

Abstract
Phthalate esters are ubiquitous environmental contaminants that are produced for a variety of common industrial and commercial purposes. We have shown that mono-(2-ethylhexyl) phthalate (MEHP), the toxic metabolite of di-(2-ethylhexyl) phthalate, induces bone marrow B cell apoptosis that is enhanced in the presence of the endogenous prostaglandin 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2). Here, studies were performed to determine if 15d-PGJ2-mediated enhancement of MEHP-induced apoptosis represents activation of an overlapping or complementary apoptosis pathway. MEHP and 15d-PGJ2 induced significant apoptosis within 8 and 5 hrs, respectively, in a pro/pre-B cell line and acted cooperatively to induce apoptosis in primary pro-B cells. Apoptosis induced with each chemical was accompanied by activation of a combination of initiator caspases (caspases-2, -8, and -9) and executed by caspase-3. Apoptosis induced with MEHP and 15d-PGJ2 was reduced in APAF1 null primary pro-B cells and accompanied by alteration of mitochondrial membranes, albeit with different kinetics, indicating an intrinsically-activated apoptosis pathway. Significant Bax translocation to the mitochondria supports its role in initiating release of cytochrome c. Both chemicals induced Bid cleavage, a result consistent with a tBid-mediated release of cytochrome c in an apoptosis amplification feedback loop; however, significantly more Bid was cleaved following 15d-PGJ2 treatment, potentially differentiating the two pathways. Indeed, Bid cleavage and cytochrome c release following 15d-PGJ2 but not MEHP treatment was profoundly inhibited by Z-VAD-FMK, suggesting that 15d-PGJ2 activates apoptosis via two pathways, Bax mobilization and protease-dependent Bid cleavage. Thus, endogenous 15d-PGJ2-mediated enhancement of environmental chemical-induced apoptosis represents activation of an overlapping but distinct signaling pathway.
Keywords: phthalate ester, 15d-PGJ2, apoptosis, B lymphocytes
Introduction
Worldwide, over 18 billion pounds of phthalate esters are produced yearly, mainly for use as plasticizers in polyvinyl chloride products such as car seats, toys, bloodbags and i.v. lines. Humans are regularly exposed to significant levels (14 and 7 μg/kg/day, respectively) of di-(2-ethylhexyl) phthalate (DEHP)3, the main plasticizer used in the production of polyvinyl chloride (1-3). Substantially higher exposures occur during acute medical procedures, resulting in blood DEHP concentrations ranging from 50-350 μM (4, 5). Furthermore, modeling studies suggest that exposure in children exceeds that in adults (6). It has been suggested that these environmental phthalate exposures have significant biologic consequences, potentially through their interaction with nuclear receptors. Mono-(2-ethylhexyl) phthalate (MEHP), the active metabolite of DEHP and an agonist of peroxisome proliferator activated receptors (PPAR) α and γ, induces PPARγ-DNA binding and recruitment of a specific subset of coactivators, selectively activates PPARγ-inducible genes and induces adipogenesis through PPARγ (7). MEHP suppresses aromatase expression in a model of reproductive toxicity (8) and induces Fas-dependent apoptosis in testis (9). DEHP/MEHP exposure results in ovarian, testicular, and developmental toxicity in rodent models (8-10).
Phthalate esters also target the immune system. Microarray analyses of liver from DEHP-treated C57BL/6 mice indicates down-regulation of complement components (11). MEHP and other monophthalates suppress ovalbumin-specific antibody production in BALB/cJ mice (12). DEHP treatment of C57Bl/6 mice induces severe thymic and splenic atrophy (13). In the spleen, treatment with another PPAR ligand, perfluorooctanoic acid, substantially reduces the percentage and absolute number of splenic CD19+ B cells by 86% (13). However, it is not clear if these latter outcomes are due to direct effects on mature B cell populations and/or on their development from bone marrow-derived precursors.
In this vein, others have shown that DEHP suppresses LPS-stimulated proliferation of splenic B cells (14), and we have shown that bone marrow B cells are susceptible to MEHP-induced cell cycle arrest and apoptosis (15). MEHP inhibited 3H-thymidine incorporation by primary murine bone marrow B cells and by a non-transformed murine pro/pre-B cell line. At moderate MEHP concentrations (150-200 μM), inhibition of 3H-thymidine incorporation resulted primarily from apoptosis as indicated by PARP cleavage and DNA fragmentation. Similarly, MEHP-induced apoptosis has been described in the monocyte cell line U937 and also is accompanied by activation of caspase-3 and DNA fragmentation (16). We observed that proliferation in either primary pro-B cells or in a pro/pre-B cell line decreased at lower MEHP concentrations (10 and 100 μM, respectively) and this growth inhibition was accompanied by p27kip up-regulation and occurred in the absence of apoptosis (14).
Perhaps of still greater physiological significance is the fact that MEHP-induced apoptosis can be enhanced in the presence of an endogenously occurring chemical, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2). 15d-PGJ2 is a PPARγ agonist (17) and a known apoptotic agent in lymphocytes (15, 18-20). We have shown that co-treatment of pro/pre-B cells with low MEHP (75 μM) and 15d-PGJ2 (1 μM) doses significantly reduced 3H-thymidine incorporation and enhanced the growth arrest seen with MEHP alone. Furthermore, co-treatment with MEHP (100 μM) and 2 μM 15d-PGJ2 enhanced the percentage of pro/pre-B cells undergoing apoptosis from 12% to 40%. (15). Importantly, 15d-PGJ2 and its precursor, prostaglandin D2, are found at high concentrations in bone marrow, potentially reaching local concentrations in the micromolar range (21). These results suggest that B cells in the bone marrow microenvironment may be particularly susceptible to phthalate-induced toxicity. Consequently, it is important to define the 15d-PGJ2-induced apoptosis pathway and to determine if bone marrow B cell apoptosis induced with the combination of 15d-PGJ2 and MEHP is mediated by distinct and complementing apoptosis signaling pathways or represents essentially the cumulative effects of two PPARγ agonists on a single death pathway. These issues were addressed here by mapping the signaling pathways activated by these two agents and determining the degree of signaling overlap.
Materials and Methods
The caspase-8-specific antibody was from Axxora (San Diego, CA). The cytochrome c-specific antibody was from BD Biosciences Clontech (Palo Alto, CA). Caspase inhibitors and 15d-PGJ2 were from Biomol (Plymouth Meeting, PA). Antibodies specific for cleaved caspases-3 and -9 and cleaved lamin were from Cell Signaling Technology (Beverly, MA). Antibodies specific for α-fodrin and caspase-2 were from Chemicon International (Temecula, CA). The α-tubulin-specific antibody was from EMD Biosciences (San Diego, CA). Plasmocin was from Invivogen (San Diego, CA). JC-1 was from Molecular Probes (Eugene, OR). The Bid-specific antibody was from R&D Systems (Minneapolis, MN). Murine rIL-7 was from Research Diagnostics (Flanders, NJ). Antibodies specific for Bax and HSP60 were from Santa Cruz Biotechnology (Santa Cruz, CA). Propidium iodide, Protease Inhibitor Cocktail for Mammalian Cells, and the β-actin-specific antibody were from Sigma Chemical Co. (St. Louis, MO). MEHP was from TCI America (Portland, OR). All other reagents were from Fisher Scientific (Suanee, GA).
Cell Culture
The stromal cell-dependent, C57BL/6-derived BU-11 cell line has been characterized previously (15, 22, 23). BU-11 cells represent B cells at the transition between the pro- and early pre-B cell stages as they are CD43+/B220+/IgM- with rearranged Ig heavy chain genes. BMS2 is a culture-dish adherent cloned bone marrow stromal cell line that supports BU-11 cell growth. Stocks of BU-11 cells were maintained on BMS2 cell monolayers in an equal mixture of DMEM and RPMI 1640 medium with 5% FBS, Plasmocin, L-glutamine and 2-mercaptoethanol. All cultures were maintained at 37° C in a humidified, 7.5% CO2 atmosphere. Cell cultures were determined to be mycoplasma negative by PCR (Mycoplasma Detection Kit; ATCC, Manassas, VA).
Primary bone marrow pro-B cell cultures were prepared from C57BL/6, B6129SF2/J, Apaf1fog/J heterozygous, Apaf1fog/J homozygous, and Ppidtm1.1Mmos mice(Jackson Laboratories, Bar Harbor, ME) as described previously (23). All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee at Boston University. Bone marrow was flushed from the femurs of 4-8 week-old mice. Red blood cells were lysed by incubation in 0.17 M NH4Cl, 10 mM KHCO3, and 1 mM EDTA at 37°C for 5 min. The remaining cells were cultured for 5-7 days in primary B cell medium (RPMI containing 10% FBS, penicillin/streptomycin, L-glutamine, 2-mercaptoethanol, and 16 ng/ml murine rIL-7). This procedure results in a B cell culture in which at least 95% of the cells express CD43 and B220.
For experiments, pro/pre-B cells were cultured (0.5-1 × 106 cells/ml medium) overnight in RPMI with 5% FBS and treated with Vh (ethanol:DMSO, 50:50, 0.1%), MEHP (150 μM), or 15d-PGJ2 (10 μM) for 0.5 - 8 hrs. Cells were pre-treated with Vh (DMSO, 0.1%) or Z-VAD-FMK (30 μM) for 30 min. Primary pro-B cells were cultured overnight (4 × 105 cells/ml medium) in primary B cell medium with 7.5% FBS and treated with Vh (ethanol:DMSO, 50:50, 0.1%), MEHP (150-200 μM) or 15d-PGJ2 (2-10 μM) for 8-32 hrs.
Analysis of Apoptosis
B cells were harvested into cold PBS containing 5% FBS and 10 μM azide. Cells were resuspended in 0.25 ml hypotonic buffer containing 50 μg/ml propidium iodide (PI), 1% sodium citrate and 0.1% Triton X-100 and analyzed with FL-2 in the log mode on a Becton Dickinson FACScan flow cytometer. The percentage of cells undergoing apoptosis was determined to be those having a weaker PI fluorescence than cells in the G0/G1 phase of the cell cycle (15, 22, 23).
Analysis of Mitochondrial Membrane Potential
Thirty min prior to harvest, JC-1 (1.4 μM, Molecular Probes, Eugene, OR) was added to each well. BU-11 cells were transferred to FACS tubes without washing and analyzed immediately by flow cytometry. Only cells in the live gate were analyzed. The percentage of cells with low mitochondrial membrane potential (ΔΨmlow) was determined to be those having an increased green fluorescence with or without a loss of red fluorescence (24).
Immunoblotting
B cells were harvested and washed once in cold PBS. For analysis of cleavage of caspases or their substrates, cytoplasmic extracts were prepared as described previously (22). For analysis of cytochrome c release, BU-11 cells were resuspended immediately in permeabilization buffer (10 mM Hepes, pH 7.4, 210 mM mannitol, 70 mM sucrose, 5 mM succinate, 0.2 mM EGTA) containing 1.4 μl/ml of a 10% digitonin solution in DMSO. Following a 5 min incubation on ice, the same volume of permeabilization buffer without digitonin was added. The mixture was vortexed briefly and then centrifuged at 14,000 rpm for 30 min. The supernatant was used to determine cytochrome c release. For analysis of Bax translocation, mitochondrial fractions were prepared as described previously (22). Protein concentrations were determined by the Bradford method.
Proteins (5-60 μg) were resolved on 6% (α-fodrin), 12% (caspases-2, -8, and -9) or 15% (Bax, Bid, caspase-3, cytochrome c, and lamin) gels, transferred to a 0.2 μm nitrocellulose membrane, and incubated with primary antibody. Primary antibodies included monoclonal mouse anti-α-fodrin (MAB1622), polyclonal rabbit anti-Bax (SC-493), polyclonal rat anti-Bid (MAB860), monoclonal rat anti-caspase-2 (MAB3501), polyclonal rabbit anti-cleaved caspase-3 (9661), polyclonal rat anti-caspase-8 (ALX-804-447), polyclonal rabbit anti-caspase-9 (9504), and polyclonal rabbit anti-cytochrome c antibody (S2050). Immunoreactive bands were detected using HRP-conjugated secondary antibodies (Biorad, Hercules, CA) followed by ECL. To control for equal protein loading, blots were re-probed with a β-actin-specific antibody (A5441), α-tubulin-specific antibody (CP06), or HSP60-specific antibody (sc-1722) and analyzed as above. To quantify changes in protein expression, band densities were determined using the UVP Bioimaging System and the Labworks 4 program (UVP, Inc., Upland, CA). The band density of the protein of interest was divided by the band density of the control protein. To normalize for differences between experiments, the band density ratio for experimental samples then was divided by the ratio in the naïve sample from that experiment.
Statistics
Statistical analyses were performed with Statview (SAS Institute, Cary, NC). Data are presented as means ± or + standard errors (SE). At least three experiments were performed in each BU-11 protocol. Experiments with primary pro-B cells were performed with a minimum of three pools of bone marrow cells, and each pool of bone marrow cells was maintained separately. One-factor ANOVAs were used to analyze the data, with the Dunnett’s or Tukey-Kramer multiple comparisons test to determine significant differences.
Results
Rapid induction of apoptosis in cultured pro/pre-B cells by MEHP and enhancement by an endogenous prostaglandin
We have shown that bone marrow B cells are susceptible to apoptosis induced by structurally diverse PPARγ agonists, including ciglitazone, GW7845, 15d-PGJ2 and MEHP (15, 22). While some studies support the hypothesis that PPARγ agonists induce apoptosis through their receptor, growing evidence suggests that many PPARγ agonists induce apoptosis via an extra-receptor mechanism and that the mechanism induced can be specific to the agonist (19, 25-27). Indeed, 15d-PGJ2 has been suggested to induce apoptosis via multiple mechanisms including inhibition of NF-κB or stimulation of reactive oxygen production (19, 28). Two PPARγ antagonists (GW9662 and T007097) had little effect on the anti-proliferative effect of MEHP or 15d-PGJ2 in the developing B cell model used here (data not shown), a result consistent with, but not proving a PPARγ-independent signaling pathway.
Interestingly, we have shown that exposure to a combination of MEHP and 15d-PGJ2 results in enhanced apoptosis in a pro/pre-B cell model (15). A higher percentage of primary pro-B cells (>95% B220+/CD43+ following culture of bone marrow cells in rIL-7 for >5 days) undergo apoptosis when co-treated with MEHP and 15d-PGJ2 than when treated with either chemical alone (Fig 1A). This enhancement could be due to convergence of independent apoptosis signaling pathways or enhanced stimulation of a common pathway. Assessing the nature of the MEHP- and PGJ2-induced apoptosis pathway(s) is particularly important since basal PGJ2 levels in the bone marrow microenvironment (21) may lower the threshold for the environmental chemical-induced apoptosis, effectively increasing the risk associated with environmental chemical exposure.
Figure 1.

MEHP and 15d-PGJ2 cooperatively induce apoptosis in cultured pro/pre-B cells and primary pro-B cells. (A) Suspension cultures of primary pro-B cells prepared from wildtype C57BL/6 mice were treated with Vh, MEHP (150 μM) and/or 15d-PGJ2 (2 μM) for 32 hr. (B) Suspension cultures of BU-11 cells were treated with ethanol:DMSO (50:50, Vh, 0.1%), MEHP (150 μM) or 15d-PGJ2 (10 μM) for the times indicated. (C-D) Suspension cultures of BU-11 cells pre-treated for 30 min with DMSO (Vh, 0.1%) or Z-VAD-FMK (30 μM) and then treated with ethanol:DMSO (50:50, Vh, 0.1%), MEHP (150 μM) or 15d-PGJ2 (10 μM) for 16 or 6 hr, respectively. Cell death was analyzed by hypotonic PI staining followed by flow cytometry. Data are presented as means ± SE from at least 3 independent experiments or 4 independently prepared and maintained pools of primary pro-B cells. *Statistically greater than Vh-treated (p<0.05, ANOVA, Dunnett’s). **Statistically different from all other treatment groups (p<0.05, ANOVA, Tukey-Kramer).
To this end we examined the kinetics with which MEHP and 15d-PGJ2 induce death in cultured pro/pre-B cells. BU-11 cells were treated with Vh (ethanol:DMSO (50:50), 0.1% final volume), MEHP (150 μM) or 15d-PGJ2 (10 μM) for up to 16 hr and analyzed for cell death by hypotonic PI staining and flow cytometry. The onset of apoptosis, as measured by the percentage of cells containing a sub-G0/G1 DNA content, was similar for MEHP and 15d-PGJ2, beginning within the first 4 hrs of treatment (Fig. 1B). However, once induced, the execution of apoptosis was significantly more rapid in cells treated with 15d-PGJ2 than MEHP. Approximately 50% of the cells had a sub-G0/G1 DNA content within 7 and 16 hr of treatment with 15d-PGJ2 and MEHP, respectively. To demonstrate that the formation of the sub-G0/G1 population was a result of apoptosis, we examined the effect of treatment with the pan-caspase inhibitor Z-VAD-FMK on MEHP- and 15d-PGJ2-induced death. BU-11 cells were pretreated for 30 min with Vh or Z-VAD-FMK (30 μM) prior to treatment with MEHP or 15d-PGJ2 for 16 or 6 hrs, respectively. Cells were analyzed for cell death by hypotonic PI staining and flow cytometry. MEHP- and 15d-PGJ2-induced formation of the sub-G0/G1 population was completely abrogated by pre-treatment with Z-VAD-FMK (Fig. 1C-D), a result consistent with the hypothesis that these chemicals induce B cell death via a protease-dependent apoptotic process.
Caspase-3 is considered to be the primary apoptosis executioner with the broadest substrate repertoire of the effector caspases and is responsible for cleaving and activating those proteins responsible for the classic nuclear features associated with apoptosis, including nuclear condensation, chromatin margination and DNA fragmentation (29). To confirm activation of caspase-3 following exposure to MEHP and 15d-PGJ2, BU-11 cells were treated as above and analyzed for caspase-3 activation by immunoblotting for cleaved, active caspase-3 and for endogenous cleaved α-fodrin, a specific caspase-3 substrate (29). MEHP and 15d-PGJ2 induced the formation of the 17 kDa active caspase-3 fragment within 2-4 hrs, and this was accompanied by cleavage of α-fodrin (Fig. 2), thereby confirming that caspase-3 was catalytically active. Thus, treatment of bone marrow B cells with either MEHP or 15d-PGJ2 results in induction of the classic, terminal features of apoptosis. Although the onset of apoptosis appears to be similar for each chemical with regard to initiating caspase-3 activation, the significantly more rapid execution of apoptosis following treatment with 15d-PGJ2 suggests different apoptotic mechanisms may be at play.
Figure 2.

MEHP (A) and 15d-PGJ2 (B) treatment activate caspase-3 in cultured pro/pre-B cells. Suspension cultures of BU-11 cells were treated with ethanol:DMSO (50:50, Vh, 0.1%), MEHP (150 μM) or 15d-PGJ2 (10 μM) for the times indicated. Cytoplasmic extracts were prepared and analyzed for formation of caspase-3 fragments (17 kDa) and cleaved α-fodrin (120, 150 kDa) by immunoblotting. Representative data from at least 3 independent experiments are presented.
Activation of caspases belonging to both the intrinsic and extrinsic apoptotic pathways by MEHP and 15d-PGJ2
Historically, caspase cascades have been assigned to one of two non-mutually exclusive pathways based on participation of a particular initiator caspase in the apoptotic process. It is increasingly being recognized that significant crossover between the intrinsic and extrinsic pathways can occur (30, 31). The activation of a full complement of caspases in the extrinsic and intrinsic pathways may enhance a weak apoptotic signal or accelerate the apoptotic process. Activation of a positive-feedback loop would be consistent with the high degree of bone marrow B cell sensitivity to these agents. Indeed, in pro/pre-B cells, the aryl hydrocarbon receptor agonist 7,12-dimethylbenz[a]anthracene and the PPARγ agonist GW7845 were found to activate initiator caspases-2 and -8 in a executioner caspase-3-dependent manner (22, 23). Therefore, studies were designed to investigate the full complement of intrinsic and extrinsic apoptosis pathway caspases activated by MEHP and 15d-PGJ2.
The likely initiator caspase in apoptosis induced by chemicals such as chemotherapeutics is caspase-9, an initiator caspase in the intrinsic pathway that is activated following permeabilization of the mitochondria (32). To examine the activation of caspase-9 in MEHP- and 15d-PGJ2-induced pro/pre-B cell apoptosis, BU-11 cells were treated with Vh (ethanol:DMSO (50:50), 0.1% final volume), MEHP (150 μM) or 15d-PGJ2 (10 μM) for up to 8 hrs and analyzed by immunoblotting for full-length and cleaved caspase-9. A loss of full-length caspase-9 and the appearance of cleaved, active caspase-9 (37, 39 kDa) were evident following treatment with either MEHP or 15d-PGJ2 (Fig. 3A-B).
Figure 3.

MEHP and 15d-PGJ2 treatment activates caspases in the intrinsic and extrinsic pathways. Suspension cultures of BU-11 cells were treated with ethanol:DMSO (50:50, Vh, 0.1%), MEHP (150 μM) or 15d-PGJ2 (10 μM) for the times indicated. Cytoplasmic extracts were prepared and analyzed for caspase-9 (A-B; 37, 39 kDa), caspase-2 (C-D; 32, 33 kDa), and caspase-8 (E-F; 43/41, 18 kDa) fragments by immunoblotting. Representative data from at least 3 independent experiments are presented.
Caspase-2 also may act as the apical caspase in the intrinsic pathway, being activated upstream of the mitochondria and leading to the release of cytochrome c, or alternatively it may be activated downstream of caspase-9 (33). In BU-11 cells treated as described above, a loss of cytoplasmic full-length caspase-2 and the appearance of cleaved caspase-2 fragments (32, 33 kDa) occurred along with the activation of caspase-9 (Fig. 3C-D).
Finally, the initiator caspase in the extrinsic pathway is caspase-8, and its activation usually occurs following ligation of a death receptor and formation of the death inducing signaling complex (34). Alternatively, active caspase-3 can cleave and activate caspase-6, which in turn may activate caspase-8 in a positive feedback loop (30, 31). BU-11 cells were treated as described above and analyzed for formation of caspase-8 fragments (43/41, 18 kDa) by immunoblotting. Caspase-8 was cleaved soon after treatment (2-3 hr) with MEHP (Fig. 3E) or 15d-PGJ2 (Fig. 3F). The lack of change in mRNA expression of several TNF receptor family ligands (TNFα, Fas ligand, and TRAIL) in BU-11 cells treated with MEHP or 15d-PGJ2 supports the hypothesis that caspase-8 activation is a downstream event in these apoptotic pathways and does not initiate an extrinsic apoptosis signaling pathway (data not shown). The concurrent activation of caspases in both the intrinsic and extrinsic apoptotic pathways suggests that a positive feedback loop stimulates apoptosis induced by these chemicals; however, the activation of a similar complement of caspases by both chemicals does not explain the difference in the rate of execution of apoptosis.
Requirement for and mechanism of cytochrome c release in MEHP- and 15d-PGJ2-induced apoptosis
The hallmark of activation of the intrinsic apoptosis pathway is the release of cytochrome c into the cytoplasm (reviewed in (34, 35)). Therefore, we examined pro/pre-B cells for alterations in the mitochondria following treatment with MEHP and 15d-PGJ2. BU-11 cells were treated with Vh (ethanol:DMSO (50:50), 0.1% final volume), MEHP (150 μM) or 15d-PGJ2 (10 μM) for up to 8 hrs and analyzed for cytochrome c release by immunoblotting cytoplasmic extracts of digitonin-permeabilized cells. As expected, cytochrome c concentrations in the cytoplasm increased following treatment with either MEHP or 15d-PGJ2 (Fig. 4A-B) and prior to (i.e. within 4 hr of treatment) formation of an apoptotic cell population, as measured by PI (see Fig 1). Further, to test the hypothesis that cytochrome c release was responsible for initiating the apoptotic cascade, the incidence of MEHP- and 15d-PGJ2-induced apoptosis was examined in primary pro-B cells that are defective in the APAF1 protein and thereby are unable to form the apoptosome that is necessary for caspase-9 activation (32). Primary pro-B cells were prepared from mice hetero- or homozygous for the APAF1fog mutation, treated with Vh, MEHP (200 μM) or 15d-PGJ2 (10 μM) for 32 or 8 hr, respectively, and analyzed for apoptosis by hypotonic PI staining and flow cytometry. A significant increase in apoptosis was observed in primary pro-B cells from APAF1fog heterozygous mice following treatment with either MEHP or 15d-PGJ2, and this was suppressed significantly in primary pro-B cells from APAF1fog homozygous mice (Fig. 4C-D). The suppression of apoptosis most likely was incomplete because APAF1fog mice still express a low level of APAF1 that allows the mice to survive to adulthood; complete abrogation of APAF1 expression results in perinatal lethality (36). The results indicate that release of cytochrome c and formation of the apoptosome is required for optimal MEHP- and 15d-PGJ2-induced apoptosis.
Figure 4.

MEHP and 15d-PGJ2 induce cytochrome c release in cultured pro/pre-B cells, and death is attenuated in APAF1fog mutant primary pro-B cells. (A-B) Suspension cultures of BU-11 cells were treated with ethanol:DMSO (50:50, Vh, 0.1%), MEHP (A; 150 μM) or 15d-PGJ2 (B; 10 μM) for the times indicated. Cytochrome c release was analyzed by immunoblotting of cytoplasmic extracts from digitonin-permeablized cells. (C-D) Suspension cultures of primary pro-B cells isolated from mice hetero- or homozygous for the APAF1fog mutation were treated with ethanol:DMSO (50:50, Vh, 0.1%), MEHP (C; 200 μM, 16hr) or 15d-PGJ2 (D; 10 μM, 8hr). Cell death was analyzed by hypotonic PI staining followed by flow cytometry. The percentage of apoptotic cells measured in a naïve population for each pool was subtracted prior to the data analysis. Data are presented as means ± SE from at least 3 independent experiments or 4 independently prepared and maintained pools of primary pro-B cells. *Statistically greater than Vh-treated (p<0.05, ANOVA, Tukey-Kramer). **Statistically less than wildtype, MEHP- or 15d-PGJ2-treated (p<0.05, ANOVA, Tukey-Kramer).
Multiple inducer-specific pathways for initiation of cytochrome c release from the mitochondrion have been hypothesized (35). One hypothesized mechanism for cytochrome c release is opening of the permeability transition pore, which leads to a loss of mitochondrial membrane potential. To begin to investigate the mechanism of cytochrome c release, BU-11 cells were treated as above for 1-16 hrs and analyzed for mitochondrial membrane potential loss by JC-1 staining and flow cytometry. Significant loss of mitochondrial membrane potential occurred within 2 and 6 hrs of treatment with MEHP and 15d-PGJ2, respectively (Fig. 5A and 5B). In order to examine the contribution of the permeability transition pore to the apoptotic process, the incidence of MEHP- and 15d-PGJ2-induced apoptosis was examined in primary pro-B cells defective in cyclophilin D, a protein component of the transition pore. Primary pro-B cells were prepared from wildtype B6129SF2/J and Ppidtm1.1Mmos mice, treated with Vh, MEHP (200 μM), or 15d-PGJ2 (10 μM) for 32 or 8 hrs, respectively, and analyzed for apoptosis by hypotonic PI staining and flow cytometry. A significant increase in apoptosis was observed in primary pro-B cells from wildtype mice following treatment with either MEHP or 15d-PGJ2 that was not significantly reduced in cyclophilin D null primary pro-B cells (Fig. 5B-C). Therefore, it is unlikely that the mitochondrial permeability transition pore plays an essential role in MEHP or 15d-PGJ2-induced cytochrome c release or apoptosis, a result consistent with those showing a role for the permeability transition pore in necrosis rather than apoptosis (37).
Figure 5.
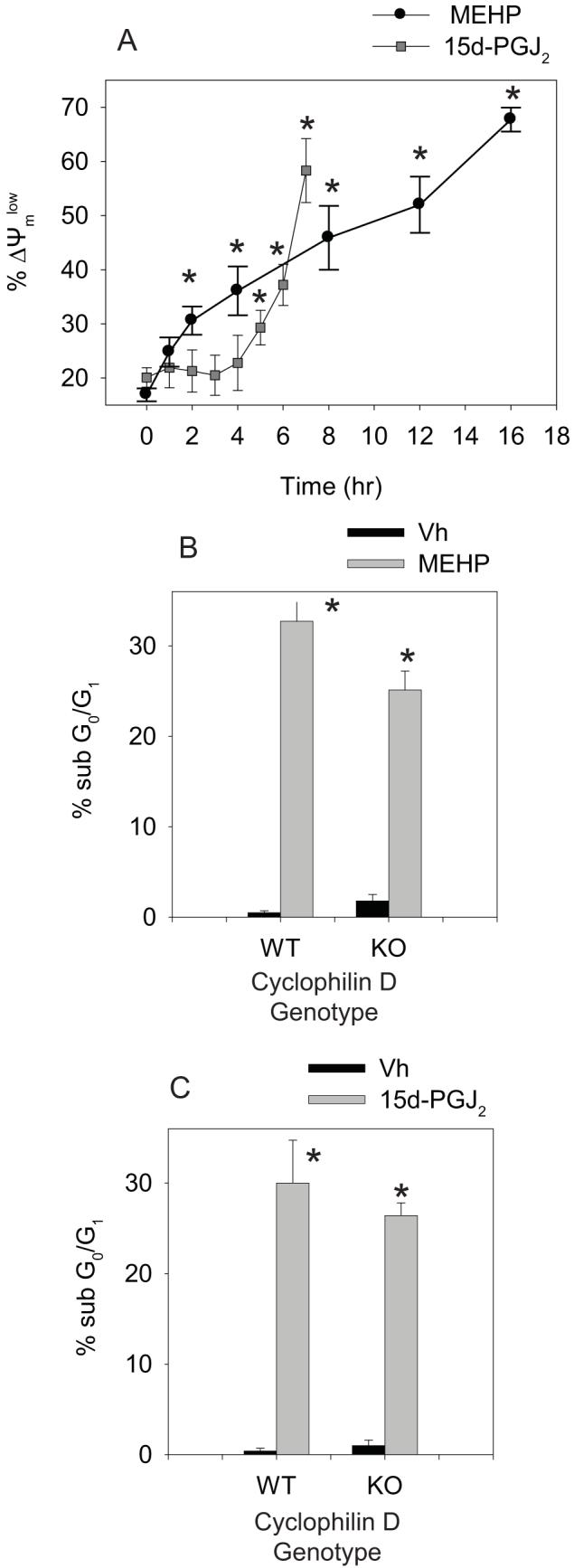
MEHP and 15d-PGJ2 induce loss of mitochondrial membrane potential, but lack of cyclophilin D does not protect against cell death. (A) Suspension cultures of BU-11 cells were treated with ethanol:DMSO (50:50, Vh, 0.1%), MEHP (150 μM) or 15d-PGJ2 (10 μM) for the times indicated. Mitochondrial membrane potential was analyzed by JC-1 staining followed by flow cytometry. (B-C) Suspension cultures of primary pro-B cells isolated from wildtype B6129SF2/J and Ppidtm1.1Mmos mice were treated with Vh, MEHP (200 μM, 16hr) or 15d-PGJ2 (10 μM, 8hr). Cell death was analyzed by hypotonic PI staining followed by flow cytometry. The percentage of apoptotic cells measured in a naïve population for each pool was subtracted prior to the data analysis. Data are presented as means ± SE from at least 3 independent experiments or 4 independently prepared and maintained pools of primary pro-B cells. *Statisically greater than Vh-treated (p<0.05, ANOVA, Dunnett).
A second hypothesized mechanism for cytochrome c release is the direct mobilization and activation of Bax, homodimerzation of which leads to pore formation in the outer mitochondrial membrane (38). To examine the distribution of Bax, BU-11 cells were treated as above for 1 - 8 hrs, and analyzed for Bax expression by immunoblotting of cytoplasmic and mitochondrial fractions prepared from digitonin-permeabilized cells. Expression of Bax in the mitochondria increased significantly following treatment with either MEHP or 15d-PGJ2, while it decreased in the cytoplasm (Fig. 6A-B), suggesting a plausible mechanism for cytochrome c release.
Figure 6.
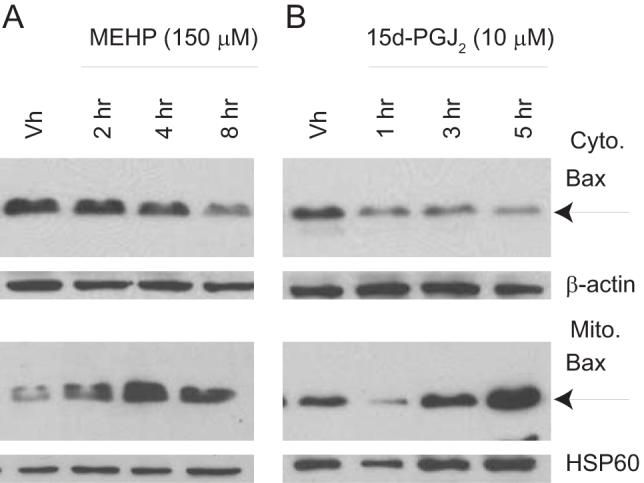
MEHP (A) and 15d-PGJ2 (B) induce translocation of Bax to the mitochondria. Suspension cultures of BU-11 cells were treated with ethanol:DMSO (50:50, Vh, 0.1%), MEHP (150 μM) or 15d-PGJ2 (10 μM) for the times indicated. Cytoplasmic and mitochondrial fractions were prepared from digitonin-permeabilized cells and analyzed for Bax expression by immunoblotting. Representative data from at least 3 independent experiments are presented.
A third hypothesized mechanism of cytochrome c release, that may occur apically or secondarily, is induction of pore formation by Bax/Bak in the outer mitochondrial membrane by Bid that has been cleaved by a protease (39). To test this possibility, BU-11 cells were treated with Vh (ethanol:DMSO (50:50), 0.1% final volume), MEHP (150 μM) or 15d-PGJ2 (10 μM) for up to 8 hrs and analyzed for full length and cleaved Bid (tBid, 15 kDa) by immunoblotting. Formation of tBid was evident following treatment with either MEHP (Fig. 7A) or 15d-PGJ2 (Fig. 7B). However, a greater proportion of the total Bid pool was cleaved following treatment with 15d-PGJ2 over time, as there was a ∼90% reduction in full length Bid following treatment with 15d-PGJ2 for 6 hrs but only a ∼40% decrease following treatment with MEHP for 8 hrs (Figs. 7A-B). These results suggest that the extent of Bid cleavage may represent a key difference in the apoptosis pathways activated by MEHP and 15d-PGJ2.
Figure 7.
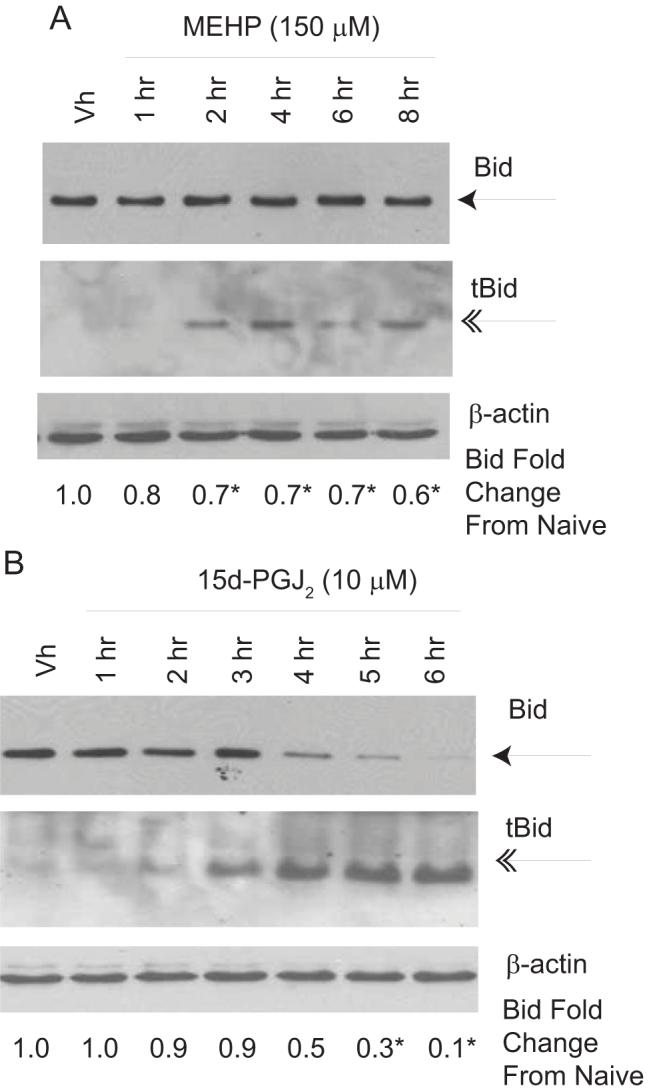
MEHP (A) and 15d-PGJ2 (B) induce Bid cleavage. Suspension cultures of BU-11 cells were treated with ethanol:DMSO (50:50, Vh, 0.1%), MEHP (150 μM) or 15d-PGJ2 (10 μM) for the times indicated. Cytoplasmic extracts were prepared and analyzed for full length (22 kDa) and cleaved Bid (15 kDa) by immunoblotting. Expression levels of full-length Bid were quantified as described in the Methods. Data are presented as means ± SE from at least 3 independent experiments. *Statistically less than Vh-treated (p<0.05, ANOVA, Dunnett’s).
Bid may be cleaved by a number of different proteases, including caspases-2, -3 and -8, calpains, cathepsins and granzyme B (39). If Bid cleavage contributes significantly to the 15d-PGJ2-induced apoptotic process, then treatment with the general caspase inhibitor, Z-VAD-FMK, should inhibit Bid cleavage and subsequent release of cytochrome c into the cytoplasm. BU-11 cells were pretreated for 30 min with Vh or Z-VAD-FMK (30 μM) prior to treatment with 15d-PGJ2 for 4-6 hrs. Cytoplasmic extracts from digitonin-permeabilized cells were analyzed for the presence of full-length Bid and mitochondrial release of cytochrome c. Z-VAD-FMK significantly suppressed both 15d-PGJ2-induced Bid cleavage and accumulation of cytoplasmic cytochrome c (Figs. 8A-B). For comparison, while Z-VAD-FMK reduced the 15d-PGJ2-induced release of cytochrome c by ∼90%, cytochrome c release induced by MEHP was reduced only ∼30% (Fig 8B). Thus, Bid cleavage and tBid-mediated release of cytochrome c most likely represent a mechanism that amplifies the apoptotic signal following treatment with 15d-PGJ2, but not MEHP.
Figure 8.
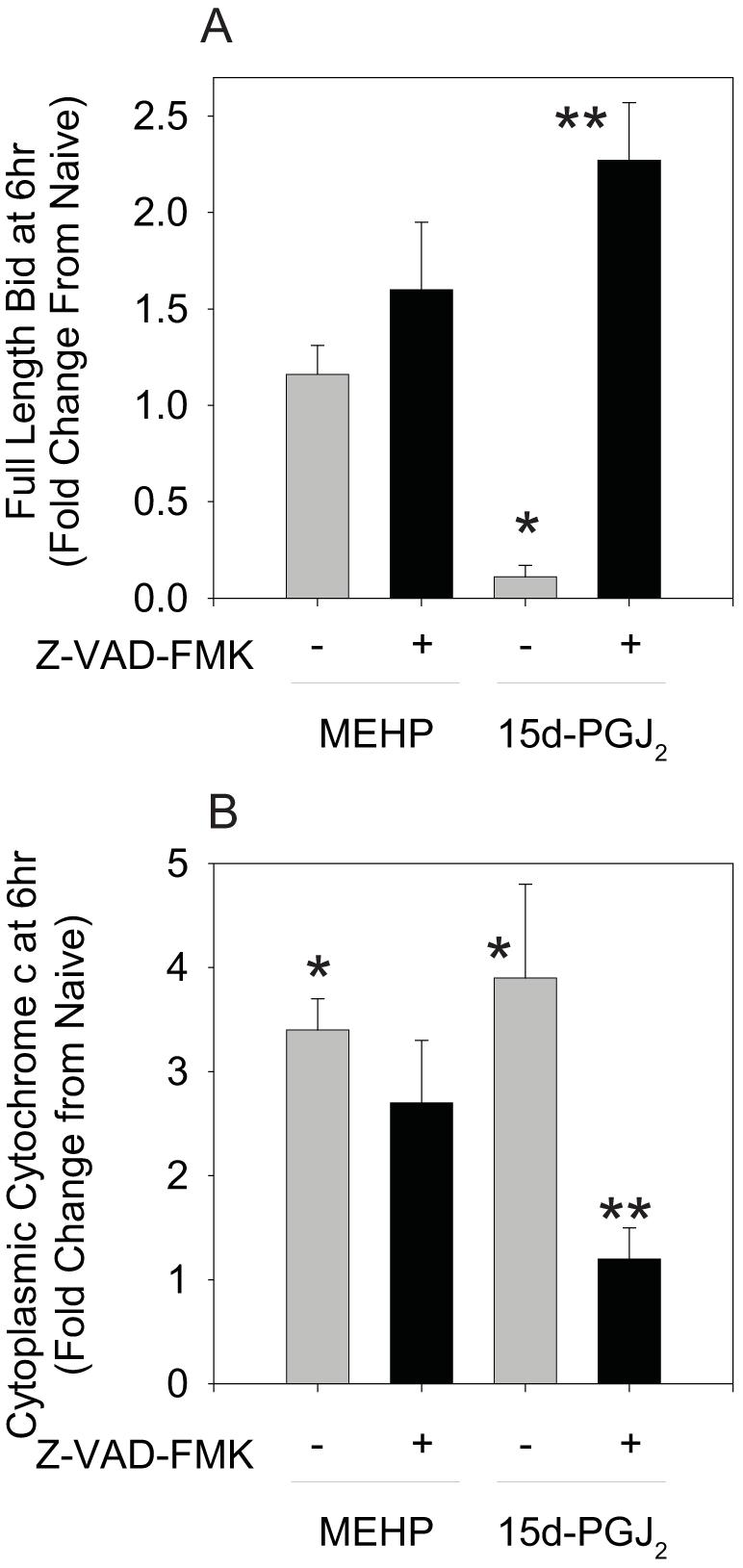
Z-VAD-FMK suppresses 15d-PGJ2-, but not MEHP-, induced Bid cleavage (A) and cytochrome c release (B). Following a 30 min pretreatment with Vh or Z-VAD-FMK (30 μM) and treatment with Vh, 15d-PGJ2 (10 μM), or MEHP for 6 hr, cytoplasmic extracts from digitonin-permeabilized cells were prepared and analyzed by immunoblotting for full-length Bid and cytochrome c. Protein expression levels were quantified as described in the Methods. Data are presented as means ± SE from at least 3 independent experiments. *Statistically different than Vh-treated (p<0.05, ANOVA, Tukey-Kramer). **Statistically different than 15d-PGJ2 alone (p<0.05, ANOVA, Tukey-Kramer).
Discussion
B cells in the developing immune system are sensitive to MEHP, a metabolite of an environmental phthalate ester. MEHP both suppresses proliferation and induces apoptosis of primary bone marrow B cells and cells of a non-transformed B cell line representing the pro/pre-B cell stage (15). We hypothesize that bone marrow B cells are particularly sensitive to apoptotic agents due to a relatively low threshold of apoptosis induction in early B cells which are deleted from the immune repertoire through apoptosis induction if their surface Ig receptors bind self antigens with high affinity (40). Indeed, we have shown that a robust and positively reinforced apoptotic pathway can be activated by a number of chemicals, as shown here for MEHP. Furthermore, the potential for chemical mixtures to amplify toxicity, as seen with the combination of MEHP and 9-cis-retinoic acid or 15d-PGJ2 (15), requires further investigation. Here we have begun to investigate the apoptotic pathways induced by both MEHP and 15d-PGJ2 in order to gain insight into the cooperative role each may play in the induction of apoptosis in the bone marrow.
As we have shown here and previously (15), both MEHP and 15d-PGJ2 induce classic terminal features of apoptosis, including caspase-3 cleavage and activity, as well as DNA fragmentation. The data are consistent with the induction of an intrinsic, mitochondria-driven apoptotic pathway by both MEHP and 15d-PGJ2 (Fig. 9, Pathway 1, solid arrows). Cytochrome c release was accompanied by caspase-9 activation following treatment with either chemical. Assembly of an apoptosome, a death complex composed of cytochrome c, APAF1, and caspase-9, was required to drive apoptosis (32), as evidenced by the significant reduction in apoptosis in primary pro-B cells from APAF1fog mutant mice. The likely mechanism of permeabilization of the mitochondrial membranes is translocation and pore formation by Bax (38, 41, 42), as both MEHP and 15d-PGJ2 induced the translocation of Bax to the mitochondria. These data support the hypothesis that stimulation of Bax-mediated cytochrome c release by both agents from the mitochondria and formation of the apoptosome results in the initial activation of caspase-9. Consequently, activation of a different complement of caspases likely does not explain the difference in the rate of execution of apoptosis seen with MEHP and 15d-PGJ2.
Figure 9.
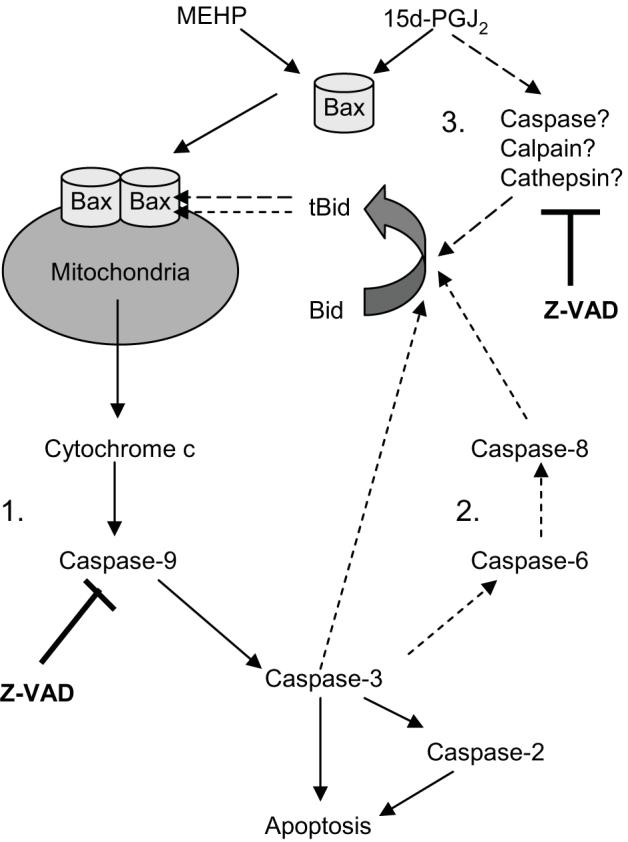
Hypothesized pathways of MEHP- and 15d-PGJ2-induced apoptosis.
Bax activation may occur through interactions with tBid, and formation of tBid was observed following treatment with MEHP or 15d-PGJ2. We hypothesize that Bid cleavage is a downstream rather than an initiating event in MEHP-induced apoptosis and may contribute to a caspase-dependent positive feedback loop (Fig. 9, pathway 2, short-dashed arrows). Studies in several systems suggest that the intrinsic mitochondrial pathway activates caspase-8 through caspases-3 and -6 (30, 31). Active caspase-8 then may cleave Bid, allowing it to translocate to the mitochondria (43). Caspase-2 can be cleaved directly by caspase-3 and may act as an amplifier by cleaving and activating Bid (33). Finally, caspase-3 itself may cleave and activate Bid (44). Activation of all of these caspases was evident following treatment with MEHP, and they likely contribute to the observed Bid cleavage.
While Bid cleavage occurred following treatment with either MEHP or 15d-PGJ2, a significantly greater portion of the Bid pool was cleaved following treatment with 15d-PGJ2. Results suggest that, while caspase-dependent cleavage of Bid, as part of a positive feedback loop, occurs with 15d-PGJ2 as with MEHP (Fig. 9, pathway 2, short-dashed arrows), another mechanism also is at work. Because the vast majority of the Bid pool was cleaved following treatment with 15d-PGJ2, we were able to experimentally intervene to investigate the role of tBid in this system. Z-VAD-FMK completely suppressed 15d-PGJ2-induced Bid cleavage and significantly, although incompletely, suppressed 15d-PGJ2-induced cytochrome c release. Two results argue against the conclusion that this cleavage of Bid results solely from activation of a caspases-9 -3, -2, -8-dependent positive feedback loop: 1) The same complement of caspases is activated following treatment with either 15d-PGJ2 or MEHP and yet Bid cleavage is significantly more substantial following treatment with 15d-PGJ2. 2) 15d-PGJ2-induced but not MEHP-induced cyctochrome c release is highly dependent upon Z-VAD-FMK-sensitive protease activation. Z-VAD-FMK commonly is used as a caspase-specific inhibitor, it also can inhibit papain-like proteases such as cathepsins, which can cleave Bid (39, 45, 46). Thus, we hypothesize that 15d-PGJ2 stimulates the cleavage of Bid via two pathways: 1) mitochondria-independent activation of a Bid-cleaving protease, potentially cathepsin (Fig. 9, pathway 3, long-dashed arrows), and 2) the subsequent activation of a positive feedback loop mediated by caspases-9, -3, -2 and -8 (Fig. 9, pathway 2, short-dashed arrows). The results with Bid found here support and extend those of Nencioni et al. (19), who found that 15d-PGJ2-induced cytochrome c release in Jurkat T cells is at least partially protease-dependent, but caspase-8-independent.
Can the apoptotic mechanisms determined here occur at concentrations of MEHP and 15d-PGJ2 that are relevant in vivo? While relatively high concentrations of a single phthalate, MEHP, were used in these in vitro studies, humans are exposed to multiple phthalates simultaneously (1). A cumulative toxic effect has been described for testis simultaneously exposed to multiple phthalates (47). Furthermore, a recent study has shown that culture conditions result in up to a 99% loss of the electrophilic 15d-PGJ2 such that μM concentrations of added 15d-PGJ2 result in only pM concentrations within the cell, concentrations in line with those shown to exhibit biological effects in vivo (48).
Co-exposure to multiple classes of toxicants within a mixture enhances toxicity. High synthesis rates of PGD2, the precursor of 15d-PGJ2, in the bone marrow may produce a significant concentration of 15d-PGJ2 in this tissue (21), providing a mechanism to enhance MEHP-induced apoptosis in the bone marrow. Further, studies have shown that co-exposure of bone marrow B cells to MEHP and 9-cis-retinoic acid, a vitamin A metabolite and RXRα ligand, results in synergistic effects on apoptosis (14). This synergy, compounded with the increases in apoptosis expected in the presence of another endogenous chemical, 15d-PGJ2, may result in significant levels of apoptosis induced by low environmental chemical doses. Beyond enhancement of MEHP toxicity by endogenous compounds, exposure to exogenous mixtures has significant potential to amplify toxicity. Exposure to DEHP/MEHP often occurs concomitant with exposure to tributyltin, a recently identified, high affinity ligand for retinoid X receptor (RXR) α and PPARγ (49). Initial studies have shown that early B cells are susceptible to tributyltin-induced apoptosis at nanomolar concentrations and that co-treatment at sub-apoptotic concentrations enhances MEHP-induced effects on proliferation4.
A source of prostaglandin in the bone marrow, the environmentally relevant multi-phthalate/PPARγ agonist/RXRα agonist mixture exposure scenario, and the potential sensitivity of bone marrow B cells to growth/death perturbing signals strongly suggest that these multiple chemical exposures have a significant effect on the development of these, if not other, bone marrow hematopoietic cells. Continued studies are required to determine if priming of the Bid pool for cleavage by exposure to 15d-PGJ2 is the mechanism by which this chemical may sensitize cells to MEHP-induced toxicity. Because evidence to date does not support a role for PPARγ in the anti-proliferative effects of its agonists, the surprising result that multiple combinations of PPARγ and RXRα agonists interact to enhance suppression of B cell proliferation requires further study.
Acknowledgements
Technical assistance was provided by the Boston University Medical Campus Flow Cytometry Facility.
Footnotes
Funding: National Institutes of Health (RO1-ES06086 to DS); National Institute of Environmental Health Sciences (P42ES007381 to JS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences or the National Institutes of Health.
- DEHP
- di-(2-ethylhexyl) phthalate
- MEHP
- mono-(2-ethylhexyl) phthalate
- PPAR
- peroxisome proliferator-activated receptor
- PI
- propidium iodide
- 15d-PGJ2
- 15-deoxy-Δ12,14-prostaglandin J2
- RXR
- retinoid × receptor
- tBid
- truncated Bid
Schlezinger and Sherr, unpublished results.
This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org
References
- 1.Blount BC, Silva MJ, Caudill SP, Needham LL, Pirkle JL, Sampson EJ, Lucier GW, Jackson RJ, Brock JW. Levels of seven urinary phthalate metabolites in a human reference population. Env. Health Persp. 2000;108:979. doi: 10.1289/ehp.00108979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koch HM, Drexler H, Angerer J. An estimation of the daily intake of di(2-ethylhexyl)phthalate (DEHP) and other phthalates in the general population. Int. J. Hyg. Environ. Health. 2003;206:77. doi: 10.1078/1438-4639-00205. [DOI] [PubMed] [Google Scholar]
- 3.Wittassek M, Heger W, Koch HM, Becker K, Angerer J, Kolossa-Gehring M. Daily intake of di(2-ethylhexyl)phthalate (DEHP) by German children -- A comparison of two estimation models based on urinary DEHP metabolite levels. Int J Hyg Environ Health. 2007;210:35. doi: 10.1016/j.ijheh.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Plonait SL, Nau H, Maier RF, Wittfoht W, Obladen M. Exposure of newborn infants to di-(2-ethylhexyl)-phthalate and 2-ethylhexanoic acid following exchange transfusion with polyvinylchloride catheters. Transfusion. 1993;33:598. doi: 10.1046/j.1537-2995.1993.33793325058.x. [DOI] [PubMed] [Google Scholar]
- 5.Tickner JA, Schettler T, Guidotti T, McCally M, Rossi M. Health risks posed by use of di-2-ethylhexyl phthalate (DEHP) in PVC medical devices: a critical review. Am. J. Ind. Med. 2001;39:100. doi: 10.1002/1097-0274(200101)39:1<100::aid-ajim10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 6.Heudorf U, Mersch-Sundermann V, Angerer J. Phthalates: toxicology and exposure. Int J Hyg Environ Health. 2007;210:623. doi: 10.1016/j.ijheh.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 7.Feige JN, Gelman L, Rossi D, Zoete V, Metivier R, Tudor C, Anghel SI, Grosdidier A, Lathion C, Engelborghs Y, Michielin O, Wahli W, Desvergne B. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis. J Biol Chem. 2007;282:19152. doi: 10.1074/jbc.M702724200. [DOI] [PubMed] [Google Scholar]
- 8.Lovekamp-Swan T, Jetten AM, Davis BJ. Dual activation of PPARα and PPARγ by mono-(2-ethylhexyl)phthalate in rat ovarian granulosa cells. Mol. Cell. Endocrinol. 2003;201:133. doi: 10.1016/s0303-7207(02)00423-9. [DOI] [PubMed] [Google Scholar]
- 9.Yao PL, Lin YC, Sawhney P, Richburg JH. Transcriptional regulation of FasL expression and participation of sTNF-alpha in response to sertoli cell injury. J Biol Chem. 2007;282:5420. doi: 10.1074/jbc.M609068200. [DOI] [PubMed] [Google Scholar]
- 10.Tomita I, Nakamura Y, Yagi Y, Tutikawa K. Fetotoxic effects of mono-2-ethyhexyl phthalate (MEHP) in mice. Env. Health Persp. 1986;65:249. doi: 10.1289/ehp.8665249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong JS, Gill SS. Gene expression changes induced in mouse liver by di(2-ethylhexyl)phthalate. Toxicol. Appl. Pharm. 2002;185:180. doi: 10.1006/taap.2002.9540. [DOI] [PubMed] [Google Scholar]
- 12.Larsen ST, Hansen JS, Thygesen P, Begtrup M, Poulsen OM, Nielsen GD. Adjuvant and immuno-suppressive effect of six monophthaltes in a subcutaneous injection model with BALB/c mice. Toxicology. 2001;169:37. doi: 10.1016/s0300-483x(01)00484-x. [DOI] [PubMed] [Google Scholar]
- 13.Yang Q, Xie Y, Depierre W. Effects of peroxisome proliferators on the thymus and spleen of mice. Clin. Exp. Immunol. 2001;122:219. doi: 10.1046/j.1365-2249.2000.01367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakazaki H, Ueno H, Nakamuro K. Estrogen receptor alpha in mouse splenic lymphocytes: possible involvement in immunity. Toxicol Lett. 2002;133:221. doi: 10.1016/s0378-4274(02)00203-5. [DOI] [PubMed] [Google Scholar]
- 15.Schlezinger JJ, Howard GJ, Hurst CH, Emberley JK, Waxman DJ, Webster T, Sherr DH. Environmental and endogenous peroxisome proliferator-activated receptor gamma agonists induce bone marrow B cell growth arrest and apoptosis: interactions between mono(2-ethylhexyl)phthalate, 9-cis-retinoic acid, and 15-deoxy-Delta12,14-prostaglandin J2. J Immunol. 2004;173:3165. doi: 10.4049/jimmunol.173.5.3165. [DOI] [PubMed] [Google Scholar]
- 16.Yokoyama Y, Okubo T, Kano I, Sato S, Kano K. Induction of apoptosis by mono(2-ethylhexyl)phthalate (MEHP) in U937 cells. Toxciol. Lett. 2003;144:371. doi: 10.1016/s0378-4274(03)00256-x. [DOI] [PubMed] [Google Scholar]
- 17.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-Δ12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR. Cell. 1995;83:803. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 18.Ray DM, Akbiyik F, Bernstein SH, Phipps RP. CD40 engagement prevents peroxisome proliferator-activated receptor gamma agonist-induced apoptosis of B lymphocytes and B lymphoma cells by an NF-kappaB-dependent mechanism. J Immunol. 2005;174:4060. doi: 10.4049/jimmunol.174.7.4060. [DOI] [PubMed] [Google Scholar]
- 19.Nencioni A, Lauber K, Grunebach F, Van Parijs L, Denzlinger C, Wesselborg S, Brossart P. Cyclopentenone prostaglandins induce lymphocyte apoptosis by activating the mitochondrial apoptosis pathway independent of external death receptor signaling. J Immunol. 2003;171:5148. doi: 10.4049/jimmunol.171.10.5148. [DOI] [PubMed] [Google Scholar]
- 20.Piva R, Gianferretti P, Ciucci A, Taulli R, Belardo G, Santoro MG. 15-Deoxy-delta 12,14-prostaglandin J2 induces apoptosis in human malignant B cells: an effect associated with inhibition of NF-kappa B activity and down-regulation of antiapoptotic proteins. Blood. 2005;105:1750. doi: 10.1182/blood-2004-04-1360. [DOI] [PubMed] [Google Scholar]
- 21.Ujihara M, Urade Y, Eguchi N, Hayashi H, Ikai K, Hayaishi O. Prostaglandin D2 formation and characterization of its synthetases in various tissues of adult rat. Arch. Biochem. Biophys. 1988;260:521. doi: 10.1016/0003-9861(88)90477-8. [DOI] [PubMed] [Google Scholar]
- 22.Schlezinger JJ, Emberley JK, Bissonnette SL, Sherr DH. An L-Tyrosine Derivative and PPARγ Agonist, GW7845, Activates a Multifaceted Caspase Cascade in Bone Marrow B Cells. Toxicol Sci. 2007;98:125. doi: 10.1093/toxsci/kfm071. [DOI] [PubMed] [Google Scholar]
- 23.Ryu HY, Emberley JK, Schlezinger JJ, Allan LL, Na S, Sherr DH. Environmental chemical-induced bone marrow B cell apoptosis: death receptor-independent activation of a caspase-3 to caspase-8 pathway. Mol Pharmacol. 2005;68:1087. doi: 10.1124/mol.105.014712. [DOI] [PubMed] [Google Scholar]
- 24.Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997;411:77. doi: 10.1016/s0014-5793(97)00669-8. [DOI] [PubMed] [Google Scholar]
- 25.Brookes PS, Morse K, Ray D, Tompkins A, Young SM, Hilchey S, Salim S, Konopleva M, Andreeff M, Phipps R, Bernstein SH. The triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid and its derivatives elicit human lymphoid cell apoptosis through a novel pathway involving the unregulated mitochondrial permeability transition pore. Cancer Res. 2007;67:1793. doi: 10.1158/0008-5472.CAN-06-2678. [DOI] [PubMed] [Google Scholar]
- 26.Schlezinger JJ, Emberley JK, Sherr DH. Activation of multiple mitogen-activated protein kinases in pro/pre-B cells by GW7845, a peroxisome proliferator-activated receptor gamma agonist, and their contribution to GW7845-induced apoptosis. Toxicol Sci. 2006;92:433. doi: 10.1093/toxsci/kfl003. [DOI] [PubMed] [Google Scholar]
- 27.Chintharlapalli S, Papineni S, Safe S. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes inhibit colon cancer cell and tumor growth through PPARgamma-dependent and PPARgamma-independent pathways. Mol Cancer Ther. 2006;5:1362. doi: 10.1158/1535-7163.MCT-06-0002. [DOI] [PubMed] [Google Scholar]
- 28.Ciucci A, Gianferretti P, Piva R, Guyot T, Snape TJ, Roberts SM, Santoro MG. Induction of apoptosis in estrogen receptor-negative breast cancer cells by natural and synthetic cyclopentenones: role of the IkappaB kinase/nuclear factor-kappaB pathway. Mol Pharmacol. 2006;70:1812. doi: 10.1124/mol.106.025759. [DOI] [PubMed] [Google Scholar]
- 29.Slee EA, Adrain C, Martin SJ. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276:7320. doi: 10.1074/jbc.M008363200. [DOI] [PubMed] [Google Scholar]
- 30.Cowling V, Downward J. Caspase-6 is the direct activator of caspase-8 in the cytochrome c-induced apoptosis pathway: absolute requirement for removal of caspase-6 prodomain. Cell Death Differ. 2002;9:1046. doi: 10.1038/sj.cdd.4401065. [DOI] [PubMed] [Google Scholar]
- 31.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 33.Zhivotovsky B, Orrenius S. Caspase-2 function in response to DNA damage. Biochem Biophys Res Commun. 2005;331:859. doi: 10.1016/j.bbrc.2005.03.191. [DOI] [PubMed] [Google Scholar]
- 34.Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4:139. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- 35.Gogvadze V, Orrenius S. Mitochondrial regulation of apoptotic cell death. Chem Biol Interact. 2006;163:4. doi: 10.1016/j.cbi.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 36.Honarpour N, Gilbert SL, Lahn BT, Wang X, Herz J. Apaf-1 deficiency and neural tube closure defects are found in fog mice. Proc Natl Acad Sci U S A. 2001;98:9683. doi: 10.1073/pnas.171283198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin XM. Bid, a BH3-only multi-functional molecule, is at the cross road of life and death. Gene. 2006;369:7. doi: 10.1016/j.gene.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 40.Lu L, Osmond DG. Apoptosis and its modulation during B lymphopoiesis in mouse bone marrow. Immunol Rev. 2000;175:158. [PubMed] [Google Scholar]
- 41.Er E, Oliver L, Cartron PF, Juin P, Manon S, Vallette FM. Mitochondria as the target of the pro-apoptotic protein Bax. Biochim Biophys Acta. 2006;1757:1301. doi: 10.1016/j.bbabio.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 42.Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006;281:21256. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 44.Slee EA, Keogh SA, Martin SJ. Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: a potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release. Cell Death Differ. 2000;7:556. doi: 10.1038/sj.cdd.4400689. [DOI] [PubMed] [Google Scholar]
- 45.Rozman-Pungercar J, Kopitar-Jerala N, Bogyo M, Turk D, Vasiljeva O, Stefe I, Vandenabeele P, Bromme D, Puizdar V, Fonovic M, Trstenjak-Prebanda M, Dolenc I, Turk V, Turk B. Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: when reaction mechanism is more important than specificity. Cell Death Differ. 2003;10:881. doi: 10.1038/sj.cdd.4401247. [DOI] [PubMed] [Google Scholar]
- 46.Jaattela M, Tschopp J. Caspase-independent cell death in T lymphocytes. Nat Immunol. 2003;4:416. doi: 10.1038/ni0503-416. [DOI] [PubMed] [Google Scholar]
- 47.Howdeshell KL, Furr J, Lambright CR, Rider CV, Wilson VS, Gray LE., Jr. Cumulative effects of dibutyl phthalate and diethylhexyl phthalate on male rat reproductive tract development: altered fetal steroid hormones and genes. Toxicol Sci. 2007;99:190. doi: 10.1093/toxsci/kfm069. [DOI] [PubMed] [Google Scholar]
- 48.Oh JY, Giles N, Landar A, Darley-Usmar V. Accumulation of 15-deoxy-Delta(12,14)-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochem J. 2008;411:297. doi: 10.1042/bj20071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, Gardiner DM, Kanno J, Iguchi T, Blumberg B. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20:2141. doi: 10.1210/me.2005-0367. [DOI] [PubMed] [Google Scholar]