buffer capacity of the red cell maintains pHi nearly constant for the duration of the experiment. Thus, pHi was estimated from the lysate pH following addition of 20 ml of 10% (v/v) Triton X-100 to the stirring cell suspension in unbuffered medium [5]. Red cell pHi was 7.09±0.06 (n=9) in Ae1+/+ and 7.08±0.04 (n=6) in Ae1-/- mice.
Vm was perturbed by changing either extracellular [K+] or [Cl-]. External [K+] was varied from 4 to 140 mM by addition of concentrated KCl to unbuffered wash medium, in the absence or presence of 1-3 μM valinomycin (Fig. 1A,B). Theoretical Vm was calculated as RT/F ln[K+]i/[K+]o. Alternatively, in the absence of valinomycin, [Cl-] was varied from 1 to 140 mM by addition of concentrated choline chloride to unbuffered isotonic sucrose/MgCl2 (Figs. 1C,D and 2). Results did not differ when Cl- was added as either the Na+ or K+ salt. In some experiments, [Cl-] was varied by equimolar substitution with the poorly permeant tartrate anion (Fig 4). In cells unexposed to DIDS or to methazolamide, Cl- has a transference number close to 1. Theoretical Vm was calculated as RT/F ln[Cl-]i/[Cl-]o.
Figure 1.
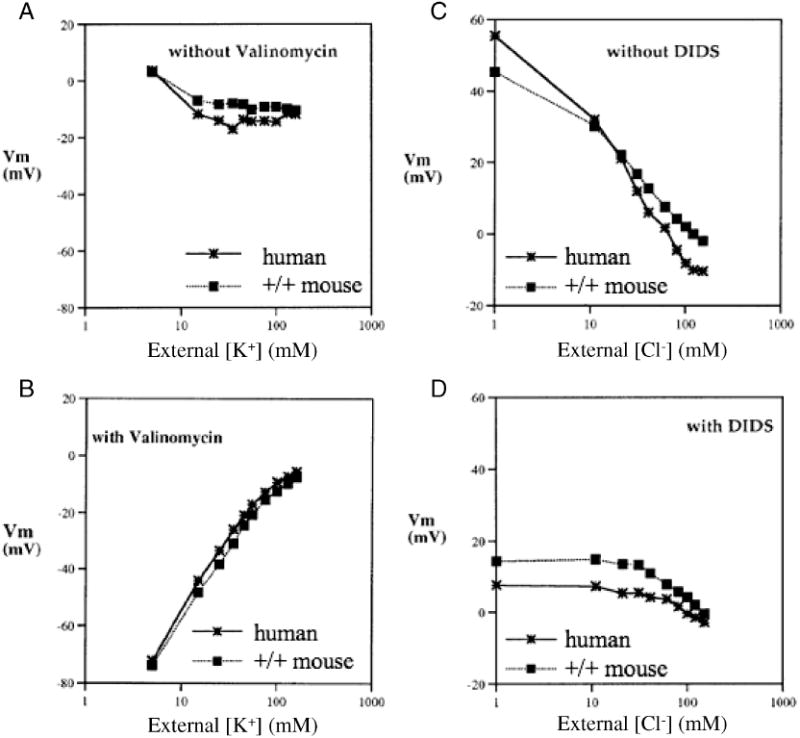
Vm measured by the CCCP method in human red cells (asterisks connected by solid lines) and in CD1 mouse red cells (filled squares connected by dotted lines). Vm was measured as a function of bath [K+] in the absence (A) or presence of 3 μM valinomycin (B, representative of 3 similar experiments), or as a function of bath [Cl-] in in the absence (C) or presence of 10 μM DIDS (D, representative of 5 similar experiments).
Figure 2.
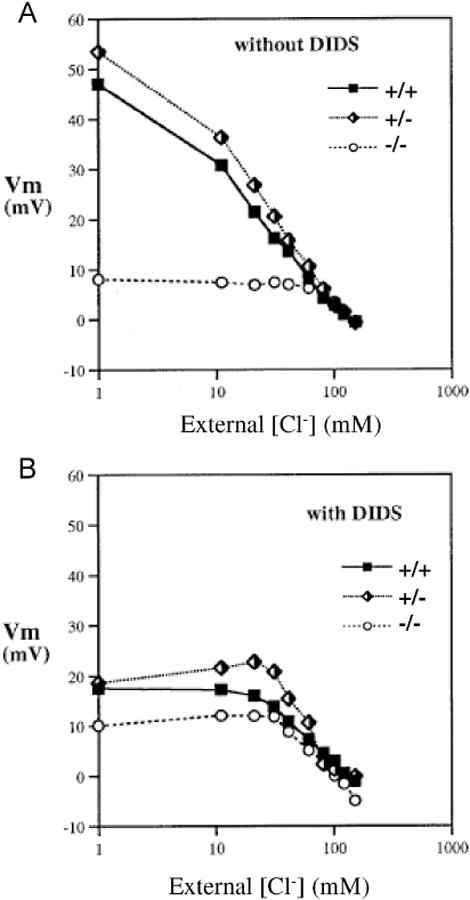
Vm measured by the CCCP method in red cells from Ae1-/-, Ae1+/-, and Ae1-/- mice as a function of bath [Cl-] in the absence (A) or presence of 10 μM DIDS (B). Representative of 5 similar experiments.
Figure 4.
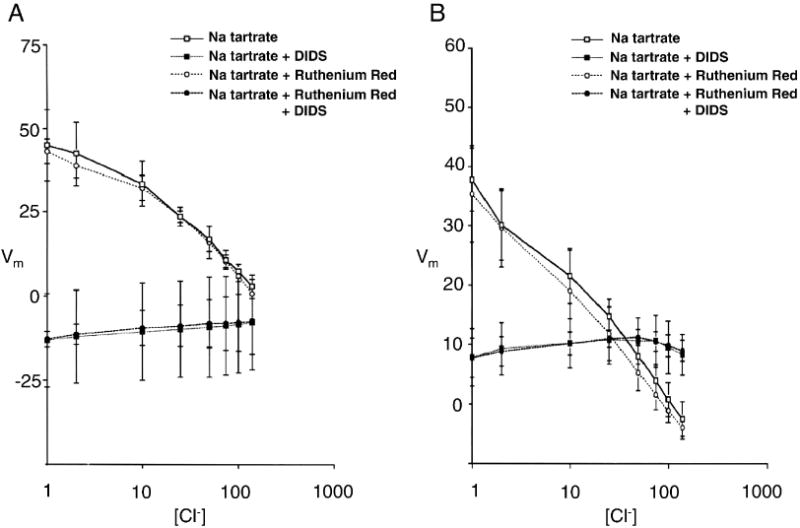
Vm was measured by the CCCP method in human red cells (A, n=4) or in CD1 mouse red cells (B, n=7) during sequential changes in bath [Cl-] with equimolar substitution by tartrate, in the absence or presence of 10 μM DIDS, 50 μM ruthenium red, or both (n=4).
Measurement of anion conductance by valinomycin-induced 86Rb efflux
Valinomycin-induced 86Rb efflux from red cells was measured as described [17], with modifications. Washed mouse red cells were preincubated at 37° for 90 min at 20% hematocrit in efflux medium containing 10 mCi/ml 86Rb+. 86Rb+-loaded red cells were resuspended at 1-2% hematocrit in efflux medium, and maintained at 37° in a shaking water bath. Valinomycin (final 1-3 μM) was added to initiate the efflux assay, and aliquots of cell suspension were taken at 30, 60, and 120 sec time points. Cells and medium were rapidly separated by centrifugation through mineral oil, and cpm of the supernatant and pellet determined by Gamma counting. Data were plotted as ln (% cpm remaining) vs. time.
Measurement of red cell anion conductance by valinomycin-activated volume decrease
Washed mouse red cells (pooled from 3-8 mice) were equilibrated at 23° in a shaking water bath at 2% hematocrit in flux medium containing (in mM) 169 NaCl, 1 KCl, 5 Na HEPES, pH 7.4. After 5 min, 1-3 μM valinomycin was added to the flask. 0.25 ml samples of suspension were aspirated at intervals into the sample port of the H*3 RTX blood analyzer to determine valinomycin-induced changes in mean cell volume (MCV) and in mean corpuscular hemoglobin concentration (CHCM), as described [16] with modification. Some cell suspensions were pretreated for 30 min with DIDS (10 μM), and used without further washing for experiments.
Nystatin-permeabilized whole cell patch clamp of red cells
Washed mouse erythrocytes were stored until use at 4° in preservation solution ((in mM 140 KCl, 10 NaCl, 1 MgCl2, 10 glucose, 1 Na-Phosphate, pH 7.40) then resuspended in bath solution (in mM, 140 N-methyl-D-gluconate chloride, 1 CaCl2, 1 MgCl2, 5 Hepes, pH 7.40). 0.5 mL of dilute red cell suspension was placed on a small coverslip and mounted in a low volume bath on the stage of an inverted microscope. Borosilicate pipettes (Corning 7052) pulled in two stages on a Narashige puller were fire-polished to resistances of 10-20 MΩ. Pipette solution contained (in mM) 140 CsCl, 1CaCl2, 1 MgCl2, 5 HEPES, pH 7.40. Whole cell access was attained with the nystatin-permeabilized patch technique [21]. The pipette was backfilled with pipette solution containing 100 μM nystatin, then front-filled with nystatin-free pipette solution. In a typical experiment, attainment of a 20 GΩ cell-attached seal was followed within ∼3 min by a decrease in pipette resistance to ∼3 GΩ, indicative of transition from cell-attached to whole cell configuration as first reported in frog erythrocytes [6] and subsequently in human [22] and mouse red cells [23].
Currents were recorded with an Axopatch 1-D amplifier, interfaced to a HP Vectra computer via a Digidata 1200 A/D board. The voltage pulse protocol was executed with Clampex software (Pclamp 6.04, Axon Instruments). Capacitance was measured by transient analysis of the current pulse elicited by stepping to 100 mV [24], yielding estimates consistent with those measured by phase-locked integration of currents elicited by sinusoidal voltage protocols [25].
Measurement of other red cell ion transport activities
K-Cl cotransport activated by hypotonic swelling was measured as extracellular Cl--dependent net K+ efflux, as previously described [18]. Gardos channel activity was measured as 1 μM A23187-activated 86Rb+ influx sensitive to 10 μM clotimazole as described [26]. Red cell cation content was measured by atomic absorption as described [18].
Immunoblot detection of red cell membrane proteins
Mouse red cell ghosts were prepared by hypotonic lysis in 5 mM Tris pH 8.0 containing 100 μg/ml phenylmethylsulfonylfluoride and solubilized in LAemmli load buffer. Samples normalized by original cell number were subjected to 4-20% gradient SDS-PAGE, then transferred to nitrocellulose. Immunoblots used either alkaline phosphatase-coupled anti-Ig (Jackson ImmunoResearch, West Grove, PA) with development by bromochloroindolyl-phenol/nitroblue tetrazolium (Kirkegaard and Perry, Gaithersburg, MD), or peroxidase-coupled anti-Ig for enhanced chemiluminescence detection (Perkin-Elmer/ NEN, Boston, MA). Primary antibodies recognizing AE1[15; 27], ankyrin [28; 29], spectrin [29], protein 4.1 [30], AQP1 [31], GLUT1 [32], mouse glycophorins [33], protein 4.2 [34] and Na+,K+-ATPase α subunit (mAb α5) [35] were previously described.
Results
Measurement of Vm by the CCCP method is valid for mouse red cells
Fig. 1A shows that neither human nor mouse cells altered Vm over a 10-fold range of [K+]. However, in the presence of valinomycin, both mouse and human red cell membranes were appropriately dominated by K+ conductance as [K+] was varied between 5 and 50 mM (Fig. 1B). Fig. 1C confirms that both human and mouse cells in the absence of valinomycin changed Vm as a function of [Cl-] nearly as predicted for a Cl--selective electrode. These data confim that the estimation of membrane potential by measurement of the proton gradient across CCCP-treated red cells is also valid for mouse red cells. DIDS treatment of red cells from either human or mouse inhibited and, below 30 mM [Cl-], nearly completely blocked membrane depolarization (Fig. 1D), as shown previously for human red cells with DIDS and methazolamide [20]. Thus, the Cl- conductance of the mouse erythrocyte, like that of the human red cell, has a large DIDS-sensitive component which predominates below 30 mM Cl- in these experimental conditions.
Ae1-/- mouse red cells lack the DIDS-sensitive component of depolarization induced by lowering bath Cl-
Vm of red cells from Ae1+/- and Ae1+/+ mice responded to varying bath Cl- nearly identically, reflecting the native Cl- conductance of the red cell membrane in the absence of valinomycin. However, Ae1-/- red cells failed to depolarize at bath [Cl-] below 60 mM, although at higher [Cl-] Vm changed normally as Cl- was varied (Fig. 2A). In the presence of DIDS (Fig. 2B), the profile of Vm vs. [Cl-] was unchanged at high [Cl-]. However, at low [Cl-], the Vm profiles of red cells from both Ae1+/+ and Ae1+/- mice resembled that of Ae1-/- cells. Thus, Ae1-/- red cells lack the DIDS-sensitive component of Cl- conductance as detected from [Cl-]-dependent changes in Vm measured by the CCCP method.
Increasing bath [K+] from 4 to 140 mM in the absence of valinomycin did not change Vm in Ae1+/+ mouse red cells, but produced a modest depolarization in Ae1-/- mouse or human red cells, suggesting that K+ conductance might be more prominent relative to anion conductance in Ae1-/- red cells than in Ae1+/+ cells. However, in the presence of valinomycin, Ae1-/- red cells were indistinguishable from Ae1+/+ red cells in their depolarizing responses to increasing bath [K+] (Fig. 3A).
Figure 3.
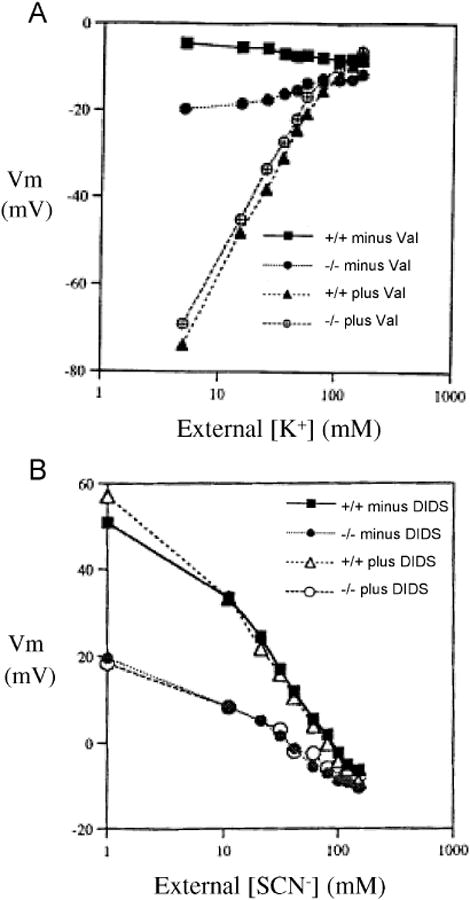
A. Vm measured by the CCCP method in red cells from Ae1-/-, Ae1+/-, and Ae1-/- mice during sequential changes in bath [K+] in the absence or presence of 3 μM valinomycin. Representative of 3 similar experiments. B. Vm measured by the CCCP method in red cells from Ae1-/-, Ae1+/-, and Ae1-/- mice as a function of bath [SCN-] in the absence of Cl-. Representative of 3 similar experiments.
The DIDS-resistant proportion of mouse red cell anion conductance during increases of bath SCN- from 1 to 140 mM was larger at negative Vm than in the presence of bath Cl-, but smaller at positive Vm than in the presence of bath Cl-. Nonetheless, across the entire range of Vm values in SCN- bath, the DIDS-sensitive conductance was absent in mouse Ae1-/- red cells (Fig. 3B).
In human red cells, Vm values above +20 mV to +40 mV activate a nonspecific cation conductance [20; 36] [37] characterized by sensitivity to inhibition by ruthenium red at concentrations as low as 10 μM [20] or by ruthenium red insensitivity with Cl--dependence (detected by tartrate substitution) [37]. However, as shown in Fig. 4, the DIDS-sensitive depolarization induced by lowering bath [Cl-] either in human (A) or in CD1 mouse red cells (B) was insensitive to 50 μM ruthenium red. Fig. 4 also shows that the CCCP method yielded similar profiles of Vm vs. bath [Cl-] when ionic strength was maintained at near constant values by tartrate substitution, as compared to the profiles of Figs. 2 and 3 in which ionic strength was not controlled during variation of [Cl-]. Interestingly, in the presence of tartrate, the DIDS-insensitive component of Cl- conductance evident at negative membrane potentials was not apparent. DIDS-sensitive depolarization was completely abolished in Ae1-/- red cells in tartrate, and the Cl--insensitive Vm in these tartrate-exposed cells was completely insensitive to ruthenium red (n=2, not shown). Thus, the depolarization induced by low bath Cl- in normal mouse red cells likely reflects anion conductance. Any possible contribution to Vm by depolarization-activated cation conductance over the range of Vm is more similar to that observed by Rodighiero et al. [37] than to that observed by Halperin et al. [20].
Valinomycin-treated Ae1-/- mouse red cells exhibit decreased DIDS-sensitive 86Rb+ efflux
After permeabilization of red cells by the K+-specific ionophore, valinomycin, the conductive efflux of K+ is limited by the cellular Cl- conductance. As shown in Fig. 5, the rate constant for 86Rb+ efflux from Ae1+/+ mouse red cells was inhibited 49±7% (n=6) by 10 μM DIDS. The rate constant for 86Rb+ efflux from Ae1+/- red cells was similarly inhibited by 45% (n=2). In contrast, Ae1-/- mouse red cells exhibited a much lower rate of 86Rb+ efflux, which was inhibited only 17±8% by DIDS (n=4, p < 0.02 compared to Ae1+/+ red cells). Thus, DIDS inhibited 50% of mouse red cell Cl- conductance as measured by valinomycin-induced 86Rb+ efflux, and ∼2/3 of that component was absent in red cells lacking Ae1.
Figure 5.
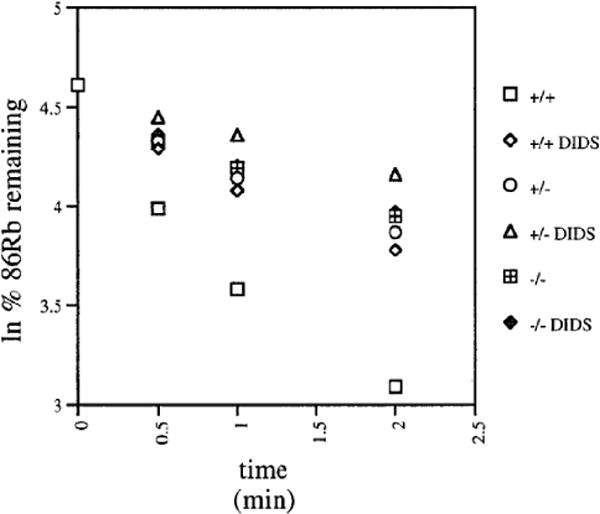
The DIDS-sensitive component of valinomycin-activated 86Rb+ efflux is inhibited in Ae1-/- mouse red cells. 3 μM valinomycin was added to 86Rb+-loaded red cells from Ae1-/-, Ae1+/-, and Ae1-/- mice, in the absence or presence of 10 μM DIDS as indicated.
Valinomycin-treated Ae1-/- mouse red cells exhibit reduced DIDS-sensitive MCV decrease and enhanced DIDS-sensitive CHCM increase
In the presence of non-rate-limiting Cl- conductance, the valinomycin-activated K+ efflux from red cells illustrated in Fig. 5 was associated with a decrease in cell volume (MCV) and a corresponding increase in intracellular hemoglobin concentration (CHCM). Fig. 6 shows that DIDS pretreatment of Ae1+/+ mouse red cells itself increased MCV by almost 5 fL, and conversely decreased CHCM by >2 g/dL. This effect of DIDS occurred as rapidly as could be measured, and was consistent with previous reports [16; 38; 39]. However, addition of valinomycin to Ae1+/+ red cells initiated a decrease in cell volume and an increase in CHCM whose rate was diminished by 10 μM DIDS. In 6 similar experiments, DIDS inhibited the rate of MCV decrease by 42±8% and the rate of CHCM increase by 56±5%. In Ae1-/- mouse red cells, in contrast, both the rapid DIDS effect on cell volume and the DIDS-sensitive components of valinomycin-induced changes in MCV (4±2%) and in CHCM (1±1%) were abolished (n=9). Red cells from Ae1+/- mice exhibited wild-type responses to DIDS, with 52±11% inhibition of valinomycin-induced CHCM increase (n=4) and 34±6% inhibition of valinomycin-induced MCV decrease (Fig. 6). Thus, DIDS inhibited ∼40-50% of valinomycin-induced changes in light scattering and hemoglobin absorbance in wildtype mouse red cells, but lack of Ae1 polypeptide was associated with absence of the DIDS-sensitive components of these hematological indices.
Figure 6.
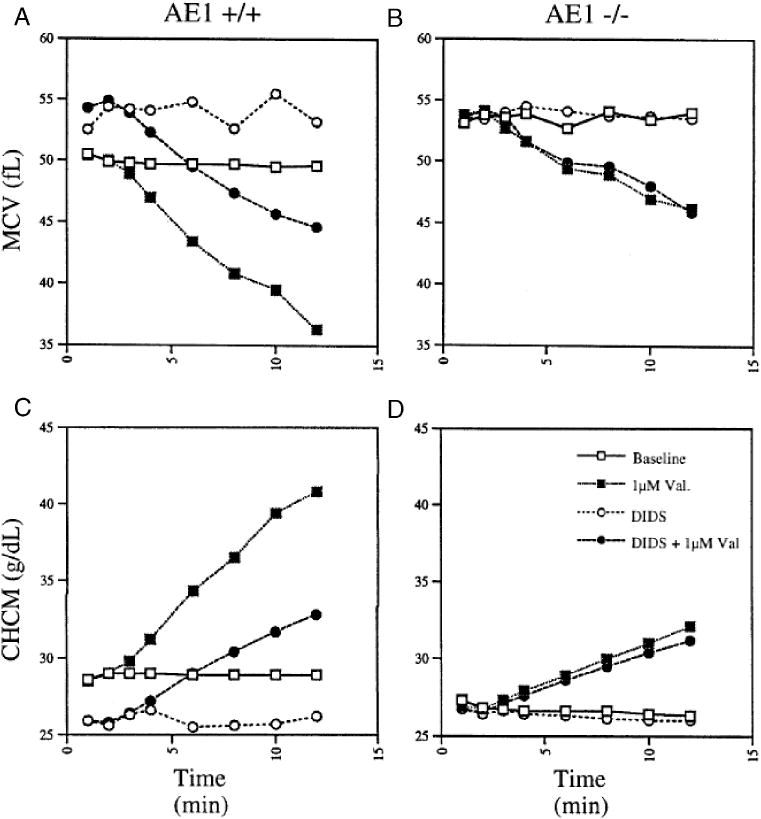
The DIDS-sensitive component of valinomycin-actvated cell shrinkage in Ae1+/+ mouse red cells (left panels) is absent in Ae1-/- red cells (right panels). Aliquots of cells incubated in the absence (squares) or presence of 10 μM DIDS (circles) were removed at the indicated times and analyzed by the H3 autoanalyzer for MCV (upper panels) and for CHCM (lower panels). Valinomycin (3 μM) was added at t=2.5 min (filled symbols) or was omitted (open symbols).
DIDS-sensitive anion currents are absent in Ae1-/- mouse red cells
To complement the above indirect measurements of red cell Cl- conductance, whole cell Cl- currents were measured directly in nystatin-permeabilized mouse red cells (Fig. 7) with Cl- as the sole permeant ion (see Methods). Since Ae1-/- red cells are smaller and have lower surface area than wild-type cells [15; 40], currents were normalized by cell capacitance: 2.7±0.8 pF for Ae1+/+ cells, and 1.7±0.5 pF for Ae1-/- cells (n=8). Ae1-/- red cells exhibited reduced normalized whole cell current magnitude, 33% that of wild-type cells measured at -100 mV and 31% at +100 mV (p=0.05, Fig. 7A and Table 1). DIDS inhibited Ae1+/+ cell current at -100 mV by 45±10% (p<0.02) and mean conductance by 45%, whereas the reduced anion current of Ae1-/- cells exhibited no sensitivity either to DIDS (Fig. 7B and Table 1) or to the anion transport inhibitor NS3623 [41] (n=4, data not shown). Unexpectedly, DIDS modestly increased conductance and current measured at +100 mV in Ae1-/- red cells (Fig. 7C).
Figure 7.
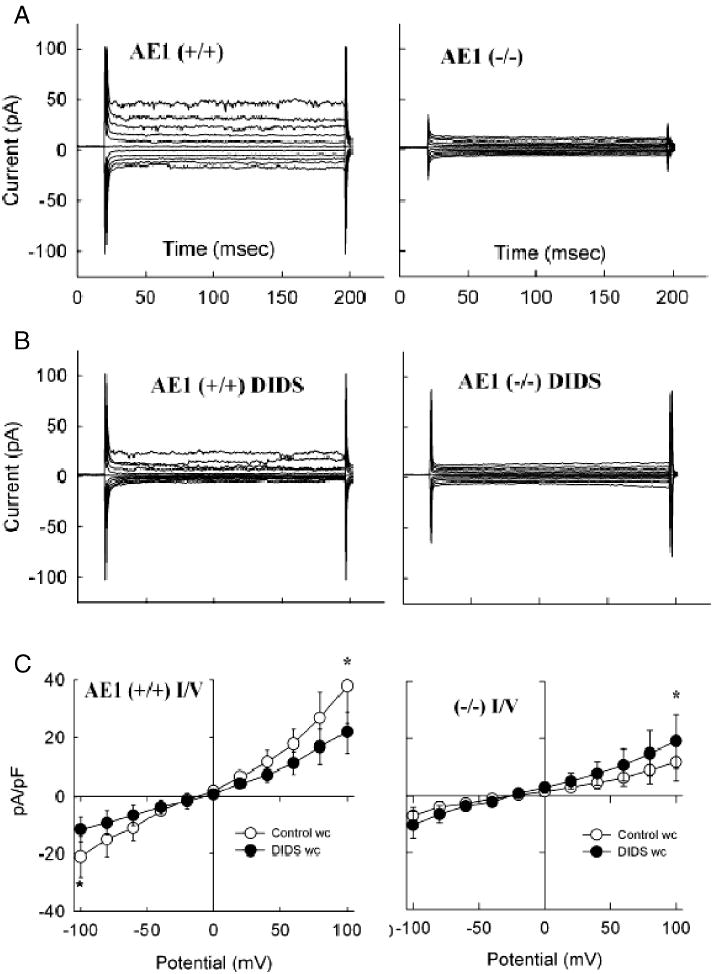
Whole cell Cl- currents recorded in the nystatin-permeabilized patch clamp configuration from Ae1+/+ (left) and from Ae1-/- mouse red cells (right). Representative family of current traces recorded during voltage steps in the absence (A) or presence of 10 μM DIDS (B). C. Mean I-V curves recorded from Ae1+/+ (n=8) and Ae1-/- red cells (n=9) in the absence (open circles) and presence of 10 μM DIDS (closed circles). Currents are capacitance-normalized.
Table 1.
DIDS-sensitive and -insensitive whole-cell Cl- currents recorded by nystatin-permeabilized patch clamp of red cells from Ae1+/+ and Ae1-/- mice.
Nystatin-permeabilized whole cell patch currents of red cells from ae1+/+ and ae1-/- mice.
| Mouse Strain | Current (pA) | Normalized Current (pA/pF) | ||
|---|---|---|---|---|
| @-100 mV | @+100 mV | @-100 mV | @+100 mV | |
| ae1+/+ (n=8) | ||||
| - DIDS | -50±25 | 92±46 | -20.5±6.9 | 38.3±13.4 |
| +DIDS | -25±15 (p=0.03) | 65±43 (p=0.01) | -11.5±4.5 (p<0.02) | 22.0±7.2 (p=0.05) |
| % inhib | 45±10% | 42±11% | 45±10% | 40±9% |
| ae1-/- (n=8) | ||||
| -DIDS | -11.4±5.4 | 12.7±5.1 | -6.7±2.9 | 12.0±6.6 |
| +DIDS | -10.9±5.3 (n.s.) | 12.9±4.5 (n.s.) | -10.2±4.8 (n.s.) | 19.0±9.3 (p<0.05) |
| % inhib | 4% | 0% | 0% | 0 % |
Values are presented as means + s.e.m.; p values are for two-tailed t tests assessing effects of DIDS.
Capacitance of AE1 +/+ cells (n=8) was 2.53±0.72 pF and 2.25±0.65 pF in absence and presence of DIDS, respectively.
Capacitance of AE1 -/- cells (n=8) was 1.73±0.47 pF and 1.06±0.36 pF in absence and presence of DIDS, respectively.
Proteins of the Ae1-/- red cell membrane
Although all the above data is consistent with the hypothesis that DIDS-sensitive Cl- conductance of the red cell is mediated by Ae1, the DIDS-sensitive Cl- conductance might be mediated by an independent polypeptide whose presence in the red cell membrane requires the Ae1-dependent integrity of normal membrane protein composition and lipid bilayer stability [15; 40]. Therefore, we evaluated the polypeptide composition of the Ae1-/- red cell membrane from mice of three ages. Fig. 8 shows, as previously reported [15], that in Ae1-/- membranes the abundance of cytoskeletal proteins ankyrin and spectrin was reduced, and protein 4.2 was completely absent, whereas protein 4.1 abundance was normal. The high Mr glycophorins of the mouse red cell were also absent, as noted previously [42], whereas the nominal glycophorins of lower Mr were reduced but not absent. Aqp1 abundance increased with age, but was substantially reduced in abundance in the absence of Ae1, with apparent loss of complex glycosylation as previously noted [43]. The α subunit of Na+,K+-ATPase was also greatly reduced in Ae1-/- red cells, in concert with the 2-3-fold elevation of intracellular Na+ content (not shown, and [15]. In contrast, abundance of Glut1 in neonatal red cells was normal, and was normally reduced at age 6 days before later disappearance from the red cell, evident at age 9 wks. Bound Ig content was elevated in Ae1-/- red cell at ages 1 and 6 days, but was undetectable in either Ae1-/- or wildtype red cells at age 9 wks.
Figure 8.
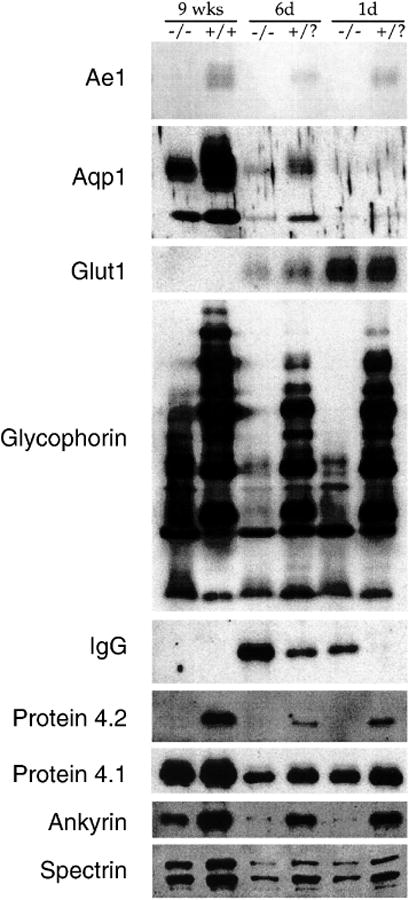
Immunoblots showing expression of the indicated membrane proteins in red cells from Ae1-/- and from Ae1+/? or Ae1+/+ mouse red cells at postnatal ages of 1 day, 6 days, and 9 wks. Representative of two to five similar experiments.
Additional altered properties of Ae1-/- red cells included a two-fold elevation of Cl--independent K+ efflux, and an 80% reduction in swelling-activated K-Cl cotransport from 6.3±0.17 (n=5) to 0.95 (n=2) mMol/(L cells × min). Moreover, Gardos channel activity mediated by Kcnn4/IK1 was increased ∼15-fold in Ae1-/- red cells from 0.064±0.014 (n=9) to 1.11 (2) mMol/(L cells × min) (data not shown). The decreased abundance or absence of multiple intrinsic and extrinsic proteins of the red cell membrane, along with altered regulation of other ion transporters, thus complicates interpretation of the absence of DIDS-sensitive Cl- conductance in red cells of the Ae1-/- mouse.
Discussion
The resting conductance of the human erythrocyte membrane is dominated by its anion conductance. This anion conductance is a therapeutic target in the adjunct treatment of sickle cell disease and other hemoglobinopathies through remediation of pathological red cell dehydration [41; 44]. The terythroid Cl- conductance has components sensitive and insensitive to inhibition by DIDS. but the molecular identities of the polypeptides mediating the two components of anion conductance remain unknown. For many years, the DIDS-sensitive component of red cell anion conductance has been attributed to a low-frequency non-exchange mode of AE1 function pharmacologically similar to AE1-mediated anion exchange. This Cl- conductance has been measured largely by net flux studies in rate-limiting conditions conferred by addition of cationophore, but the mechanism of the hypothesized AE1-mediated conductance has been unclear. As evidence favoring a “slippage” mechanism has not been forthcoming, a “tunneling” mechanism has been proposed [7]. In the time since, uncoupled leak conductances accompanying ion transport have been identified for cotransporters of amino acids, neurotransmitters, and metals, and most recently (in the presence of polyatomic anions) in CLC Cl-/H+ exchangers [45] and SLC26 Cl-/anion exchangers [46]. Among AE1 polypeptides, however, only trout AE1-mediated anion exchange is accompanied by an anion conductance that can be altered by mutagenesis of individual amino acid residues whose mutation does not necessarily alter anion exchange [13; 14].
We have confirmed that anion conductance also dominates the resting conductance of the mouse red cell membrane, and that approximately half of that anion conductance is DIDS-sensitive. We show by four methods that the DIDS-sensitive component of mouse red cell Cl- conductance is greatly reduced or absent in Ae1-/- red cells. These data are consistent with the hypothesis that Ae1 mediates the DIDS-sensitive Cl- conductance in mouse red cells. However, the profoundly altered protein composition of the fragile Ae1-/- red cell membrane allows for the continued possibility that the Ae1-/- red cell membrane has lost one or more anion channel polypeptides that mediate(s) DIDS-sensitive Cl- conductance, possibly but not necessarily in collaboration with the Ae1 polypeptide.
Ae1 as mediator of DIDS-sensitive anion conductance in the red cell
In human red cells, several experimental findings have challenged the hypothesis that DIDS-sensitive Cl- conductance is mediated by AE1 “slippage” (normal operation of the anion exchanger with an occasional conformational half-cycle of the carrier without bound substrate). As summarized by Frohlich [7], Cl- conductance (measured as valinomycin-triggered net K efflux) did not require extracellular Cl-, and increased without saturation proportionate to increasing symmetrical [Cl-] or with either variant of unilateral increase in the presence of fixed contralateral [Cl-]. Anion selectivity (as rank order of rates) differed for conductance and exchange, although anion selectivity of DIDS-sensitive and DIDS-insensitive components of conductance was indistinguishable. Conductance activation energy was much lower than the activation energy for anion exchange. In addition, anion conductance and exhange differed in sensitivity to pharmacological inhibition. Thus, NAP-taurine and phloretin each were more effective inhibitors of anion exchange than of anion conductance. However, phloretin at concentrations ineffective for inhibition of conductance blocked conductance inhibition by DNDS.
We observed the DIDS-sensitive component of anion conductance in mouse red cells (40-50%) to be slightly lower than the 59-65% reported for DIDS- or DNDS-sensitive anion conductance in human red cells [2; 9; 10; 16]. Membrane potential measured by the CCCP method revealed a higher proportion of DIDS-sensitive anion conductance at more positive values of Vm than at more negative values. This observation was consistent with the smaller proportion of DIDS-sensitive anion conductance detected by valinomycin-activated 86Rb+ efflux under hyperpolarizing conditions. It was similarly consistent with the outward rectification of the DIDS-sensitive component of current recorded by nystatin-permeabilized whole cell patch clamp in Ae1+/+ red cells. The DIDS-sensitive component of anion conductance was greatly reduced in Ae1-/- red cells as measured indirectly by the CCCP method (Figs. 2,3), by valinomycin-activated 86Rb+ efflux (Fig. 5), by autoanalyzer measurement of MCV and CHCM (Fig. 6), and directly by nystatin-permeabilized patch clamp (Fig. 7). In the presence of tartrate, the proportional contribution to total anion conductance of the DIDS-sensitive component was greatly increased (Fig. 4), and was absent in Ae1-/- red cells, suggesting that tartrate permeability of the DIDS-insensitive conductance is minimal. Ruthenium red-sensitive cation conductance did not apparently contribute to low Cl--induced depolarization at the most positive values of Vm (Fig. 4).
Effects of lack of Ae1 on other membrane proteins
As noted previously [15; 43], the absence of Ae1 from the mouse red cell membrane is accompanied by major deficits of several cytoskeletal and transport proteins (Fig. 8). In addition, K-Cl cotransport was downregulated while Cl--independent K+ efflux was increased. The Cl--independent component may reflect in part the increased activity of the clotrimazole-sensitive Ca2+-activated K+ permeability (Gardos channel) activity assigned to the IK1/Kcnn4 polypeptide [47; 48; 49] likely resulting in turn from the elevated resting intracellular [Ca2+] in Ae1-/- red cells [50]. This remarkable biochemical remodeling of the red cell membrane in the absence of Ae1, in concert with increased membrane fragility and shape instability, is likely caused by the lack of the intrinsic stabilization of membrane lipid and other protein contributed by Ae1 as the major intrinsic protein of the red cell membrane [15], substantially independent of the Ae1-Ank1 linkage [51]. This instability of membrane protein composition challenges the conclusion that Ae1 mediates both DIDS-sensitive anion conductance and anion exchange, since one or more DIDS-sensitive Cl- channels may be among the many polypeptides lost from the fragile plasma membrane of the Ae1-/- mouse red cell.
The relationship between the mediator(s) of DIDS-sensitive anion conductance in the human and mouse red cells, and the anion conductance associated with wildtype trout AE1 [13] remains unclear. Engineered removal of transmembrane spans 6 and 7 from human AE1 elicited DIDS-sensitive anion conductance in parallel with a decrease in anion exchange activity [52]. In contrast, several individual missense mutations in AE1 associated with spherocytic or ovalocytic variants of stomatocytosis elicited apparent cation conductance of variable stilbene sensitivity in parallel with decreased anion exchange activity [53; 54]. As evident from mutagenesis studies of the ClC superfamily, some of which are Cl- channels and some electrogenic Cl-/H+ exchangers [55], small changes in structure may produce apparently large changes in function and mechanism.
The still-mysterious rapid effect of DIDS on MCV and CHCM was clearly not dependent on the presence of Ae1 in the membrane, nor was it dependent on the presence of the intact membrane protein profile of the Ae1+/+ red cell. The original reports of the DIDS-induced MCV increase in human red cells [39] noted the increase within seconds to tens of seconds, with maximal effect at nominal 0.1-0.2 μM DIDS, and without accompanying detectable cation influx. However, other components of the only ∼80% pure DIDS preparations, such as IADS [56] may act on red cells rapidly and potently enough to contribute to this effect. This AE1-independent effect of “DIDS” is a reminder that even in the red cell, 10 μM DIDS cannot be considered a highly specific drug.
Possible identity of mediator(s) of the DIDS-insensitive anion conductance
As true for the DIDS-sensitive conductance, the molecular identity of the one or more mediators of DIDS-insensitive conductance of the human and mouse red cell remains unknown. Forskolin-activated anion currents insensitive to DIDS but sensitive to glibenclamide have been recorded in Plasmodium falciparum-infected human red cells, and these currents are absent from plasmodium-infected cells from cystic fibrosis patients homozygous for the ΔF508 mutation [57]. Similar currents reported in uninfected human red cells share mechanosensitive gating, halide permselectivity of (I- > Br- > Cl-), and ATP permeability [58], distinguishing them from CFTR expressed in recombinant systems or in most native epithelia. Such currents have not been recorded in uninfected or Plasmodium-infected mouse erythrocytes. Apparently CFTR-dependent ATP release from both human and mouse red cells has been reported [59; 60], but subsequent observations have not detected immunological evidence for CFTR expression in human erythrocytes [61].
Swelling-activated, voltage-dependent, and oxidation-sensitive anion conductance has also been observed in Plasmodium falciparum-infected human red cells [62]. Similar currents recorded in Plasmodium berghei-infected mouse erythrocytes were absent from plasmodium-infected Clcn2-/- red cells [23]. The relationship between this current and the 80 pS outwardly rectifying anion conductance upregulated in red cells of cystic fibrosis patients [58] remains unclear.
Estimates of total red cell conductance vary according to method
Past measurements of human red cell conductance have proven dependent on experimental technique. Use of valinomycin to make native Cl- conductance limiting for K+ efflux yielded whole cell conductance values of 1-4 × 10-5 S · cm-2. Measurements with voltage-sensitive fluorescent dyes produced lower estimates of 1-3 × 10-6 S · cm-2 (summarized in [63]). Microelectrode measurements of membrane impedance at low frequencies extrapolated to values ≤ 5-7 × 10-6 S · cm-2 [64]. With assumptions of mouse red cell intracellular [K+] of 160 mM [65] and a mouse red cell surface area of 90 cm2 [66]), the observed rate of valinomycin-induced 86Rb+ efflux (Fig. 5) corresponds to a mouse red cell conductance of 6.9 × 10-5 S · cm-2. The valinomycin-triggered (light scattering-defined) volume change in mouse red cells (Fig. 6) provides a lower estimate of whole cell conductance of 3 × 10-6 S · cm-2. Both of these estimates are within or near the range of human red cell conductance values previously reported for each method.
Our nystatin-permeabilized on-cell patch recordings of -50 pA inward current at -100 mV in NMDG Cl bath (Fig. 7, Table 1) indicate a directly measured mouse red cell conductance of 5.5 × 10-4 S · cm-2. With an assumed human red cell surface area of 135 μm2 [67; 68], conventional whole cell patch clamp of normal human red cells in symmetrical NaCl yielded similar values of 4.0 × 10-4 S · cm-2 in the negative voltage range and 9.6 × 10-4 S · cm-2 in the positive range [69]. Lower human red cell conductance values were reported by Desai et al. [70] at <7 × 10-5 S · cm-2, and by Huber and colleagues [56; 62; 71; 72] at 3.7, 5.2, and 9.1 × 10-5 S · cm-2 in asymmetrical NaClo/Na gluconatei, increasing to 1-2 × 10-4 S · cm-2 in symmetrical Na gluconate [71; 72]. Rodighiero et al. reported the highest values of 7-13 × 10-3 S · cm-2 [37].
Thus, our value of mouse red cell conductance directly measured by nystatin-permeabilized on-cell patch clamp lies in the exponential mid-range of previously reported human red cell conductance values measured by conventional whole cell patch clamp. These values, extending across 2.5 orders of magnitude, were obtained with borosilicate pipets of similar tip resistance (8-20 MΩ) producing comparable Gigohm seals, and without apparent relationship to the use of symmetrical or asymmetrical anion solutions. The similar values for proportional DIDS sensitivity of whole cell anion conductance observed across the wide range of conductance values obtained by the several analytical methods applied in the current work indicates that the higher conductance values from patch clamp measurements include both DIDS-sensitive and -insensitive components of anion conductance, and cannot be adequately explained either by pipet edge leak or by nystatin leak from the pipet into adjacent membrane. The mechanical stress imposed on the red cell membrane by pipet contact, seal formation, and (for conventional whole cell recording) either electrical pulse or suction, or (for permeabilized patch) potential lipid bilayer perturbation by ionophore each might contribute to the distinctly higher values of whole red cell conductance recorded by patch clamp.
Conclusion
DIDS-sensitive anion conductance in Ae1-/- mouse red cells is reduced or absent as determined by four technical approaches. The data are consistent with mouse Ae1-mediated anion conductance by a “tunneling” mechanism, but in themselves are not informative about conductance mechanism. More importantly, the ability to assign DIDS-sensitive anion conductance to Ae1 function is confounded by the decreased abundance or loss of multiple proteins from fragile Ae1-/- red cell membranes. Definitive implication of wildtype Ae1 in mediation of anion conductance will require techniques allowing acute abrogation of Ae1 function while leaving most if not all other membrane proteins intact during the assay period. Such approaches might include chromophore-assisted [73] or fluorophor-assisted laser inactivation [74].
Acknowledgments
We thank Michelle Rotter, Alan K. Stuart-Tilley, Tammy Nguyen, and Babette Gwynn for technical assistance, Doug Fambrough, Dennis Brown, Morris Birnbaum, Marilyn Farquhar, and Carl Cohen for antibodies, and Sam Lux for encouragement.
This work was supported by NIH grants DK43495 (SLA), HL73112 (DHV), HL64885 (LP), HL15157 (The Boston Sickle Cell Center to CB and SLA), and DK34854 (The Harvard Digestive Diseases Center). SLA was an Established Investigator of the American Heart Association during the time these experiments were performed.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stewart AK, Kurschat CE, Alper SL. The AE Anion Exchanger Gene Family. In: Alpern RJ, Hebert SC, editors. The Kidney: Physiology and Pathophysiology. Elsevier; NY: 2007. pp. 1499–1503. [Google Scholar]
- 2.Knauf PA, Fuhrmann GF, Rothstein S, Rothstein A. The relationship between anion exchange and net anion flow across the human red blood cell membrane. J Gen Physiol. 1977;69:363–86. doi: 10.1085/jgp.69.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunter MJ. Human erythrocyte anion permeabilities measured under conditions of net charge transfer. J Physiol. 1977;268:35–49. doi: 10.1113/jphysiol.1977.sp011845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macey RI, Adorante JS, Orme FW. Erythrocyte membrane potentials determined by hydrogen ion distribution. Biochim Biophys Acta. 1978;512:284–95. doi: 10.1016/0005-2736(78)90253-5. [DOI] [PubMed] [Google Scholar]
- 5.Vestergaard-Bogind B, Bennekou P. Calcium-induced oscillations in K+ conductance and membrane potential of human erythrocytes mediated by the ionophore A23187. Biochim Biophys Acta. 1982;688:37–44. doi: 10.1016/0005-2736(82)90576-4. [DOI] [PubMed] [Google Scholar]
- 6.Hamill O. Potassium and chloride channels in red blood cells. In: Sakmann B, N E, editors. Single Channel Recording. Plenum Press; NY: 1983. [Google Scholar]
- 7.Frohlich O. The “tunneling” mode of biological carrier-mediated transport. J Membr Biol. 1988;101:189–98. doi: 10.1007/BF01872834. [DOI] [PubMed] [Google Scholar]
- 8.Knauf PA, Law FY, Marchant PJ. Relationship of net chloride flow across the human erythrocyte membrane to the anion exchange mechanism. J Gen Physiol. 1983;81:95–126. doi: 10.1085/jgp.81.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaplan JH, Pring M, Passow H. Band-3 protein-mediated anion conductance of the red cell membrane. Slippage vs ionic diffusion. FEBS Lett. 1983;156:175–9. doi: 10.1016/0014-5793(83)80272-5. [DOI] [PubMed] [Google Scholar]
- 10.Frohlich O. Relative contributions of the slippage and tunneling mechanisms to anion net efflux from human erythrocytes. J Gen Physiol. 1984;84:877–93. doi: 10.1085/jgp.84.6.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson N, Sacher A, Nelson H. The significance of molecular slips in transport systems. Nat Rev Mol Cell Biol. 2002;3:876–81. doi: 10.1038/nrm955. [DOI] [PubMed] [Google Scholar]
- 12.DeFelice LJ, Goswami T. Transporters as channels. Annu Rev Physiol. 2007;69:87–112. doi: 10.1146/annurev.physiol.69.031905.164816. [DOI] [PubMed] [Google Scholar]
- 13.Martial S, Guizouarn H, Gabillat N, Pellissier B, Borgese F. Consequences of point mutations in trout anion exchanger 1 (tAE1) transmembrane domains: evidence that tAE1 can behave as a chloride channel. J Cell Physiol. 2006;207:829–35. doi: 10.1002/jcp.20631. [DOI] [PubMed] [Google Scholar]
- 14.Martial S, Guizouarn H, Gabillat N, Pellissier B, Borgese F. Importance of several cysteine residues for the chloride conductance of trout anion exchanger 1 (tAE1) J Cell Physiol. 2007;213:70–8. doi: 10.1002/jcp.21093. [DOI] [PubMed] [Google Scholar]
- 15.Peters LL, Shivdasani RA, Liu SC, Hanspal M, John KM, Gonzalez JM, Brugnara C, Gwynn B, Mohandas N, Alper SL, Orkin SH, Lux SE. Anion exchanger 1 (band 3) is required to prevent erythrocyte membrane surface loss but not to form the membrane skeleton. Cell. 1996;86:917–27. doi: 10.1016/s0092-8674(00)80167-1. [DOI] [PubMed] [Google Scholar]
- 16.Freedman JC, Novak TS, Bisognano JD, Pratap PR. Voltage dependence of DIDS-insensitive chloride conductance in human red blood cells treated with valinomycin or gramicidin. J Gen Physiol. 1994;104:961–83. doi: 10.1085/jgp.104.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jennings ML. Rapid electrogenic sulfate-chloride exchange mediated by chemically modified band 3 in human erythrocytes. J Gen Physiol. 1995;105:21–47. doi: 10.1085/jgp.105.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armsby CC, Brugnara C, Alper SL. Cation transport in mouse erythrocytes: role of K(+)-Cl- cotransport in regulatory volume decrease. Am J Physiol. 1995;268:C894–902. doi: 10.1152/ajpcell.1995.268.4.C894. [DOI] [PubMed] [Google Scholar]
- 19.Brugnara C, Kopin AS, Bunn HF, Tosteson DC. Regulation of cation content and cell volume in hemoglobin erythrocytes from patients with homozygous hemoglobin C disease. J Clin Invest. 1985;75:1608–17. doi: 10.1172/JCI111867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halperin JA, Brugnara C, Tosteson MT, Van Ha T, Tosteson DC. Voltage-activated cation transport in human erythrocytes. Am J Physiol. 1989;257:C986–96. doi: 10.1152/ajpcell.1989.257.5.C986. [DOI] [PubMed] [Google Scholar]
- 21.Mahaut-Smith MP, Sage SO, Rink TJ. Rapid ADP-evoked currents in human platelets recorded with the nystatin permeabilized patch technique. J Biol Chem. 1992;267:3060–5. [PubMed] [Google Scholar]
- 22.Schwarz W, Grygorczyk R, Hof D. Recording single-channel currents from human red cells. Methods Enzymol. 1989;173:112–21. doi: 10.1016/s0076-6879(89)73009-3. [DOI] [PubMed] [Google Scholar]
- 23.Huber SM, Duranton C, Henke G, Van De Sand C, Heussler V, Shumilina E, Sandu CD, Tanneur V, Brand V, Kasinathan RS, Lang KS, Kremsner PG, Hubner CA, Rust MB, Dedek K, Jentsch TJ, Lang F. Plasmodium induces swelling-activated ClC-2 anion channels in the host erythrocyte. J Biol Chem. 2004;279:41444–52. doi: 10.1074/jbc.M407618200. [DOI] [PubMed] [Google Scholar]
- 24.Schwiebert EM, Kizer N, Gruenert DC, Stanton BA. GTP-binding proteins inhibit cAMP activation of chloride channels in cystic fibrosis airway epithelial cells. Proc Natl Acad Sci U S A. 1992;89:10623–7. doi: 10.1073/pnas.89.22.10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spruce AE, Iwata A, White JM, Almers W. Patch clamp studies of single cell-fusion events mediated by a viral fusion protein. Nature. 1989;342:555–8. doi: 10.1038/342555a0. [DOI] [PubMed] [Google Scholar]
- 26.Brugnara C, De Franceschi L, Alper SL. Ca(2+)-activated K+ transport in erythrocytes. Comparison of binding and transport inhibition by scorpion toxins. J Biol Chem. 1993;268:8760–8. [PubMed] [Google Scholar]
- 27.Alper SL, Stuart-Tilley AK, Biemesderfer D, Shmukler BE, Brown D. Immunolocalization of AE2 anion exchanger in rat kidney. Am J Physiol. 1997;273:F601–14. doi: 10.1152/ajprenal.1997.273.4.F601. [DOI] [PubMed] [Google Scholar]
- 28.Korsgren C, Cohen CM. Associations of human erythrocyte band 4.2. Binding to ankyrin and to the cytoplasmic domain of band 3. J Biol Chem. 1988;263:10212–8. [PubMed] [Google Scholar]
- 29.Alper SL, Stuart-Tilley A, Simmons CF, Brown D, Drenckhahn D. The fodrin-ankyrin cytoskeleton of choroid plexus preferentially colocalizes with apical Na+K(+)-ATPase rather than with basolateral anion exchanger AE2. J Clin Invest. 1994;93:1430–8. doi: 10.1172/JCI117120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Armsby CC, Stuart-Tilley AK, Alper SL, Brugnara C. Resistance to osmotic lysis in BXD-31 mouse erythrocytes: association with upregulated K-Cl cotransport. Am J Physiol. 1996;270:C866–77. doi: 10.1152/ajpcell.1996.270.3.C866. [DOI] [PubMed] [Google Scholar]
- 31.Sabolic I, Valenti G, Verbavatz JM, Van Hoek AN, Verkman AS, Ausiello DA, Brown D. Localization of the CHIP28 water channel in rat kidney. Am J Physiol. 1992;263:C1225–33. doi: 10.1152/ajpcell.1992.263.6.C1225. [DOI] [PubMed] [Google Scholar]
- 32.Hudson AW, Ruiz M, Birnbaum MJ. Isoform-specific subcellular targeting of glucose transporters in mouse fibroblasts. J Cell Biol. 1992;116:785–97. doi: 10.1083/jcb.116.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dolci ED, Palade GE. Ontogenetic expression of the murine erythrocyte glycophorins. J Cell Sci. 1989;93(Pt 1):191–7. doi: 10.1242/jcs.93.1.191. [DOI] [PubMed] [Google Scholar]
- 34.Peters LL, Jindel HK, Gwynn B, Korsgren C, John KM, Lux SE, Mohandas N, Cohen CM, Cho MR, Golan DE, Brugnara C. Mild spherocytosis and altered red cell ion transport in protein 4. 2-null mice. J Clin Invest. 1999;103:1527–37. doi: 10.1172/JCI5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lebovitz RM, Takeyasu K, Fambrough DM. Molecular characterization and expression of the (Na+ + K+)-ATPase alpha-subunit in Drosophila melanogaster. Embo J. 1989;8:193–202. doi: 10.1002/j.1460-2075.1989.tb03364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christophersen P, Bennekou P. Evidence for a voltage-gated, non-selective cation channel in the human red cell membrane. Biochim Biophys Acta. 1991;1065:103–6. doi: 10.1016/0005-2736(91)90017-3. [DOI] [PubMed] [Google Scholar]
- 37.Rodighiero S, De Simoni A, Formenti A. The voltage-dependent nonselective cation current in human red blood cells studied by means of whole-cell and nystatin-perforated patch-clamp techniques. Biochim Biophys Acta. 2004;1660:164–70. doi: 10.1016/j.bbamem.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 38.Hoefner DM, Blank ME, Davis BM, Diedrich DF. Band 3 antagonists, p-azidobenzylphlorizin and DIDS, mediate erythrocyte shape and flexibility changes as characterized by digital image morphometry and microfiltration. J Membr Biol. 1994;141:91–100. doi: 10.1007/BF00232877. [DOI] [PubMed] [Google Scholar]
- 39.Blank ME, Hoefner DM, Diedrich DF. Morphology and volume alterations of human erythrocytes caused by the anion transporter inhibitors, DIDS and p-azidobenzylphlorizin. Biochim Biophys Acta. 1994;1192:223–33. doi: 10.1016/0005-2736(94)90122-8. [DOI] [PubMed] [Google Scholar]
- 40.Southgate CD, Chishti AH, Mitchell B, Yi SJ, Palek J. Targeted disruption of the murine erythroid band 3 gene results in spherocytosis and severe hAemolytic anAemia despite a normal membrane skeleton. Nat Genet. 1996;14:227–30. doi: 10.1038/ng1096-227. [DOI] [PubMed] [Google Scholar]
- 41.Bennekou P, de Franceschi L, Pedersen O, Lian L, Asakura T, Evans G, Brugnara C, Christophersen P. Treatment with NS3623, a novel Cl-conductance blocker, ameliorates erythrocyte dehydration in transgenic SAD mice: a possible new therapeutic approach for sickle cell disease. Blood. 2001;97:1451–7. doi: 10.1182/blood.v97.5.1451. [DOI] [PubMed] [Google Scholar]
- 42.Hassoun H, Hanada T, Lutchman M, Sahr KE, Palek J, Hanspal M, Chishti AH. Complete deficiency of glycophorin A in red blood cells from mice with targeted inactivation of the band 3 (AE1) gene. Blood. 1998;91:2146–51. [PubMed] [Google Scholar]
- 43.Bruce LJ, Beckmann R, Ribeiro ML, Peters LL, Chasis JA, Delaunay J, Mohandas N, Anstee DJ, Tanner MJ. A band 3-based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood. 2003;101:4180–8. doi: 10.1182/blood-2002-09-2824. [DOI] [PubMed] [Google Scholar]
- 44.Brugnara C, De Franceschi L, Bennekou P, Alper SL, Christophersen P. Novel therapies for prevention of erythrocyte dehydration in sickle cell anemia. Drug News Perspect. 2001;14:208–20. doi: 10.1358/dnp.2001.14.4.858404. [DOI] [PubMed] [Google Scholar]
- 45.Nguitragool W, Miller C. Uncoupling of a CLC Cl-/H+ exchange transporter by polyatomic anions. J Mol Biol. 2006;362:682–90. doi: 10.1016/j.jmb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 46.Shcheynikov N, Wang Y, Park M, Ko SB, Dorwart M, Naruse S, Thomas PJ, Muallem S. Coupling modes and stoichiometry of Cl-/HCO3- exchange by slc26a3 and slc26a6. J Gen Physiol. 2006;127:511–24. doi: 10.1085/jgp.200509392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vandorpe DH, Shmukler BE, Jiang L, Lim B, Maylie J, Adelman JP, de Franceschi L, Cappellini MD, Brugnara C, Alper SL. cDNA cloning and functional characterization of the mouse Ca2+-gated K+ channel, mIK1. Roles in regulatory volume decrease and erythroid differentiation. J Biol Chem. 1998;273:21542–53. doi: 10.1074/jbc.273.34.21542. [DOI] [PubMed] [Google Scholar]
- 48.Begenisich T, Nakamoto T, Ovitt CE, Nehrke K, Brugnara C, Alper SL, Melvin JE. Physiological roles of the intermediate conductance, Ca2+-activated potassium channel Kcnn4. J Biol Chem. 2004;279:47681–7. doi: 10.1074/jbc.M409627200. [DOI] [PubMed] [Google Scholar]
- 49.Hoffman JF, Joiner W, Nehrke K, Potapova O, Foye K, Wickrema A. The hSK4 (KCNN4) isoform is the Ca2+-activated K+ channel (Gardos channel) in human red blood cells. Proc Natl Acad Sci U S A. 2003;100:7366–71. doi: 10.1073/pnas.1232342100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akel A, Wagner CA, Kovacikova J, Kasinathan RS, Kiedaisch V, Koka S, Alper SL, Bernhardt I, Wieder T, Huber SM, Lang F. Enhanced suicidal death of erythrocytes from gene-targeted mice lacking the Cl-/HCO(3)(-) exchanger AE1. Am J Physiol Cell Physiol. 2007;292:C1759–67. doi: 10.1152/ajpcell.00158.2006. [DOI] [PubMed] [Google Scholar]
- 51.Stefanovic M, Markham NO, Parry EM, Garrett-Beal LJ, Cline AP, Gallagher PG, Low PS, Bodine DM. An 11-amino acid beta-hairpin loop in the cytoplasmic domain of band 3 is responsible for ankyrin binding in mouse erythrocytes. Proc Natl Acad Sci U S A. 2007;104:13972–7. doi: 10.1073/pnas.0706266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parker MD, Young MT, Daly CM, Meech RW, Boron WF, Tanner MJ. A conductive pathway generated from fragments of the human red cell anion exchanger AE1. J Physiol. 2007;581:33–50. doi: 10.1113/jphysiol.2007.128389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bruce LJ, Robinson HC, Guizouarn H, Borgese F, Harrison P, King MJ, Goede JS, Coles SE, Gore DM, Lutz HU, Ficarella R, Layton DM, Iolascon A, Ellory JC, Stewart GW. Monovalent cation leaks in human red cells caused by single amino-acid substitutions in the transport domain of the band 3 chloride-bicarbonate exchanger, AE1. Nat Genet. 2005;37:1258–63. doi: 10.1038/ng1656. [DOI] [PubMed] [Google Scholar]
- 54.Guizouarn H, Martial S, Gabillat N, Borgese F. Point mutations involved in red cell stomatocytosis convert the electroneutral anion exchanger 1 to a nonselective cation conductance. Blood. 2007;110:2158–65. doi: 10.1182/blood-2006-12-063420. [DOI] [PubMed] [Google Scholar]
- 55.Dutzler R. The ClC family of chloride channels and transporters. Curr Opin Struct Biol. 2006;16:439–46. doi: 10.1016/j.sbi.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 56.Stumpf A, Almaca J, Kunzelmann K, Wenners-Epping K, Huber SM, Haberle J, Falk S, Duebbers A, Walte M, Oberleithner H, Schillers H. IADS, a decomposition product of DIDS activates a cation conductance in Xenopus oocytes and human erythrocytes: new compound for the diagnosis of cystic fibrosis. Cell Physiol Biochem. 2006;18:243–52. doi: 10.1159/000097671. [DOI] [PubMed] [Google Scholar]
- 57.Verloo P, Kocken CH, Van der Wel A, Tilly BC, Hogema BM, Sinaasappel M, Thomas AW, De Jonge HR. Plasmodium falciparum-activated chloride channels are defective in erythrocytes from cystic fibrosis patients. J Biol Chem. 2004;279:10316–22. doi: 10.1074/jbc.M311540200. [DOI] [PubMed] [Google Scholar]
- 58.Decherf G, Bouyer G, Egee S, Thomas SL. Chloride channels in normal and cystic fibrosis human erythrocyte membrane. Blood Cells Mol Dis. 2007;39:24–34. doi: 10.1016/j.bcmd.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 59.Abraham EH, Sterling KM, Kim RJ, Salikhova AY, Huffman HB, Crockett MA, Johnston N, Parker HW, Boyle WE, Jr, Hartov A, Demidenko E, Efird J, Kahn J, Grubman SA, Jefferson DM, Robson SC, Thakar JH, Lorico A, Rappa G, Sartorelli AC, Okunieff P. Erythrocyte membrane ATP binding cassette (ABC) proteins: MRP1 and CFTR as well as CD39 (ecto-apyrase) involved in RBC ATP transport and elevated blood plasma ATP of cystic fibrosis. Blood Cells Mol Dis. 2001;27:165–80. doi: 10.1006/bcmd.2000.0357. [DOI] [PubMed] [Google Scholar]
- 60.Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. Participation of cAMP in a signal-transduction pathway relating erythrocyte deformation to ATP release. Am J Physiol Cell Physiol. 2001;281:C1158–64. doi: 10.1152/ajpcell.2001.281.4.C1158. [DOI] [PubMed] [Google Scholar]
- 61.Hoffman JF, Dodson A, Wickrema A, Dib-Hajj SD. Tetrodotoxin-sensitive Na+ channels and muscarinic and purinergic receptors identified in human erythroid progenitor cells and red blood cell ghosts. Proc Natl Acad Sci U S A. 2004;101:12370–4. doi: 10.1073/pnas.0404228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huber SM, Uhlemann AC, Gamper NL, Duranton C, Kremsner PG, Lang F. Plasmodium falciparum activates endogenous Cl(-) channels of human erythrocytes by membrane oxidation. Embo J. 2002;21:22–30. doi: 10.1093/emboj/21.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoffman JF. Estimates of the elecctrical conductance of the red cell membrane. In: Bamberg E, P H, editors. Progress in Cell Research. Elsevier; Amsterdam: 1992. pp. 173–178. [Google Scholar]
- 64.Takashima S, Asami K, Takahashi Y. Frequency domain studies of impedance characteristics of biological cells using micropipet technique. I. Erythrocyte. Biophys J. 1988;54:995–1000. doi: 10.1016/S0006-3495(88)83037-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benos DJ. Intracellular analysis of sodium, potassium, and chloride in mouse erythrocytes. J Cell Physiol. 1980;105:185–7. doi: 10.1002/jcp.1041050120. [DOI] [PubMed] [Google Scholar]
- 66.Waugh RE, Sarelius IH. Effects of lost surface area on red blood cells and red blood cell survival in mice. Am J Physiol. 1996;271:C1847–52. doi: 10.1152/ajpcell.1996.271.6.C1847. [DOI] [PubMed] [Google Scholar]
- 67.Evans E, Fung YC. Improved measurements of the erythrocyte geometry. Microvasc Res. 1972;4:335–47. doi: 10.1016/0026-2862(72)90069-6. [DOI] [PubMed] [Google Scholar]
- 68.Gifford SC, Derganc J, Shevkoplyas SS, Yoshida T, Bitensky MW. A detailed study of time-dependent changes in human red blood cells: from reticulocyte maturation to erythrocyte senescence. Br J HAematol. 2006;135:395–404. doi: 10.1111/j.1365-2141.2006.06279.x. [DOI] [PubMed] [Google Scholar]
- 69.Browning JA, Staines HM, Robinson HC, Powell T, Ellory JC, Gibson JS. The effect of deoxygenation on whole-cell conductance of red blood cells from healthy individuals and patients with sickle cell disease. Blood. 2007;109:2622–9. doi: 10.1182/blood-2006-03-001404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Desai SA, Bezrukov SM, Zimmerberg J. A voltage-dependent channel involved in nutrient uptake by red blood cells infected with the malaria parasite. Nature. 2000;406:1001–5. doi: 10.1038/35023000. [DOI] [PubMed] [Google Scholar]
- 71.Myssina S, Huber SM, Birka C, Lang PA, Lang KS, Friedrich B, Risler T, Wieder T, Lang F. Inhibition of erythrocyte cation channels by erythropoietin. J Am Soc Nephrol. 2003;14:2750–7. doi: 10.1097/01.asn.0000093253.42641.c1. [DOI] [PubMed] [Google Scholar]
- 72.Lang KS, Myssina S, Tanneur V, Wieder T, Huber SM, Lang F, Duranton C. Inhibition of erythrocyte cation channels and apoptosis by ethylisopropylamiloride. Naunyn Schmiedebergs Arch Pharmacol. 2003;367:391–6. doi: 10.1007/s00210-003-0701-z. [DOI] [PubMed] [Google Scholar]
- 73.Hoffman-Kim D, Diefenbach TJ, Eustace BK, Jay DG. Chromophore-assisted laser inactivation. Methods Cell Biol. 2007;82:335–54. doi: 10.1016/S0091-679X(06)82011-X. [DOI] [PubMed] [Google Scholar]
- 74.Marks KM, Braun PD, Nolan GP. A general approach for chemical labeling and rapid, spatially controlled protein inactivation. Proc Natl Acad Sci U S A. 2004;101:9982–7. doi: 10.1073/pnas.0401609101. [DOI] [PMC free article] [PubMed] [Google Scholar]