

Abstract
In many studies, particularly in the field of systems biology, it is essential that identical protein sets are precisely quantified in multiple samples such as those representing differentially perturbed cell states. The high degree of reproducibility required for such experiments has not been achieved by classical mass spectrometry-based proteomics methods. In this study we describe the implementation of a targeted quantitative approach by which predetermined protein sets are first identified and subsequently quantified at high sensitivity reliably in multiple samples. This approach consists of three steps. First, the proteome is extensively mapped out by multidimensional fractionation and tandem mass spectrometry, and the data generated are assembled in the PeptideAtlas database. Second, based on this proteome map, peptides uniquely identifying the proteins of interest, proteotypic peptides, are selected, and multiple reaction monitoring (MRM) transitions are established and validated by MS2 spectrum acquisition. This process of peptide selection, transition selection, and validation is supported by a suite of software tools, TIQAM (Targeted Identification for Quantitative Analysis by MRM), described in this study. Third, the selected target protein set is quantified in multiple samples by MRM. Applying this approach we were able to reliably quantify low abundance virulence factors from cultures of the human pathogen Streptococcus pyogenes exposed to increasing amounts of plasma. The resulting quantitative protein patterns enabled us to clearly define the subset of virulence proteins that is regulated upon plasma exposure.
A key element of the experimental framework for systems biology is the comprehensive, quantitative measurement of whole biological systems in differentially perturbed states (1). Among the different types of measurements possible, protein quantification is particularly informative because proteins catalyze or control the majority of cellular functions. Currently the most widely applied quantitative proteome analysis technologies consist of the labeling of the samples by stable isotopes, the reproducible separation of complex peptide mixtures, usually by capillary LC, and the identification and quantification of selected peptides by tandem mass spectrometry and sequence database searching (2, 3). Relative quantitative values are generated by these methods if two or more samples are being compared, and absolute quantification can be achieved if suitable, calibrated reference samples are available (4). Using such shotgun methods, in each measurement only a fraction of the analytes present in a complex sample is identified and quantified. Peptide ions are selected by the mass spectrometer automatically based on precursor ion signal intensities. Due to a multitude of factors, including interference between analytes and variations in precursor ion spectra, the selection of peptides is not reproducible in consecutive runs in particular for peptides of lower signal intensities. As a critical consequence of this undersampling effect comprehensive analyses of whole systems are not supported by these technologies rendering them poorly suitable for systems biology and other experiments that depend on the comparison of complete or at least reproducible data sets.
To overcome these fundamental technical limitations confronting proteomics we have suggested in the past a substantially different approach that emulates successful genomics strategies (5–7). It depends on the generation of deep, ideally complete proteome maps followed by the targeted analysis of peptides that collectively represent the proteins that constitute the system under investigation. We have termed peptides that are typically observed in a mass spectrometer and that uniquely identify a particular protein “proteotypic peptides” (PTPs)1 (8). Significant progress has been achieved with several aspects of this approach. First, we have initiated the PeptideAtlas project (9), a central repository of high quality MS/MS-based peptide identifications, and associated software tools supporting diverse types of queries including the identification of PTPs. At present extensive LC-MS data sets for the yeast Saccharomyces cerevisiae (10), Drosophila melanogaster (11), human (11), and human plasma (12) have been integrated in the PeptideAtlas. Second, multiple reaction monitoring (MRM) has emerged as the method of choice for the targeted detection and quantification of peptides and phosphopeptides in complex mixtures (13–16). Exquisite sensitivity and a large dynamic range have been achieved using triple quadrupole mass spectrometers operated in MRM mode. The two-stage filtering of MRM, first at the level of the precursor ions and second at the level of the fragment ions, significantly reduces the noise level, thus increasing the signal-to-noise ratio. Recently the number of MRM transitions and thus the number of peptides analyzed in a single LC-MS/MS run has been significantly extended by the introduction of time constraints for the transitions (scheduled MRM) (15). Collectively these characteristics make MRM ideally suited for the multiplexed quantification of low abundance proteins in complex samples.
Despite these advances several technical limitations at present preclude the routine application of the targeted proteomics technology described above. These are mainly centered around the issue of target selection and validation. For the success of the measurement it is critical that for each protein in the targeted protein set the optimal peptides are chosen and that for these peptides the optimal MRM transitions are identified and validated. To support this process, we developed a suite of software tools termed TIQAM (Targeted Identification for Quantitative Analysis by MRM). Starting with a protein set in question the system uses the PeptideAtlas and other sources of information to select the PTPs and generate a list of transitions. These are used for MRM experiments triggering MS2 spectra acquisition. TIQAM integrates the acquired data to support the selection of the best performing transitions whose specificity has been confirmed by matching MS2 spectra. In this study we introduce TIQAM and describe, for the first time, the implementation of the full targeted proteomics strategy in a single experiment. First, the proteome in question was mapped using peptide IEF and LC-MS/MS, and a PeptideAtlas instance with extensive proteome coverage was generated. Second, based on this proteome map, PTPs were selected, and specific transitions were validated using TIQAM. Third, these optimized transitions were used to quantify the protein set in several biological samples.
We applied the targeted proteomics strategy to study the dynamic behavior of virulence factors of the Gram-positive bacterium Streptococcus pyogenes. S. pyogenes is a common colonizer of skin and the upper respiratory tract where it causes relatively mild clinical conditions such as impetigo or pharyngitis, respectively (17). However, invasive strains can penetrate into deeper tissues and cause severe and potentially life-threatening conditions such as necrotizing fasciitis and sepsis. A recent study funded by the World Health Organization reported that S. pyogenes causes an annual prevalence of over 600 million cases of pharyngitis and 111 million cases of streptococcal skin infections worldwide (18). In addition, at least 517,000 deaths occur each year due to severe S. pyogenes infections, thus emphasizing that the pathogen is an important cause of morbidity and mortality. Although the pathogenesis of S. pyogenes has been extensively studied and several virulence factors that facilitate bacterial colonization, immune system evasion, and spread have been identified, a comprehensive understanding of the mechanisms active during early infection is lacking (19). Upon colonization of throat or skin, the human host leaks plasma into the sites of infection to antagonize the bacteria. However, S. pyogenes has developed the ability to resist antibodies and other antimicrobial components present in plasma. To uncover the underlying mechanisms recent studies have analyzed the transcriptional and proteomic changes upon plasma exposure of S. pyogenes (20). However, so far only a few of the virulence factors could be detected and quantified on the proteome level. Therefore, we applied the targeted proteomics technology described above to investigate how S. pyogenes regulates the expression of virulence factor proteins when exposed to increasing concentrations of human plasma. We identified a subset of virulence factors that is clearly induced upon contact with plasma and presumably is of particular importance during the early infection stages of S. pyogenes.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Growth Conditions, and Sample Preparation—
The SF370 strain used in this study is the ATCC 700294 strain. 500-μl overnight cultures of SF370 were grown in 50 ml of Todd-Hewitt broth (Difco) in parallel with Todd-Hewitt broth supplemented with 1, 5, 10, or 20% human plasma at 37 °C in a 5% CO2 atmosphere. The optical density at 620 nm was measured at given intervals of 30 min until the bacterial cultures reached midexponential growth phase (A620 = 0.5). The cultures were harvested, washed three times in ice-cold PBS, and diluted in sterile water. Following lysis of the bacteria with Gentra Systems cell lysis protocol for Gram-positive bacteria in a FastPREP machine (Savant Machines Inc.), samples were briefly incubated with DNase and RNase and resuspended in urea buffer (6 m urea, 200 mm Tris, pH 8.3, 5 mm EDTA, 0.2% Triton).
Isoelectric Focusing—
The bacterial proteins were precipitated with methanol/chloroform (21). The protein solution was diluted with 4 volumes of methanol, 1 volume of chloroform, and 3 volumes of water; mixed; and centrifuged. The upper organic phase was discarded without disturbing the interphase containing the proteins. 3 volumes of methanol were added, and the sample was pelleted by another spin. The samples were resolubilized in 6 m urea, 100 mm HEPES at pH 8.5. The proteins were reduced with 5 mm DTT for 45 min at 37 °C and alkylated with 25 mm iodoacetamide for 45 min in the dark before diluting the sample with 100 mm HEPES at pH 8.5 to a final urea concentration below 1.5 m. Proteins were digested by incubation with trypsin (1:100, w/w) for at least 6 h at 37 °C. The peptides were cleaned up by C18 reversed-phase spin columns according to the manufacture's instructions (Harvard Apparatus). Half of the dried down peptides were resolubilized to a final concentration of 1 mg/ml in free flow electrophoresis separation buffer containing 8 m urea, 250 mm mannitol, 23% Prolyte mixture pH 4–7 (FFE Weber Inc., now BD Diagnostics). The separation in the pH range of 4–7 was carried out as described previously (22) at a sample loading rate of 1 mg/h. The other half of the peptides was resolubilized in off-gel electrophoresis buffer containing 6.25% glycerol and 1.25% IPG buffer (GE Healthcare). The peptides were separated on pH 3–10 IPG strips (GE Healthcare) with a 3100 OFFGEL fractionator (Agilent) as described previously (23) using a protocol of 1 h of rehydration at a maximum of 500 V, 50 μA, and 200 milliwatts followed by the separation at a maximum of 8000 V, 100 μA, and 300 milliwatts until 50 kV-h were reached. After isoelectric focusing the fractions were concentrated and cleaned up by C18 reversed-phase spin columns according to the manufacture's instructions (Harvard Apparatus).
Reversed-phase LC-MS Analysis—
The setup of the microcapillary reversed-phase LC-MS system was as described previously (22, 24). ESI-based LC-MS/MS (LTQ, Thermo Finnigan) analyses were carried out using an Agilent 1100 series (Agilent Technologies) on a 75-μm × 10.5-cm fused silica microcapillary reversed-phase column. After sample loading, the samples were separated by a 65-min linear gradient of 5–35% acetonitrile in water containing 0.1% formic acid with a flow rate of 200 nl/min. The HPLC system was coupled to an LTQ linear ion trap mass spectrometer equipped with a nano-LC electrospray ionization source (both from Thermo Electron, San Jose, CA). The data acquisition mode was set to one full MS scan (from 400 to 1600 m/z range) followed by three MS/MS events using data-dependent acquisition where the three most intense ions from a given MS scan were subjected to CID if the intensity of the precursor ion peak exceeded 10,000 ion counts. The electrospray voltage was set to 2.0 kV, and the specific m/z value of the peptide fragmented by CID was excluded from reanalysis for 2 min with a repeat count of 2.
The hybrid LTQ-FT-ICR mass spectrometer (Thermo Finnigan) was interfaced to a nanoelectrospray ion source. Chromatographic separation of peptides was achieved on an Agilent Series 1100 LC system (Agilent Technologies) equipped with an 11-cm fused silica emitter, 100-μm inner diameter (BGB Analytik), packed in house with a Magic C18 AQ 5-μm resin (Michrom Bioresources). Peptides were separated by a 65-min linear gradient of 5–40% acetonitrile in water containing 0.1% formic acid with a flow rate of 0.95 μl/min. Three MS/MS spectra were acquired in the linear ion trap per each FT-MS scan that was acquired at 100,000 full-width half-maximum nominal resolution settings with an overall cycle time of ∼1 s. Charge state screening was used to select for ions with at least two charges and reject ions with undetermined charge state. Only signals exceeding 150 counts were chosen for MS2 analysis and then dynamically excluded from triggering MS2 scans for 15 s. The LTQ-FT mass spectrometer was set to accumulate 106 ions for MS1 scans over no more than 1 s and 104 ions over a maximum of 0.5 s for MS2 scans.
Data Processing and PeptideAtlas Build—
The raw files from the LTQ and LTQ-FT (Xcalibur versions 1.0 and 2.0, respectively) were converted to mzXML by ReAdW (version 1.0, default parameters). Peak extraction was performed by MzXML2Search (version November 9, 2006, default parameters).
MS/MS spectra were searched using SEQUEST (version 27, revision 0) (25) against the predicted proteome from S. pyogenes M1 GAS, complete genome National Center for Biotechnology Information (NCBI) genome number NC_002737, consisting of 1697 proteins as well as known contaminants such as porcine trypsin and human keratins (Non-Redundant Protein Database, National Cancer Institute Advanced Biomedical Computing Center, 2004). For the samples exposed to human plasma the S. pyogenes M1 GAS protein database was combined with the Human International Protein Index (IPI) 3.26 protein database. The search was performed with semitryptic cleavage specificity with unlimited missed cleavages, mass tolerance of 3 Da for the precursor ions and 0.5 Da for the fragment ions, methionine oxidation as variable modification, and cysteine carbamidomethylation as fixed modification. The database search results were further processed using the PeptideProphet program (26) modified to further discriminate correct from incorrect peptides using the difference between theoretical and observed peptide pI values (22). The protein redundancy in the data set was eliminated by the ProteinProphet program (27).
The S. pyogenes PeptideAtlas build was constructed using the sequence search results of 196,432 MS/MS spectra. Peptides were included in the atlas if their PeptideProphet probability was p ≥ 0.9. By selecting peptides in this way, the combined false discovery rate for spectra in the atlas is 0.9%, and the combined sensitivity is estimated at 90%. Of the total 50,000 spectra identified with false discovery rate of 0.9% PeptideProphet estimates 438 incorrect assignments. The identified spectra were coalesced into 7181 distinct peptide sequences of which 5360 were observed at least twice. The majority (>90%) of the 438 incorrect assignments will be among the peptides only observed once. The 7181 identified peptides map to a list of 964 non-redundant proteins of which 132 were only identified with one or more singly observed peptides (supplemental Data 1). The results of this analysis were loaded into the PeptideAtlas database as described previously (9) and are available for browsing and downloading. There all identified proteins and peptides can be viewed along with the corresponding MS/MS scans, protein coverage, precursor m/z, charge, and score.
Isotope-coded Protein Labeling (ICPL)—
The ICPL labeling was performed as described previously (28) with the following modifications: 200 μg of S. pyogenes protein lysate was methanol/chloroform-precipitated as described above (21) and resolubilized in 25 μl of 6 m urea, 100 mm HEPES, pH 8.5. The proteins were reduced and alkylated as described above with the exception that DTT was replaced with tris(2-carboxyethyl)phosphine hydrochloride. The urea concentration was reduced to below 1.5 m by the addition of 100 mm HEPES, pH 8.5, and the proteins were digested with trypsin (1:100, w/w) for at least 6 h at 37 °C. Every sample was labeled with ICPL light, and a pool of all samples was labeled with ICPL heavy. After labeling, the samples were combined at a 1:1 ratio and cleaned up by C18 reversed-phase spin columns according to the manufacture's instructions (Harvard Apparatus).
ICPL labeled samples were analyzed on the LTQ mass spectrometer as described above. MS/MS spectra were analyzed using the Comet (29) search engine with the following parameters: semitryptic cleavage specificity with maximally two missed cleavage sites, peptide mass tolerance of 3.0 Da, ICPL modification at the N terminus and at lysine as variable modification, and carbamidomethylation of cysteine as fixed modification. The search results were processed by PeptideProphet and ProteinProphet as described above.
TIQAM Software Suite—
The TIQAM software suite consists of three modules. TIQAM PeptideAtlasClient is a client for retrieving PTPs from the PeptideAtlas. TIQAM Digestor integrates PeptideAtlasClient output, pepXML files, and other data to generate transition lists according to user preferences. TIQAM Viewer provides a graphical environment to analyze MRM-triggered MS2 experiments and validate transitions. TIQAM was programmed in Java using the Eclipse software framework. It is available in versions for Windows, Linux, and Mac OS X. TIQAM Viewer requires an active MySQL installation for storing data. The TIQAM modules are available for download (Seattle Proteome Center).
Transition Validation—
Based on the PeptideAtlas data we generated MRM transitions specific for the proteotypic peptides using TIQAM. We restricted the peptides to those in the mass range of 800–2400 Da and not containing methionine. For each precursor we calculated the transitions with precursor charges 2+ and 3+ and the four smallest y-ions with m/z > precursor m/z + 30. Collision energies (CE) were calculated according to the following formulas: CE = 0.044 × m/z + 5.5 (2+) and CE = 0.051 × m/z + 0.5 (3+). Then MRM-triggered MS/MS experiments were performed with 100 transitions per run (dwell time: 20 ms/transition) on a pool of all 10 samples. Results were imported into TIQAM, and the three transitions with the best signal-to-noise ratio were selected for quantitative analysis if the corresponding MS/MS spectrum was in accordance with the targeted peptide. The validated transitions are listed in supplemental Data 3.
MRM-based Quantification—
For quantitative analysis we spiked an ICPL heavy labeled pool of all samples as internal standard to each sample in a 1:1 ratio. For each transition we calculated the corresponding transition of the heavy labeled peptide. This resulted in six transitions per peptide and in total 282 transitions for 48 peptides of nine virulence and 12 housekeeping proteins (for three peptides only four transitions each were selected). The housekeeping proteins were included in the analysis for normalization of sample loading amount. By restricting the acquisition of each transition to a 2–3-min window around its elution time, the time scheduling feature of the acquisition software enabled the analysis of all 282 transitions in a single run per sample without compromising on sensitivity.
LC-MS Setup for MRM Measurements—
MRM analyses were performed on a hybrid triple quadrupole/linear ion trap mass spectrometer (4000 QTRAP) operated with a beta release of Analyst 1.4.2 supporting scheduled experiments (Applied Biosystems/MDS Sciex). The instrument was coupled to a Tempo nano-LC system (Applied Biosystems/MDS Sciex) for peptide separation using a 30-min gradient from 5 to 30% acetonitrile (0.1% formic acid) at 300 nl/min flow rate. A fused silica emitter of 75-μm inner diameter was packed in house with 13 cm of Reprosil-Pur 120 ODS-3.3 μm (Dr. Maisch GmbH). For validation runs we had the MRM traces trigger MS/MS spectrum acquisition on the two highest transitions. MS/MS spectra were acquired in the trap mode (enhanced product ion) with 100- or 300-ms fixed fill time (housekeeping proteins: 20 ms) with Q0 trapping enabled, Q1 resolution low (1.5 m/z half-maximum peak width), scan speed of 4000 amu/s, m/z range of 300–1300, and two scans summed. Quantitative analyses in MRM mode were performed with Q1 and Q3 operated in unit resolution (0.7 m/z half-maximum peak width).
Statistical Analysis—
For quantification peak height was determined with Multiquant software (version 1.0.0.1, Applied Biosystems/MDS Sciex) after confirming for each peptide the coelution of all transitions. Peptides with unfavorable elution profile (bad resolution) or interfering noise in the heavy or light transitions were excluded from further data analysis and from the transition table (supplemental Data 3). No individual outlier data points were removed. We performed four steps of normalization. 1) The ratio of (peak height light transition)/(peak height heavy transition) was calculated to correct for spray efficiency and ionization differences between runs. 2) Because the internal standard was derived from a pool of all samples, the mean of a particular transition over the 10 analyzed samples should be equal to 1. To adjust for systematic shifts we normalized every transition to the mean of the transition over all samples. 3) To correct for systematic errors from uneven total protein starting material in the samples, we calculated for every sample the median of all peptides from the housekeeping proteins (supplemental Fig. 3) and used the resulting vector for normalizing the data. 4) The obtained response variables were finally log-transformed to fulfill the linear mixed-effects model assumption of residual normality and to stabilize their variance.
Given previous findings, we hypothesized that exposure to various amounts of human plasma during infection induces virulence factor expression changes in S. pyogenes. We analyzed the present data using linear mixed-effects models. The calculations were carried out using SAS version 9.1 (SAS Institute Inc., Cary, NC). Specifically we examined mean changes of the nine virulence factor and 12 housekeeping proteins in association with the amount of plasma. The underlying experimental design is essentially a two-way design with repeated measures on protein abundance. We have the factors plasma concentration (Factor A) with five levels (0, 1, 5, 10, and 20%) and within nested biological sample (Factor D) with two levels. For each sample, we observe repeated measures on the J∈{1,2,3,4} levels of proteotypic peptides (Factor B) and on the K∈{2,3} levels of the within nested MRM transitions (Factor C). Notice that the duplicate measurements for each MRM transition were averaged. For each model, the amount of plasma, the proteotypic peptide, and MRM transition were considered to be fixed effects, whereas the biological sample was a random effect. Denoting the quantitative measure of protein abundance as Y, the model can be written as
 |
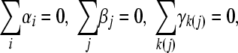 |
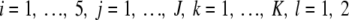 |
Model 1 where all δl(i) and error terms ɛijkl are assumed jointly independent and normally distributed with zero mean and common but unknown variance σbetw2 and σe2, respectively. The grand mean μ, αi, βj, and γk(j) are fixed effects. Thus the problem of differential protein abundance upon contact with different amounts of plasma can be formulated as H0: α1 = … = α5 = 0 versus H1: αi ≠ 0 for at least two i.
RESULTS
S. pyogenes PeptideAtlas
To provide the basis for the targeted proteomics strategy we first generated a S. pyogenes PeptideAtlas. For this we performed two-dimensional peptide separation of tryptic digests of S. pyogenes protein extracts by IEF and reversed-phase liquid chromatography. The IEF-based separations were carried out by continuous peptide separation in a free flow electrophoresis unit (22) or by IPG strip-based off-gel electrophoresis (23). The resulting IEF fractions were analyzed by reversed-phase liquid chromatography coupled online to an LTQ or LTQ-FT mass spectrometer. From a total of more than 190,000 searched spectra, 7181 distinct peptide sequences were identified with a PeptideProphet probability value of ≥0.9 corresponding to a false discovery rate of 0.9% (22, 26). The 7181 identified peptides resulted in the identification of 964 distinct proteins with a ProteinProphet probability value of ≥0.9 (supplemental Data 1) (27). This represents 57% of the predicted ORFs (n = 1697) and an average of 4.2 peptides identified per protein (ranging from one to 26 unique peptides per protein). Of the 964 proteins, 832 were identified by peptides detected at least twice, decreasing the probability of a false positive identification. In the S. pyogenes proteome 1054 proteins have a known or putative function, whereas 643 proteins are still labeled as hypothetical proteins. In this data set we now present protein data confirming the expression of 241 (37%) proteins previously annotated as hypothetical, thereby considerably extending the confirmed proteome of S. pyogenes (Fig. 1a). The 241 hypothetical proteins were identified with 810 unique peptides corresponding to an average of 3.36 peptides per protein. Of the 1054 proteins with known or putative function we identified 723 proteins, corresponding to an identification rate of almost 70% (Fig. 1a). The whole data set was assembled in a PeptideAtlas in a manner described previously (9, 12) and is available for browsing or downloading.
Fig. 1.

Characterization of the S. pyogenes proteome. Multidimensional peptide separation was coupled to MS identification to characterize the S. pyogenes proteome. a, coverage of the hypothetical, confirmed, or total predicted proteins by the proteome map. b, distribution of the detected proteins across the cellular component ontology groups.
Analysis of gene ontology groups of the identified proteins revealed that all the major cellular component ontology groups were represented with the majority of identified proteins being annotated as either cytoplasmic or membrane (Fig. 1b), reflecting the overall gene ontology distribution of the S. pyogenes proteome (as shown in supplemental Fig. 1a). Similar trends were also found for the biological process and molecular function gene ontologies (supplemental Fig. 1, b and c).
Definition of the Target Protein Set
Of the 1697 genes, only a limited number encode proteins that play a role in virulence. Based on the current knowledge we assembled a list of known (17) and putative virulence factors (supplemental Data 2). Quantitative and repeated analysis of these virulence factors upon plasma exposure would provide valuable information regarding their role in the process of bacterial survival in a host environment. Therefore we chose these proteins as targets for our analysis.
Generation and Validation of MRM Transitions for Proteotypic Peptides
In contrast to typical LC-MS/MS shotgun experiments, in MRM measurements no complete fragment ion spectra are recorded. Instead signal intensities for selected predefined MRM transitions are acquired over time. Each transition is defined by a combination of a Q1 and a Q3 m/z value resulting in a filtering at the peptide and fragment ion level. The selective targeting in MRM measurements yields high duty cycles for each transition resulting in high detection sensitivity; exquisite selectivity is achieved because of the two-stage signal filtering. However, the optimal choice of transitions is critical for the sensitivity and selectivity of an MRM experiment. Furthermore the targeted peptides should ionize well under ESI conditions and uniquely identify the protein, i.e. they should be proteotypic. The selection of PTPs and the validation and optimization of specific transitions are therefore critical steps in targeted proteomics. We developed a suite of software tools termed “TIQAM” to support our targeted proteomics strategy (Fig. 2) in four steps as follows.
Fig. 2.

The TIQAM work flow. Starting with a protein list of interest, tryptic peptides are generated by TIQAM. Several sources of information might be integrated to prioritize peptides for which specific transitions are generated. Upon performing MRM-triggered MS/MS experiments, the data are displayed in a protein/peptide-centered view to validate the transitions. The user may then decide to perform additional rounds of validation or export a final list of validated transitions for quantitative experiments.
Selection of Proteotypic Peptides—
Only a subset of the peptides of a tryptic digest is observed by mass spectrometry in a proteomics experiment. The selection of these PTPs uniquely identifying the proteins of interest is of critical importance for a targeted proteomics approach. TIQAM interfaces to PeptideAtlas to prioritize the peptides based on the number of previous observations that indicate good MS compatibility. In addition, TIQAM supports the upload of results from locally performed MS experiments, thereby assisting the user to take full advantage of both internal and external information. In cases where no experimental data are available it has been proposed that proteotypic peptides may be predicted based on physicochemical properties (8, 30, 31). By integrating such information TIQAM supports the selection of peptides with higher probability of detection from proteins which so far have remained undetectable by MS.
Transition Selection—
In addition to selecting the target peptides, specific precursor ions (charge states) and fragment ions need to be chosen for an MRM-based experiment. Based on the preferences of the user TIQAM generates a list of transitions for the targeted analysis by MRM.
Validation and Optimization of Transitions—
Although the two-stage selection of MRM-based experiments results in a highly specific analysis, transitions should be confirmed by MS2 spectrum acquisition to avoid false identification and quantification due to unspecific signals derived from more abundant peptides in the same m/z but different retention time window. For this validation process, the MS instrument is operated in a mixed mode where the sensitive detection of transitions in MRM mode is used to trigger the acquisition of full MS/MS spectra. TIQAM maps the resulting MRM traces and associated MS/MS spectra back to the list of targeted proteins and peptides. In addition, MS/MS database search results statistically validated by PeptideProphet (26) may be imported. The combined information of MRM traces, MS/MS spectra with overlaid theoretical fragment ion spectra, and associated probabilities helps to validate the peptide identifications. In addition to the validated peptides, the respective retention times and the corresponding MRM signal intensities are stored by TIQAM. The best performing transitions are then selected for subsequent quantitative analysis.
Additional Validation Cycles—
An important conceptual difference in a targeted proteomics project compared with a quantitative shotgun experiment is the option of performing additional validation cycles before starting quantification. In particular, proteins of low abundance might require several attempts until specific transitions are confidently identified. TIQAM supports the targeting of additional peptides for proteins not identified in the first validation cycle. This facilitates an efficient validation process where a few peptides are targeted in the first round of validation and only the lowest abundance proteins require the testing of more peptides. Once sufficient transitions have been validated, the proteotypic transitions may be exported for quantitative analysis of multiple samples.
Targeting Virulence Factors Using TIQAM
The temporal regulation of virulence factors has been proposed as a major factor for the pathogenicity of particular S. pyogenes strains. However, a broad analysis of the response of virulence factor proteins to external stimuli was so far not feasible. Therefore we applied TIQAM to specifically target virulence factors based on the current literature (supplemental Data 2). By the extensive proteome mapping we identified 14 of these virulence factor proteins that were expressed under the experimental conditions. Based on this information accessible in the PeptideAtlas we used TIQAM to select PTPs and generate transitions specific for the four singly charged y-fragment ions with an m/z above the precursor ion for doubly and triply charged precursor ions, respectively. Subsequently we performed MRM-triggered MS/MS experiments to validate these transitions by comparing theoretical and observed MS/MS spectra using TIQAM (Fig. 3, a and b). We were able to identify nine virulence factors by this approach in the unfractionated, pooled sample (supplemental Data 2). In addition, we validated transitions specific for 12 housekeeping proteins for normalization purposes. Of all manually validated transitions, we selected for each peptide the three transitions with the best signal-to-noise ratio resulting in a total of 141 transitions for 48 peptides (for three peptides we found only two intense transitions) (supplemental Data 3) that we used to quantify the virulence factors in the biological samples.
Fig. 3.

Validation and quantitative measurements. Validation using TIQAM is shown. The MRM traces (a) and an MS/MS spectrum (b) of a peptide (AGEQAIFVR) of the virulence factor protein “surface lipoprotein” are displayed. c, quantitative analysis. Upon successful validation each sample was analyzed by MRM. The use of prototype “scheduled MRM” software enabled the acquisition of 282 transition traces in one LC-MS run without compromising sensitivity or sampling rate.
Quantitative Analysis
To quantify the targeted virulence factors and the control proteins, each sample was spiked with a heavy labeled pool of all samples as internal standard. The 282 resulting transitions (141 each for light and heavy) were analyzed in a single LC-MS run per sample. This was possible by using prototype instrument acquisition software that features the acquisition of transitions restricted to retention time windows so that at a particular time only a fraction of the transitions is analyzed (15). Hence a large number of total transitions may be analyzed without the need to compromise on dwell time or sampling rate (Fig. 3c).
To characterize the virulence factor expression changes upon contact with plasma, we cultured S. pyogenes in medium supplemented with five different amounts of plasma ranging from 0 to 20%. Two replicate experiments were performed for all the plasma concentrations investigated. The resulting 10 samples were analyzed twice by MRM. We obtained quantitative data for each targeted peptide from all 10 samples demonstrating the strength of the MRM approach for reliable quantification of multiple samples (supplemental Data 4). In Fig. 4A we display the relative peptide intensities of the 10 biological experiments as the average (±S.D.) over all available transitions of the duplicate analysis. The plot reveals the relatively minor deviation in the MRM analysis compared with the variation detected for the biological replicate experiments (different colors). To determine which proteins change significantly in abundance due to the change in plasma concentration, we fitted a linear mixed-effects model to the data for each targeted virulence factor. Fig. 4B visualizes the 95% confidence intervals for the population marginal means of expression for the investigated amounts of human plasma (total mean and plasma effect). Four proteins were significantly regulated: putative endopeptidase O (p < 0.0001), collagen-like surface protein A (p = 0.0003), C5a peptidase (p = 0.0044), and streptopain (p = 0.0099). The first three were induced, whereas streptopain was repressed upon plasma stimulation. Further these plots indicate that three proteins (putative serine protease, M protein type 1, and glyceraldehyde-3-phosphate dehydrogenase) were not regulated. Surface lipoprotein and C3-degrading proteinase seem to show a (statistically non-significant) up-regulation trend upon plasma exposure. The quantitative data obtained over five different conditions allows the distinction of two types of response characteristics. For putative endopeptidase O and C5a peptidase we found a steady increase in protein amount with increasing plasma concentration starting at 1% and up to 20%. In contrast, the collagen-like surface protein A showed an additional strong increase in expression when the plasma concentration was above 5%.
Fig. 4.

Quantification of potential virulence factors by MRM analysis. S. pyogenes was incubated with medium containing 0, 1, 5, 10, or 20% human plasma. Two independent experiments were performed for each concentration, resulting in a total of 10 samples. Every sample was quantitatively analyzed twice by MRM. Each peptide was targeted by three (or two) specific transitions, resulting in six (or four) measurements per peptide and sample (heavy and light). A, peptide averages and S.D. (error bars) of the six (or four) measurements. The replicate experiments at each concentration are distinguished by color. The one to four peptides per protein are plotted next to each other. Therefore, a protein with three proteotypic peptides is plotted with three red and three blue values at each concentration. B, 95% confidence intervals (error bars) for the mean relative protein abundance with dependence on plasma concentration (derived using linear mixed-effects models). n.s., not significant.
We applied the same statistical analysis to the 12 proteins included in the MRM measurements for normalization. Interestingly part of these housekeeping proteins also showed a statistically significant change in protein abundance (supplemental Fig. 3). This indicates that, beside the specific response in virulence factor expression, cell metabolism in S. Pyogenes is partly altered upon environmental changes.
Comparison of Targeted Versus Shotgun Proteomics Approaches
Systems biology experiments in particular require the reproducible acquisition of quantitative data for specific sets of proteins from multiple samples. To compare the performance of our targeted approach with a conventional shotgun approach, we analyzed five samples (one of each investigated plasma concentration) also in data-dependent acquisition mode on a linear ion trap (LTQ) after confirming good instrument performance (supplemental Fig. 2). We then scored how many of the targeted virulence proteins were reliably identified by database searching. Using the targeted strategy we obtained quantitative data for all nine validated proteins from each of the five samples. In contrast, only two proteins of the targeted virulence factors were consistently detected using the shotgun approach (Table I). Three additional proteins were identified in a fraction of the samples. No evidence could be obtained in any of the samples for the remaining four proteins. Similar results were obtained by analyzing these samples on the LTQ-FT mass spectrometer (data not shown). This demonstrates the power of the targeted MRM-based approach to identify and subsequently reliably quantify proteins beyond the detection limit of a conventional shotgun approach.
Table I.
Comparison of protein identification using the targeted approach and conventional shotgun proteomics
Potential virulence factors were targeted using TIQAM. The table lists proteins with MRM transitions successfully validated by MS/MS spectra. For all of these, quantitative values were successfully retrieved by MRM in all 10 samples analyzed. To compare this with a shotgun proteomics approach, five samples, one of each experimental condition, were analyzed by LC-MS/MS on a linear ion trap mass spectrometer (LTQ). ProteinProphet scores above 0.9 corresponding to a false discovery error rate of below 1% are reported. Dashes, not identified.
 |
DISCUSSION
This study describes a novel proteomics work flow for targeted quantitative analysis. First the proteome is mapped out, and subsequently specific proteins of interest are targeted for quantification using the highly sensitive and selective MRM technique. To support the novel aspects of this approach we developed TIQAM, an easy to use software suite with graphical user interface. We applied this strategy to study the change of virulence protein expression of S. pyogenes upon exposure to plasma.
S. pyogenes expresses an array of proteins that facilitate bacterial colonization, survival, and spread (17). To identify novel preventive and therapeutic opportunities it is crucial to understand how S. pyogenes changes its protein expression in the different environments encountered during infection. Conventional proteomics methods used for the detection and quantification of bacterial proteins have limited sensitivity and dynamic range and are impaired by excessive amounts of contaminating human proteins. Therefore, we applied our novel strategy to quantify the expression of virulence factors by S. pyogenes exposed to increasing amounts of human plasma. We found in this study that four of the nine targeted virulence factors are significantly regulated in response to human plasma, underlining the adaptive ability of S. pyogenes (Fig. 4). Plasma acts as a major reservoir for antibacterial components (e.g. immunoglobulins and complement factors) that the bacteria must evade to survive. Interestingly one of the up-regulated proteins, C5a peptidase, targets the complement system: C5a is an anaphylatoxin and acts as an effective leukocyte chemoattractant, causing the accumulation of white blood cells. The C5a peptidase has been identified previously as a promising vaccine candidate (32, 33), and its potential in this context is further emphasized by the present data. These results demonstrate that S. pyogenes specifically increases the expression of blood-related virulence factors upon plasma exposure. It remains to be elucidated how the expression pattern of virulence proteins differs in S. pyogenes colonizing different human sites of infection. Future targeted quantitative analyses using MRM may help to shed light on the adaptation mechanisms pathogens have evolved to overcome the immune system.
The key for the successful reproducible quantitative analysis of these streptococcal samples was the application of MRM technology. Triple quadrupole mass spectrometers operated in MRM mode have been widely used in the small molecule field for quantitative analysis. It has been demonstrated before that MRM can be likewise used to quantify peptides (4) or specifically target potential phosphorylation sites (34, 35). In particular the analysis of serum samples, which is hampered by the large range of protein concentration, benefited remarkably from applying MRM-based technology (15, 36, 37). However, despite the fact that sensitivity, selectivity, and dynamic range are key factors for successful quantitative proteomics analysis, MRM has so far not been applied widely in proteomics. One reason for this discrepancy is the effort required for every single protein to establish a quantitative MRM assay. In the conventional discovery proteomics work flow the samples are analyzed by MS, and the obtained data are subsequently mined by specialized software under qualitative (identification) and quantitative aspects. In contrast, before the quantitative analysis may be started in an MRM-based work flow it is necessary to identify proteotypic peptides, to generate and optimize peptide-specific transitions, and to validate these transitions. So far this process is not supported by the available software applications. Available software, like MIDAS, features the generation of MRM transition from proteins or peptides (34). However, unlike the TIQAM software suite, this tool does not take into account additional information to prioritize the PTPs. By incorporating PTP information from the PeptideAtlas, local LC-MS experiments, or predictions TIQAM reduces the required instrument time significantly thereby supporting the application of MRM for projects with a large number of target proteins. Yet the generation of the MRM transition lists is only the first step in an MRM experiment. The subsequent analysis of the validation experiments and the organization of the associated data pose the major challenges for projects involving a larger number of proteins. Previously different software products had to be used to 1) view MRM traces and MS2 spectra, 2) view database search results, and 3) keep track of targeted proteins, the validated peptides and retention times, and the associated best performing transitions. TIQAM is built on a database structure to combine these different types of information and keep track of user input. By displaying all this in a graphical user interface the process of peptide validation and transition selection is streamlined in TIQAM. This is the key for applying the targeted MRM-based approach also to larger projects of hundred proteins and more. Applying a three-step strategy of validation, optimization by scheduled MRM, and quantification we estimate a time requirement of 2–3 days to set up validated and optimized MRM transitions for 100–200 proteins provided that PTPs are available in a database like PeptideAtlas. This will allow users to quickly establish an increasingly comprehensive list of proteotypic transitions covering all the relevant proteins in their respective fields. Combining those in a global initiative will eventually result in organism-specific proteome-wide proteotypic transition databases that would eliminate or reduce time-intensive method development. Once optimized proteotypic transitions are available, the highly reproducible and sensitive quantitative analysis by MRM can be started with a throughput of about 15 samples per day whereby in each sample at present up to 1000 transitions can be measured via scheduled MRM.
In conclusion, we demonstrate here a novel work flow for quantitative proteomics that we applied to accurately quantify potential virulence factors in crude cell extracts of Streptococcus pyogenes. Our strategy combines a shotgun proteomics approach to identify proteotypic peptides with MRM-based techniques for the highly sensitive and selective quantification of targeted proteins. We developed TIQAM that significantly facilitates and accelerates the generation and validation of transitions for the MRM-based quantification. Although we restricted our study to the analysis of nine virulence factor proteins, TIQAM opens up the possibility to efficiently target large numbers of proteins by an MRM-based approach. Thereby we established an efficient work flow for the more sensitive and selective quantification of proteins of biological or medical interest.
Supplementary Material
Acknowledgments
We thank Alexander Schmidt for advice on the ICPL labeling and help with the LTQ-FT-ICR-MS analysis.
Footnotes
Published, MCP Papers in Press, April 13, 2008, DOI 10.1074/mcp.M800032-MCP200
The abbreviations used are: PTP, proteotypic peptide; ICPL, isotope-coded protein labeling; MRM, multiple reaction monitoring; TIQAM, Targeted Identification for Quantitative Analysis by MRM.
This work was supported, in whole or in part, by National Institutes of Health Grant N01-HV-28179 from the NHLBI (to N. K. and E. W. D.). This work was also supported by SNF Grant 31000-10767 and a grant from F. Hoffmann-La Roche Ltd. (Basel, Switzerland) (to the Competence Center for Systems Physiology and Metabolic Disease) and by the Swiss National Science Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate his fact.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Ideker, T., Thorsson, V., Ranish, J. A., Christmas, R., Buhler, J., Eng, J. K., Bumgarner, R., Goodlett, D. R., Aebersold, R., and Hood, L. ( 2001) Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292, 929–934 [DOI] [PubMed] [Google Scholar]
- 2.Ong, S. E., and Mann, M. ( 2005) Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 1, 252–262 [DOI] [PubMed] [Google Scholar]
- 3.Aebersold, R., and Mann, M. ( 2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 4.Gerber, S. A., Rush, J., Stemman, O., Kirschner, M. W., and Gygi, S. P. ( 2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U. S. A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aebersold, R. ( 2003) Constellations in a cellular universe. Nature 422, 115–116 [DOI] [PubMed] [Google Scholar]
- 6.Domon, B., and Aebersold, R. ( 2006) Mass spectrometry and protein analysis. Science 312, 212–217 [DOI] [PubMed] [Google Scholar]
- 7.Kuster, B., Schirle, M., Mallick, P., and Aebersold, R. ( 2005) Scoring proteomes with proteotypic peptide probes. Nat. Rev. Mol. Cell Biol. 6, 577–583 [DOI] [PubMed] [Google Scholar]
- 8.Mallick, P., Schirle, M., Chen, S. S., Flory, M. R., Lee, H., Martin, D., Ranish, J., Raught, B., Schmitt, R., Werner, T., Kuster, B., and Aebersold, R. ( 2007) Computational prediction of proteotypic peptides for quantitative proteomics. Nat. Biotechnol. 25, 125–131 [DOI] [PubMed] [Google Scholar]
- 9.Desiere, F., Deutsch, E. W., King, N. L., Nesvizhskii, A. I., Mallick, P., Eng, J., Chen, S., Eddes, J., Loevenich, S. N., and Aebersold, R. ( 2006) The PeptideAtlas project. Nucleic Acids Res. 34, D655–D658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King, N. L., Deutsch, E. W., Ranish, J. A., Nesvizhskii, A. I., Eddes, J. S., Mallick, P., Eng, J., Desiere, F., Flory, M., Martin, D. B., Kim, B., Lee, H., Raught, B., and Aebersold, R. ( 2006) Analysis of the Saccharomyces cerevisiae proteome with PeptideAtlas. Genome Biol. 7, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desiere, F., Deutsch, E. W., Nesvizhskii, A. I., Mallick, P., King, N. L., Eng, J. K., Aderem, A., Boyle, R., Brunner, E., Donohoe, S., Fausto, N., Hafen, E., Hood, L., Katze, M. G., Kennedy, K. A., Kregenow, F., Lee, H., Lin, B., Martin, D., Ranish, J. A., Rawlings, D. J., Samelson, L. E., Shiio, Y., Watts, J. D., Wollscheid, B., Wright, M. E., Yan, W., Yang, L., Yi, E. C., Zhang, H., and Aebersold, R. ( 2004) Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol. 6, R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deutsch, E. W., Eng, J. K., Zhang, H., King, N. L., Nesvizhskii, A. I., Lin, B., Lee, H., Yi, E. C., Ossola, R., and Aebersold, R. ( 2005) Human Plasma PeptideAtlas. Proteomics 5, 3497–3500 [DOI] [PubMed] [Google Scholar]
- 13.Domon, B., and Aebersold, R. ( 2006) Challenges and opportunities in proteomics data analysis. Mol. Cell. Proteomics 5, 1921–1926 [DOI] [PubMed] [Google Scholar]
- 14.Wolf-Yadlin, A., Hautaniemi, S., Lauffenburger, D. A., and White, F. M. ( 2007) Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. U. S. A. 104, 5860–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stahl-Zeng, J., Lange, V., Ossola, R., Eckhardt, K., Krek, W., Aebersold, R., and Domon, B. ( 2007) High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol. Cell. Proteomics 6, 1809–1817 [DOI] [PubMed] [Google Scholar]
- 16.Qu, J., Jusko, W. J., and Straubinger, R. M. ( 2006) Utility of cleavable isotope-coded affinity-tagged reagents for quantification of low-copy proteins induced by methylprednisolone using liquid chromatography/tandem mass spectrometry. Anal. Chem. 78, 4543–4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cunningham, M. W. ( 2000) Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13, 470–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carapetis, J. R., Steer, A. C., Mulholland, E. K., and Weber, M. ( 2005) The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694 [DOI] [PubMed] [Google Scholar]
- 19.Musser, J. M., and DeLeo, F. R. ( 2005) Toward a genome-wide systems biology analysis of host-pathogen interactions in group A Streptococcus. Am. J. Pathol. 167, 1461–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson, B. P., Levander, F., von Pawel-Rammingen, U., Berggard, T., Bjorck, L., and James, P. ( 2005) The protein expression of Streptococcus pyogenes is significantly influenced by human plasma. J. Proteome Res. 4, 2302–2311 [DOI] [PubMed] [Google Scholar]
- 21.Wessel, D., and Flugge, U. I. ( 1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138, 141–143 [DOI] [PubMed] [Google Scholar]
- 22.Malmstrom, J., Lee, H., Nesvizhskii, A. I., Shteynberg, D., Mohanty, S., Brunner, E., Ye, M., Weber, G., Eckerskorn, C., and Aebersold, R. ( 2006) Optimized peptide separation and identification for mass spectrometry based proteomics via free-flow electrophoresis. J. Proteome Res. 5, 2241–2249 [DOI] [PubMed] [Google Scholar]
- 23.Heller, M., Ye, M., Michel, P. E., Morier, P., Stalder, D., Junger, M. A., Aebersold, R., Reymond, F., and Rossier, J. S. ( 2005) Added value for tandem mass spectrometry shotgun proteomics data validation through isoelectric focusing of peptides. J. Proteome Res. 4, 2273–2282 [DOI] [PubMed] [Google Scholar]
- 24.Yi, E. C., Lee, H., Aebersold, R., and Goodlett, D. R. ( 2003) A microcapillary trap cartridge-microcapillary high-performance liquid chromatography electrospray ionization emitter device capable of peptide tandem mass spectrometry at the attomole level on an ion trap mass spectrometer with automated routine operation. Rapid Commun. Mass Spectrom. 17, 2093–2098 [DOI] [PubMed] [Google Scholar]
- 25.Eng, J. K., Mccormack, A. L., and Yates, J. R. ( 1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 26.Keller, A., Nesvizhskii, A. I., Kolker, E., and Aebersold, R. ( 2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 27.Nesvizhskii, A. I., Keller, A., Kolker, E., and Aebersold, R. ( 2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 28.Schmidt, A., Kellermann, J., and Lottspeich, F. ( 2005) A novel strategy for quantitative proteomics using isotope-coded protein labels. Proteomics 5, 4–15 [DOI] [PubMed] [Google Scholar]
- 29.Keller, A., Eng, J., Zhang, N., Li, X. J., and Aebersold, R. ( 2005) A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 2005 0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang, H., Arnold, R. J., Alves, P., Xun, Z., Clemmer, D. E., Novotny, M. V., Reilly, J. P., and Radivojac, P. ( 2006) A computational approach toward label-free protein quantification using predicted peptide detectability. Bioinformatics 22, e481–e488 [DOI] [PubMed] [Google Scholar]
- 31.Lu, P., Vogel, C., Wang, R., Yao, X., and Marcotte, E. M. ( 2007) Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 25, 117–124 [DOI] [PubMed] [Google Scholar]
- 32.Pandiripally, V., Gregory, E., and Cue, D. ( 2002) Acquisition of regulators of complement activation by Streptococcus pyogenes serotype M1. Infect. Immun. 70, 6206–6214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shet, A., Kaplan, E., Johnson, D., and Cleary, P. P. ( 2004) Human immunogenicity studies on group A streptococcal C5a peptidase (SCPA) as a potential vaccine against group A streptococcal infections. Indian J. Med. Res. 119, (suppl.) 95–98 [PubMed] [Google Scholar]
- 34.Unwin, R. D., Griffiths, J. R., Leverentz, M. K., Grallert, A., Hagan, I. M., and Whetton, A. D. ( 2005) Multiple reaction monitoring to identify sites of protein phosphorylation with high sensitivity. Mol. Cell. Proteomics 4, 1134–1144 [DOI] [PubMed] [Google Scholar]
- 35.Cox, D. M., Zhong, F., Du, M., Duchoslav, E., Sakuma, T., and McDermott, J. C. ( 2005) Multiple reaction monitoring as a method for identifying protein posttranslational modifications. J. Biomol. Tech. 16, 83–90 [PMC free article] [PubMed] [Google Scholar]
- 36.Liao, H., Wu, J., Kuhn, E., Chin, W., Chang, B., Jones, M. D., O'Neil, S., Clauser, K. R., Karl, J., Hasler, F., Roubenoff, R., Zolg, W., and Guild, B. C. ( 2004) Use of mass spectrometry to identify protein biomarkers of disease severity in the synovial fluid and serum of patients with rheumatoid arthritis. Arthritis Rheum. 50, 3792–3803 [DOI] [PubMed] [Google Scholar]
- 37.Anderson, L., and Hunter, C. L. ( 2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.