

Abstract
Synthesis of cysteinyl-tRNACys in methanogenic archaea proceeds by a two-step pathway in which tRNACys is first aminoacylated with phosphoserine by phosphoseryl-tRNA synthetase (SepRS). Characterization of SepRS from the mesophile Methanosarcina mazei by gel filtration and nondenaturing mass spectrometry shows that the native enzyme exists as an α4 tetramer when expressed at high levels in Escherichia coli. However, active site titrations monitored by ATP/PPi burst kinetics, together with analysis of tRNA binding stoichiometry by fluorescence spectroscopy, show that the tetrameric enzyme binds two tRNAs and that only two of the four chemically equivalent subunits catalyze formation of phosphoseryl adenylate. Therefore, the phenomenon of half-of-the-sites activity, previously described for synthesis of 1 mol of tyrosyl adenylate by the dimeric class I tyrosyl-tRNA synthetase, operates as well in this homotetrameric class II tRNA synthetase. Analysis of cognate and noncognate reactions by ATP/PPi and aminoacylation kinetics strongly suggests that SepRS is able to discriminate against the noncognate amino acids glutamate, serine, and phosphothreonine without the need for a separate hydrolytic editing site. tRNACys binding to SepRS also enhances the capacity of the enzyme to discriminate among amino acids, indicating the existence of functional connectivity between the tRNA and amino acid binding sites of the enzyme.
The synthesis of aminoacyl-tRNA by tRNA synthetases embodies the genetic code by faithfully pairing amino acids with the subset of cognate tRNA isoacceptors. In recent years, the textbook understanding by which each of the standard 20 amino acids is matched with tRNA by a canonical tRNA synthetase has been modified to include a number of important exceptions (1). Among these is the synthesis of cysteinyl-tRNACys in a subset of archaebacteria that contain most or all of the enzymes in the methanogenesis pathway (2). In these organisms, tRNACys is first aminoacylated with the nonstandard amino acid phosphoserine by phosphoseryl-tRNA synthetase (SepRS).2 The misacylated Sep-tRNACys is then converted to Cys-tRNACys through the action of the pyridoxal phosphate-dependent enzyme Sep-tRNA:Cys-tRNA synthase. This indirect pathway to aminoacyl-tRNA is conceptually similar to the two-step routes to glutaminyl-tRNA and asparaginyl-tRNA synthesis in many organisms, which also involve misacylation followed by a subsequent tRNA-dependent modification reaction (3).
Sequence comparisons show that SepRS is a class II aminoacyl-tRNA synthetase and that it is most similar to the α subunit of phenylalanyl-tRNA synthetase (PheRS) (4). PheRS is one of three tRNA synthetases, all members of the class II family, that have been previously shown to exist as tetramers. PheRS and (in some organisms) glycyl-tRNA synthetase (GlyRS) possess a heterotetrameric (α2β2) quaternary organization (5, 6). In contrast, alanyl-tRNA synthetase (AlaRS) from Escherichia coli is a homotetramer in solution (7). SepRS is encoded by a single gene (2), and crystal structures of the enzyme from Methanococcus maripaludis, Methanococcus jannaschii, and Archaeoglobus fulgidus reveal an α4 homotetramer in each case (8, 9). Further, sedimentation equilibrium analyses of the M. jannaschii and A. fulgidus enzymes revealed that each exists as a tetramer in solution (8). Therefore, it appears that, although the SepRS sequences are most similar to those of PheRS, the quaternary organization of the enzyme instead matches that of E. coli AlaRS. Interestingly, the unusual pyrrolysyl-tRNA synthetase, which exists only in the archaeal Methanosarcina family and in the Gram-positive bacterium Desulfitobacterium hafniense, may also form an α4 tetramer (10). The crystal structure of a truncated pyrrolysyl-tRNA synthetase enzyme from Methanosarcina mazei revealed a dimeric organization, but molecular modeling based on structural similarity with SepRS suggests that a tetrameric structure for the native enzyme is conceivable (11).
Structure-function relationships for the tetrameric tRNA synthetases PheRS, GlyRS, AlaRS, and SepRS are among the least well characterized in the family, in part due to the very large sizes of the enzymes. Crystal structures of GlyRS have been solved only for enzymes that function as homodimers, whereas structural information for AlaRS is limited to a catalytically active monomeric fragment of the enzyme from Aquifex aeolicus (12–14). The best characterized tRNA-bound complex among tetrameric enzymes is that of Thermus thermophilus PheRS; this structure reveals that the α2β2 tetramer binds two tRNA molecules, with each tRNA making contact with all four enzyme monomers (15). Based on structural and phylogenetic comparisons among PheRS, GlyRS, and other class II tRNA synthetases, it appears that the α2β2 tetrameric structure can be considered operationally as a variant of the class II homodimer structure, with the α-subunit possessing catalytic motifs, whereas the β-subunit contributes more to tRNA binding (16, 17). Because neither the α nor β subunit of α2β2 tRNA synthetases is independently capable of catalysis, a stoichiometry of two active sites per tetramer is easily rationalized without the need to consider the concept of half-of-the-sites activity (18). In the half-of-the-sites mechanism, which has been most thoroughly described for the synthesis of tyrosyl adenylate by dimeric TyrRS, only half of the active sites present in a tRNA synthetase oligomer function in catalysis (19).
In contrast to PheRS and GlyRS, the α4 E. coli AlaRS homotetramer may possess unique oligomeric features. Deletion analysis has shown that removal of the 176 C-terminal amino acids from the 875-amino acid enzyme disrupts the tetrameric structure to give monomers that retain catalytic activity in both adenylate synthesis and alanylation of tRNA (20). As is the case generally for class II tRNA synthetases, the class-defining motif 1 sequence in E. coli AlaRS is present in the N-terminal portion of the sequence, and by analogy with other class II enzymes, it would be expected to be the key determinant that forms the quaternary interface (21). Thus, the role of the C-terminal E. coli AlaRS sequence in tetramerization suggests that the oligomeric assembly of homotetrameric tRNA synthetases possess unique features not present among homodimers or heterotetramers. Despite this, the crystal structures of SepRS do reveal substantial structural similarity with PheRS. However, although the A. fulgidus SepRS-tRNACys cocrystal structure revealed that two tRNAs were bound per tetramer, interactions of the enzyme with tRNA were made only with the anticodon loop (8). Because the 3′ acceptor end of the tRNA does not bind in the catalytic site, this structure clearly represents a nonproductive complex from which inferences regarding at least some functional characteristics must be made with caution. No information has been available regarding the stoichiometry of tRNA binding to any homotetrameric tRNA synthetase in solution.
Other biochemical studies on SepRS have shown that tRNA identity elements include the discriminator base U73 and all three anticodon nucleotides, as is also the case for canonical CysRS enzymes (22, 23). A crystal structure of the A. fulgidus enzyme with phosphoserine bound has also shown that recognition is accomplished, perhaps somewhat unexpectedly, by hydrogen bonding with polar but uncharged side chains, including Ser231, Ser233, and Asn325 (8). Mutagenesis of the equivalent and conserved residues in M. maripaludis SepRS produces decreases of 103–104-fold in kcat/Km for activation of phosphoserine (9), suggesting that the crystal structure correctly identifies the site of amino acid binding.
To begin a more detailed study of the catalytic properties of SepRS, we have chosen to study the enzyme from Methanosarcina mazei. An experimental genetic system has been established for this mesophilic methanogen, permitting correlative in vivo studies (24). Further, M. mazei (and Methanosarcina barkeri) possess both redundant pathways for synthesis of Cys-tRNACys and three distinct tRNACys isoacceptors in their genomes (25, 26), suggesting that the properties of SepRS from these organisms may be of particular interest.
To facilitate study of M. mazei SepRS, we have cloned and expressed the enzyme in a recombinant bacterial system and purified it to homogeneity. Analysis of burst kinetics for pyrophosphate exchange, together with tRNA binding by equilibrium fluorescence, shows that the homotetrameric enzyme exhibits half-of-the-sites reactivity, a phenomenon previously documented only for homodimeric tRNA synthetases. Further investigation of the amino acid specificity suggests that SepRS can discriminate against structurally similar amino acids without the need for an editing activity, a finding consistent with the absence of significant homology with domains of PheRS that edit noncognate aminoacyl-tRNA synthesized by that enzyme. Finally, we show that efficient phosphoserylation by SepRS requires methylation of tRNACys at the N1 position of G37 in the anticodon loop, providing a rare example of the need for post-transcriptional modification as a prerequisite for tRNA synthetase function.
EXPERIMENTAL PROCEDURES
Expression and Purification of M. mazei SepRS—M. mazei genomic DNA was purchased from the American Type Culture Collection (Manassas, VA) and used as template for PCR amplification of the SepRS open reading frame. The resulting DNA fragment was digested with NdeI and BamHI and inserted into pEt16b for expression of His-tagged protein in E. coli Rosetta2(DE3) pLysS cells. Cultures were grown aerobically at 37 °C in LB medium supplemented with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol. When the cultures reached an A600 of 0.5, expression of His-tagged enzyme was induced by adding isopropyl-β-d-thiogalactoside to a final concentration of 0.6 mm. Cultures were then grown for a further 5 h at 37 °C prior to harvesting.
Cells expressing SepRS were resuspended in a buffer containing 50 mm NaH2PO4 (pH 7.8), 150 mm NaCl, 1 mm β-mercaptoethanol (βME), and a Roche Applied Science EDTA-free Complete protease inhibitor tablet and were disrupted by sonication. The enzyme was purified by Ni2+-nitrolotriacetic acid-agarose chromatography (Qiagen), as suggested by the provider. The pEt16b vector contains a Factor Xa-cleavable His tag that was removed according to the manufacturer's instructions, by incubation with Factor Xa overnight at 4 °C. The cleaved protein was run over a second Ni2+-nitrilotriacetic acid column, and the flow-through containing active protein was pooled and dialyzed into 50 mm Hepes-HCl (pH 8.0), 50 mm NaCl, 1 mm βME, and 50% glycerol for storage at –20 °C. SepRS was recovered at better than 99% purity as judged by SDS-polyacrylamide gel electrophoresis at yields of ∼5 mg of protein/liter of culture.
Preparation and Methylation of tRNA—To prepare tRNA for kinetic analysis, the M. mazei tRNACys2 gene was transcribed from a duplex DNA template constructed from complementary synthetic oligonucleotides containing a 17-bp overlapping region, which were then extended using the Klenow fragment of E. coli DNA polymerase (27). 2′-O-methyl sugars were used at the two 5′ nucleotides of the noncoding strand to reduce 3′ heterogeneity in the transcripts (27). The oligonucleotides used were 5′-AATTCCTGCAGTAATACGACTCACTATAGCCAAGATGGCGGAGCGGCTACGCAATCGCCT-3′ and 5′-mUmGGAGCCAAGATCCGGATTCGAACCGGAATGGTATCGCTCTGCAGGCGATTGC-3′.2′-O-Methyl nucleotides are represented by mU and mG, and the underlined portions represent the overlap region. Boldface type indicates the T7 RNA polymerase promoter.
Transcription reactions were carried out as described (27). RNA was recovered by ethanol precipitation, resuspended in 10 mm Tris-HCl (pH 7.5), and heated to 85 °C for 3 min. MgCl2 was then added to 2 mm final concentration, and the reactions were slow-cooled to room temperature. The tRNA transcript was methylated at position m1G37 by M. jannaschii Trm5 methylase in a buffer consisting of 50 mm Tris-HCl (pH 8.0), 0.05 mm EDTA, 2 mm DTT, 3 mm MgCl2, 50 mm KCl, and 0.012 mg/ml bovine serum albumin (28). Trm5 was purified from an overexpression construct in E. coli, as described (29). The concentrations of S-adenosylmethionine and Trm5 used were 50 and 5 μm, respectively, and the reactions were incubated at 45 °C for 1 h. Over 50% of transcripts were typically methylated in this reaction, as judged by control reactions employing 3H-labeled S-adenosylmethionine. The reactions were stopped by phenol/chloroform/isoamyl alcohol extraction, and the tRNAs were recovered by ethanol precipitation.
To remove unmethylated transcripts, the recovered tRNA was resuspended in a solution containing 10 mm Tris (pH 7.5), 1 mm EDTA, and 20 mm MgCl2. 16-mer oligodeoxynucleotides complementary to the tRNA sequence flanking nucleotide G37 were then added at a molar ratio of 1:1 with tRNA. The oligonucleotide used was 5′-mGmGmUmAmUmCmGmCTCTGmCmAmGmG-3′ (where “m” indicates a 2′-O-methyl ribose modification). Reactions were heated to 85 °C for 10 min and then slowly cooled for 10 min to ambient temperature. The solution was then adjusted to 50 mm Tris (pH 8.3), 75 mm KCl, 3 mm MgCl2, 10 mm DTT. RNase H (Promega) was then added at 0.1 units/μl to initiate the cleavage reaction. Reactions were incubated for 30 min at 37 °C and stopped by phenol/chloroform/isoamyl alcohol extraction, and the RNA was recovered by ethanol precipitation. In these reactions, the m1G37 modification protects the DNA-tRNA hybrid from RNase H cleavage in the RNA strand at the methylation site.
Recovered tRNA was resuspended in a urea loading buffer consisting of 8 m urea, 20 mm EDTA, 5 mm Tris, pH 7.5, and 0.05% (v/v) dye. To separate uncleaved, methylated tRNA from cleaved tRNA half-molecules, ∼2–3 mg of tRNA was loaded on a prerun 1-mm-thick 12% (w/v) acrylamide, 7 m urea gel and run for ∼4–6 h at 45 watts. The dye standards in the gel were xylene cyanol and bromphenol blue. The band containing full-length tRNA was excised by UV shadowing and eluted by a modified crush-and-soak method (30). After butanol extraction and ethanol precipitation, the pellet was resuspended in 10 mm Tris-HCl (pH 7.4) and stored in aliquots at –80 °C.
Determination of Quaternary Structure by Gel Filtration and Mass Spectrometry—The oligomeric structure of SepRS was determined by analytical size exclusion chromatography. The enzyme was analyzed by Superdex 200 column chromatography on an fast protein liquid chromatography system (Amersham Biosciences), run at a flow rate of 0.50 ml/min at ambient temperature in 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 5 mm MgCl2, 5 mm βME. The molecular mass markers (Bio-Rad) for calibration were as follows: thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), equine myoglobin (17 kDa), and vitamin B12 (1.4 kDa).
The quaternary structure of SepRS was further verified by nondenaturing mass spectrometry (31). 20 μm SepRS was dialyzed against 10 mm ammonium acetate (pH 6.5) using Slide-A-Lyser Mini Dialysis Units with a 10,000 molecular weight cut-off. The dialysis buffer was changed three or more times to ensure removal of all glycerol. Dilute protein was then desalted by running over a Bio-Rad Bio-Spin 6 column prewashed with 10 mm ammonium acetate (pH 6.5). The final protein concentration was between 1 and 10 μm. Approximately 5–10 μl of sample was then loaded into a gold-coated borosilicate capillary suitable for electrospray. Mass spectra were obtained on a modified quadrupole time-of-flight mass spectrometer described in detail elsewhere (32). The voltages and pressures in each part of the instrument were optimized to preserve noncovalent interactions. The mass spectra were calibrated using a standard solution of sodium iodide and cesium iodide. The protocol described by Hernandez and Robinson was used to calculate the analyte mass corresponding to the charge states present in the mass spectrum (33).
tRNA Binding Affinity and Stoichiometry by Intrinsic Tryptophan Fluorescence—Equilibrium fluorescence titrations were performed at room temperature with 0.25 μm SepRS in 20 mm Tris-HCl (pH 7.5), 50 mm NaCl, 2 mm MgCl2, and 5 mm βME. Tryptophan fluorescence was excited at 295 nm, and the emission was monitored from 300 to 400 nm. After excitation at 295 nm, the emission intensities at 345 nm were recorded and corrected for dilution and for intrinsic quenching due to RNA absorbance at the excitation and emission wavelengths according to the formula, Fc = Fobs × antilog((A295 + A345)/2) (34). Control solutions of bovine serum albumin or of tryptophan showed no fluorescence response to tRNA. The tRNA was titrated in the concentration range of 0.05–8 μm. The Kd values were derived by fitting the data to the following quadratic binding equation using Scientist software,
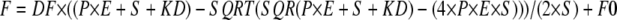 |
(Eq. 1) |
where F represents the measured fluorescence, DF is the change in fluorescence over the tRNA concentration range, E is the initial concentration of enzyme, S is the concentration of tRNA, F0 is the initial fluorescence before the addition of enzyme, P is a parameter that evaluates the data quality and is set initially to 1.0, and KD is the dissociation constant.
The tRNA·SepRS binding stoichiometry was determined by monitoring the quenching of intrinsic tryptophan fluorescence. 1 μm SepRS tetramer was incubated in a buffer containing 20 mm Tris-HCl (pH 7.5), 50 mm to 1 m NaCl, 2 mm MgCl2, and 5 mm βME. tRNA concentrations were varied from 0 to 8 μm. After excitation at 295 nm, the emission intensities at 345 nm were recorded and corrected for the inner filter effect, as described above. Emission intensities were used to generate the percentage of quenching observed, and these were then plotted versus tRNA concentration.
Active Site Titration—The number of SepRS active sites was determined by measuring ATP depletion during the formation of phosphoseryl adenylate at 37 °C as described (19). [γ-32P]ATP (5 μm,15 μCi/ml) and phosphoserine (3 mm) were incubated in 100 mm Tris-HCl (pH 7.5), 20 mm MgCl2, 25 mm KCl, and 4 mm DTT in the presence of pyrophosphatase (2 units/ml). Before adding SepRS (1 μm) to initiate the reaction, 5 μl was removed for the zero time reading. The number of active sites was determined by fitting the decay of ATP over time to an exponential decay curve,
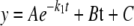 |
(Eq. 2) |
where A represents the burst amplitude related to formation of the enzyme-bound aminoacyl adenylate intermediate, B is the “steady state” decay of aminoacyl adenylate, and C is the ordinate intercept of the linear portion of the fit, which can be used to find the number of active sites using the equation,
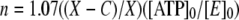 |
where X represents the number of counts/min before the addition of enzyme, C is the ordinate intercept, and [ATP]0 and [E]0 represent the initial ATP and enzyme concentrations, respectively (30).
ATP-PPi Exchange Assay—The reaction mixture for the ATP-PPi exchange assay consisted of 100 mm Tris-HCl (pH 7.5), 20 mm MgCl2, 25 mm KCl, 4 mm DTT, 1 mm [32P]PPi (10–50 μCi/ml), and 5 mm ATP. Km and kcat for phosphoserine and phosphothreonine were determined by varying their concentrations from 0.01 to 1 mm and from 0.05 to 30 mm, respectively. For the noncognate substrates serine and glutamate, Km and kcat were determined by varying concentrations of these substrates from 0.025 to 1.2 m. The concentrations of SepRS used were 60 nm for phosphoserine activation reactions and 1000 nm for reactions of other amino acid substrates. Stock solutions of all amino acids were adjusted to neutral pH. The reactions were stopped by adding 1.5 μl of the reaction mixture to 3.0 μl of a solution containing 400 mm sodium acetate (pH 5.0), 0.1% SDS. 2.0 μl of this mixture was spotted on polyethyleneimine cellulose plates that had been prewashed in water. Thin layer chromatography, using 750 mm KH2PO4 (pH 3.5) and 4 m urea as developing buffer, was employed for separation of [32P]ATP from [32P]PPi and [32P]Pi, as previously described (35). The dried chromatogram was exposed overnight to an image plate and was quantified on a Storm 840 Phosphor-Imager (Amersham Biosciences). Kinetic parameters were obtained from at least two independent measurements. Initial velocities obtained by time course analyses were plotted against substrate concentration and fit to the Michaelis-Menten equation with KaleidaGraph. Km and kcat were determined directly from these plots.
Aminoacylation Assay and Steady-state Kinetics—SepRS aminoacylation assays were carried out by a recently described highly sensitive assay (35–37). tRNACys2 ending in -CC75 prepared by in vitro transcription was 32P-labeled at the 3′-terminal internucleotide linkage using α-32P-labeled ATP and the addition reaction of tRNA nucleotidyltransferase (35). Upon the addition of enzyme, the reaction was incubated at 37 °C for 2 min followed by phenol/chloroform/isoamyl alcohol extraction. Unincorporated label was removed by passing the reaction mixture over two consecutive Bio-Rad Micro Bio-Spin 30 chromatography columns preequilibrated in nuclease-free water. To prepare the tRNA sample for use in kinetic assays, labeled and unlabeled substrate were mixed to the appropriate final concentration, heated at 85 °C for 3 min, and then cooled at room temperature for 10 min in the presence of 2.0 mm MgCl2. All reactions were quenched in a buffer containing 0.1% SDS, 0.15 m sodium acetate (pH 5.2). P1 nuclease digestions were then performed by adding 1.5 μl of the reaction mixture to a microtiter well containing 3.0 μl of 0.1 mg/ml P1 nuclease (Fluka), 0.15 m sodium acetate (pH 5.2) and incubating for 10 min at ambient temperature. Aminoacylated tRNA (as 3′-aminoacylated A76) and nonreacted substrate (as unmodified AMP) were separated by TLC in 10% ammonium acetate, 5% acetic acid and quantitated by phosphorimaging analysis, as described previously (35).
Reactions to determine plateau aminoacylation levels were performed in 6 mm ATP, 50 mm Tris (pH 7.5), 20 mm KCl, 10 mm MgCl2, and 5 mm DTT over a time course of 5–10 min. Enzyme was kept in 3-fold excess over tRNA in all plateau reactions; typically, 3 μm tRNA and 10 μm enzyme were used. Plateau aminoacylation levels were determined directly by the ratio of aminoacyl-A76 and unmodified A76 after P1 nuclease treatment and TLC separation (35). All steady-state kinetic reactions were performed at 37 °C in a buffer containing 6 mm ATP, 5 mm DTT, 50 mm Tris (pH 7.5), 20 mm KCl, and 10 mm MgCl2. Concentration ranges used for phosphoserine and phosphothreonine were 0.01–1 mm and 0.5–20 mm, respectively. The tRNACys2 concentration was kept at 40 μm, which was verified to be saturating for all reactions. Initial velocities obtained by time course analyses were plotted against substrate concentration and fit to the Michaelis-Menten equation with KaleidaGraph. Km for the amino acid and kcat were determined directly from these plots. The mean and S.D. values of 2–4 measurements for each derived parameter are reported in Tables 1 and 2.
TABLE 1.
ATP-PPi exchange kinetics
| Phosphoserine (WT) | phosphothreonine (WT) | Phosphoserine (T307S) | phosphothreonine (T307S) | Serine (WT) | Glutamate (WT) | |
|---|---|---|---|---|---|---|
| kcat (s-1) | 10.6 ± 2.4 | (1.4 ± 0.2) × 10-2 | 11.2 ± 2.8 | (2.1 ± 0.1) × 10-2 | (7.0 ± 1.4) × 10-2 | (5.9 ± 2.2) × 10-2 |
| Km (amino acid) (mm) | 0.27 ± 0.04 | 2.2 ± 0.6 | 0.15 ± .08 | 0.93 ± .25 | 45.3 ± 4.4 | 35.1 ± 9.2 |
| kcat/Km (s-1m-1) | 3.9 × 104 | 6.4 | 7.5 × 104 | 22.6 | 1.5 | 1.7 |
TABLE 2.
Steady-state kinetic parameters for aminoacylation WT SepRS
| Phosphoserine | Phosphothreonine | Serine | Glutamate | |
|---|---|---|---|---|
| Plateau aminoacylation (%) | 72.7 ± 10 | 27.2 ± 5 | 8.0 ± 2 | 9.5 ± 3 |
| kcat (s-1) | 0.45 ± 0.10 | (5.4 ± 0.2) × 10-3 | NDa | ND |
| Km (amino acid) (mm) | 0.04 ± 0.01 | 8.4 ± 4.0 | ND | ND |
| kcat/Km (amino acid) (s-1m-1) | 1.13 × 104 | 0.64 | ND | ND |
Plateau aminoacylation levels are too low to permit accurate determination of kinetic parameters.
RESULTS
Oligomeric Structure and tRNA Binding Stoichiometry of SepRS—Gel filtration analysis of M. mazei SepRS yields a molecular mass of ∼250 kDa, corresponding to a tetrameric structure for the 539-amino acid enzyme (calculated subunit Mr = 60,909) (Fig. 1A). Because tetrameric aminoacyl-tRNA synthetases are often found as monomers or dimers in some organisms (21), we sought further verification using nondenaturing mass spectrometry (Fig. 1B) (31). Using the method described by Hernandez and Robinson (33), we confirmed Mr of 68,992 ± 10 for the SepRS monomer (data not shown) and derived an Mr of 255,442 ± 180 for the distribution between 8000 and 9000 m/z. This confirms a tetrameric organization for the enzyme. Unlike the diversity exhibited by PheRS, GlyRS, and AlaRS (21), thus far all SepRS enzymes for which the quaternary structure has been examined have been found to be homotetrameric (8).
FIGURE 1.

Characterization of M. mazei SepRS. A, gel filtration of SepRS (0.4 nmol of tetramer) on Superdex 200 calibrated by known molecular weight markers (see “Experimental Procedures”). B, electrospray mass spectrometry showing peaks corresponding to the α4 SepRS enzyme. The net positive charge (z) and the precise value of m/z for each peak are indicated. The size of the tetramer is obtained by multiplying the m/z value for each peak by the corresponding charge. Each peak provides independent information confirming the size of the protein at ∼256 kDa, corresponding to a tetrameric quaternary structure.
To determine whether all four chemically identical SepRS active sites are functional, we performed active site titration by monitoring ATP exhaustion in the amino acid activation step, in the presence of inorganic pyrophophatase. An initial burst of aminoacyl adenylate was observed, followed by a slow steady-state rate of conversion (Fig. 2A). The burst shows that product release is rate-limiting for SepRS-catalyzed ATP-PPi exchange in the absence of tRNA, as observed for many other tRNA synthetases (19, 38). Extrapolation of the linear portion of the fit allows calculation of 2.3 active sites per tetramer. This is consistent with a half-of-the-sites model for catalysis, as previously observed for the homodimeric tyrosyl-tRNA synthetase (39, 40).
FIGURE 2.

tRNA binding affinity and stoichiometry of SepRS. A, rapid initial depletion of ATP by SepRS, in the first step reaction forming phosphoseryl adenylate from phosphoserine and ATP, performed in the presence of inorganic pyrophosphatase. B, quenching of intrinsic tryptophan fluorescence of SepRS by tRNA binding. Shown are emission scans taken as tRNACys. The concentration of tRNA was titrated from 0 (maximum emission at 340 nm) to 8 μm. The fluorescence intensity on the ordinate is given in arbitrary units. C, determination of the stoichiometry of tRNA binding to SepRS by tryptophan fluorescence quenching.
We next examined tRNA binding to SepRS using fluorescence spectroscopy. Titration of the enzyme with increasing concentrations of tRNACys2 (up to 8 μm), with correction for tRNA absorption (inner filter effect), showed that the intrinsic tryptophan fluorescence is quenched by tRNA binding (λex = 295 nm) (Fig. 2B). The fluorescence intensity versus tRNA concentration was fit to a quadratic binding formula (see “Experimental Procedures”), yielding a Kd of 0.16 ± 0.04 μm, in the range reported for other cognate synthetase-tRNA interactions (41, 42).
This relatively high affinity for tRNA enabled a direct determination of the tRNA binding stoichiometry by fluorescence, in a concentration range that permits correction for the inner filter effect. High concentrations of enzyme (1 μm tetramer) were used to shift the binding equilibria in the direction of the enzyme-tRNA complex. The concentration of tRNA was then titrated in the range 0.5–8 μm. This produced a biphasic quenching of the enzyme fluorescence (Fig. 2C). An initial steep phase was observed for tRNA concentrations up to 2.0 μm (19% quenching); this was followed by a second much flatter phase up to 8.0 μm tRNA (31.5% quenching). Correction for inner filter effect is not possible at higher tRNA concentrations. Quenching was unaffected by ionic strength in the range of 0.2–1.0 m NaCl (data not shown). We interpret these data to mean that two tRNAs bind specifically to a SepRS tetramer, with the further fluorescence quenching at higher tRNA stoichiometries probably due to nonspecific formation of higher order complexes that are insensitive to salt. The results are consistent with the active site titration experiment (Fig. 2A) and imply an asymmetric structural organization in which adenylate synthesis in two of the four subunit active sites is followed by transfer of phosphoserine to tRNA in those subunits. The origin of the enzyme asymmetry, which is evidently present even in the absence of tRNA, is unknown.
Amino Acid Discrimination by SepRS—A fundamental property of all tRNA synthetases is the capacity to discriminate among amino acids. Some of the enzymes are capable of sufficient selectivity directly in the synthetic reactions, whereas others possess a separate active site in which the aminoacyl ester bond of misacylated tRNAs is hydrolyzed. Among class IIc tRNA synthetases, it is known that PheRS catalyzes hydrolytic editing of misacylated Tyr-tRNAPhe, although it would seem that tyrosine should be readily excluded on steric grounds (43). Phosphoserine is nearly isosteric with glutamate, which is generally present in vivo in millimolar concentrations due to its central metabolic role and which cannot be easily rejected by the SepRS active site on steric grounds. Given the known editing reaction of the related PheRS and the relatively high concentrations of glutamate in vivo, it appeared possible that SepRS might misacylate tRNACys with glutamate and thus require an editing activity as well. We also chose to examine discrimination against serine, because that comparison allows experimental determination of the role of the phosphate group in providing selectivity. Finally, we examined the capacity of SepRS to activate and transfer phosphothreonine to tRNA. Although this amino acid is not a naturally occurring metabolite in its free form, phosphothreonylation of tRNA is of considerable interest in the context of engineered genetic code expansion. There have been no prior studies examining the amino acid discrimination properties of any SepRS enzyme.
The amino acid selectivity of SepRS was examined at both the amino acid activation and aminoacyl transfer steps of the reaction. First, the rate of exchange of 32P-labeled pyrophosphate into ATP was measured in the presence of phosphoserine, glutamate, serine, and phosphothreonine (Fig. 3 and Table 1). Significantly impaired synthesis of all noncognate (NC) adenylates was observed. Km for phosphothreonine was increased by 8-fold as compared with phosphoserine, whereas kcat was decreased by over 103-fold. Km for serine and glutamate were further elevated, with increases of over 102-fold compared with Ser(P). Overall, the selectivity in adenylate synthesis, as measured by the ratio (kcat/Km)Ser(P)/(kcat/Km)NC, varied from 2.5 × 104-fold (serine) to 5.8 × 103-fold (phosphothreonine).
FIGURE 3.

ATP-PPi exchange by SepRS. Thin layer chromatogram of a reaction time course showing separation of [32P]PPi from [32P]ATP and [32P]Pi. The first lane on the left is a control reaction in which SepRS was omitted.
tRNA aminoacylation by M. mazei SepRS was measured using an assay based on 32P labeling at the 3′-internucleotide linkage of the tRNA (37). This assay has been applied extensively to steady-state and pre-steady-state kinetic analysis of E. coli GlnRS (35, 36, 44) and allows use of very high concentrations of the (unlabeled) amino acid when required. It was found that unmodified transcripts of all M. mazei tRNACys isoacceptors are poor substrates for SepRS, with plateau aminoacylation levels (measured in reactions using a molar excess of enzyme) in the range of only 10–20% (Fig. 4A). This does not suffice for rigorous enzymological studies, particularly in an indirect aminoacylation pathway, where examination of a second enzyme requiring aminoacylated tRNA as substrate is also desired (2). However, we found that enzymatic methylation of tRNACys transcripts at the N1 moiety of nucleotide G37, using the M. jannaschii tRNA methylase Trm5 (28), increased plateau phosphoserylation levels to 65–80% (Fig. 4A and Table 2). Analysis of the crystal structure of A. fulgidus SepRS bound to tRNACys reveals that N1 of G37 is directly contacted by the backbone carbonyl group of the strictly conserved Gly443 (8). The m1G37 requirement for efficient aminoacylation then suggests that the interactions of G37 differ in the context of modified tRNA, and/or in the M. mazei complex (a homolog of M. jannaschi Trm5 exists in M. mazei). Because methylation by Trm5 employs the sulfur-containing cofactor S-adenosylmethionine, this requirement appears to link the unusual SepRS/Sep-tRNA:Cys-tRNA synthase pathway to more global aspects of sulfur metabolism in this methanogen (4). Using methylated transcripts, we also found that phosphothreonine could be aminoacylated to plateau levels of 25–30%, sufficient to determine steady-state kinetic parameters. SepRS attaches serine and glutamate to tRNACys to plateau levels of only 9–10%, too low for accurate measurement of steady-state parameters (Table 2). Initial rates of serylation and glutamylation of tRNACys are lower than for phosphothreonylation (data not shown).
FIGURE 4.

Steady-state aminoacylation by SepRS. A, time course for plateau phosphoserylation by SepRS in the absence or presence of m1G37 in tRNACys. The inset shows the imaged TLC plate at the respective time points. The percentage of tRNA that is aminoacylated (plateau value) is determined directly by the ratio of intensities for Sep-AMP and AMP. B, dependence of reaction rate on amino acid concentration, used to derive kcat and Km for the cognate SepRS reaction. C, the active site of A. fulgidus SepRS showing amino acids that interact directly with the substrate phosphoserine (8).
Measurement of kcat and Km for phosphoserylation of M. mazei tRNACys2 by SepRS shows that Km for phosphoserine is 6-fold lower than observed in ATP-PPi exchange, suggesting that SepRS undergoes a conformational change upon tRNA binding that improves the complementarity of the amino acid binding pocket (Fig. 4B and Table 2). Further, Km for phosphothreonine is increased by 4-fold in the two-step aminoacylation reaction as compared with activation alone, indicating that the presence of tRNA also decreases the complementarity for phosphothreonine. The kcat of 0.45 s–1 measured for cognate phosphoserylation is 5-fold higher than that reported for heterologous aminoacylation of an unmodified M. jannaschii tRNACys transcript by M. maripaludis SepRS and is similar to values for cognate aminoacylation reactions measured for other tRNA synthetases, such as E. coli CysRS (46, 47). Overall discrimination against phosphothreonine at the level of kcat/Km is 1.7 × 104-fold, and the low plateau aminoacylation levels and slower initial rates of aminoacylation for glutamate and serine strongly suggest that the discrimination against these (biologically relevant) amino acids will be even greater. Even serine and glutamate concentrations of up to 1.2 m, far above in vivo levels, failed to support more efficient aminoacylation. The discrimination observed is above the overall error rate of protein synthesis of 1 in 103 to 104, suggesting strongly that SepRS does not require an editing active site (48). This conclusion is underlined by the observation that, in several class II tRNA synthetases that do perform hydrolytic editing, the rate of noncognate amino acid activation is only 40–300-fold below that for cognate amino acids (49, 50). Finally, the sequence of SepRS does not align well with that of the β subunit of PheRS, which contains the B3/B4 domain known to be responsible for editing. Together, these data demonstrate clearly that SepRS is able to reject noncognate amino acids without the need for editing.
The crystal structure of A. fulgidus SepRS bound to tRNA and phosphoserine shows that the phosphate moiety of the amino acid substrate is contacted by several conserved residues, including a bidentate interaction with Asn325 (Fig. 4C) (8). A pair of conserved tyrosines, Tyr273 and Tyr274, form hydrogen bonds with the carboxyl and amino group, respectively. The only other contacts to phosphoserine are made by the strictly conserved residue Thr305. The side chain hydroxyl group of Thr305 donates a hydrogen bond to the carboxylate group of phosphoserine and approaches to within 3.9 Å of the side-chain β-carbon. Because Thr305 is positioned to exclude phosphothreonine on steric grounds, we reasoned that mutation to a smaller group might promote phosphothreonylation. To test this hypothesis, the equivalent Thr307 residue in M. mazei SepRS (supplemental Fig. 1) was mutated to serine. This alteration is most appropriate, because the increased pocket volume required to accommodate the additional methyl group on the phosphothreonine substrate is precisely matched by the removal of a methyl group from the enzyme; by contrast, mutation to alanine would probably have deleterious effects due to the removal of a hydrogen bond. Kinetic analysis of the purified T307S mutant indeed revealed a 3.2-fold improvement in kcat/Km for phosphothreonyl adenylate synthesis, as compared with wild-type SepRS (Table 1). However, the mutant was unable to transfer phosphothreonine to tRNACys at greater than 10% plateau levels (data not shown), significantly less than the 27% plateau observed for phosphothreonylation by wild-type SepRS (Table 2). These observations provide further evidence that tRNA binding to SepRS improves the capacity of the enzyme to discriminate among amino acids. From the perspective of genetic code engineering, these data also suggest that modification of the SepRS amino acid pocket to enhance the phosphothreonylation capacity of the enzyme will probably require much more extensive mutagenesis in and around the active site.
DISCUSSION
Half-of-the-sites Activity—Formation of phosphoseryl adenylate by M. mazei SepRS is characterized by rapid depletion of ATP followed by a slower steady-state rate. This is a feature of the overall aminoacylation reaction that is typical of both class I and class II tRNA synthetases, as first shown for the reaction catalyzed by B. stearothermophilus TyrRS (19). The burst provides information regarding the first catalytic turnover on the enzyme; in particular, the stoichiometry of product formation derived from extrapolation of the subsequent steady-state rate to the ordinate reveals the number of functional active sites (Fig. 2A). This analysis shows that only two of the four chemically equivalent SepRS active sites catalyze phosphoseryl adenylate formation. Since both gel filtration and nondenaturing mass spectrometry reveal that M. mazei SepRS is tetrameric, it follows that the enzyme exhibits half-of-the-sites activity with respect to amino acid activation. Half-of-the-sites activity for the adenylate formation reaction has previously been demonstrated for both TyrRS and TrpRS from B. stearothermophilus (19). Both of these class I tRNA synthetases are homodimers that catalyze (at least under some conditions), synthesis of approximately 1 mol of aminoacyl adenylate. It has also been shown that the class II M. jannaschii ProRS and E. coli HisRS homodimers rapidly synthesize only 1 mol of aminoacyl adenylate per dimer (51–53). Our data now extend the half-of-the-sites reactivity phenomenon to also encompass a homotetrameric enzyme; thus, the phenomenon appears to be generally present in multimeric tRNA synthetases of both classes. The only other homotetrameric tRNA synthetase that has been extensively studied is E. coli AlaRS (7), but no information regarding the stoichiometries of adenylate synthesis or tRNA binding is available for that enzyme.
tRNA binding measured by quenching of intrinsic tryptophan fluorescence shows that the SepRS homotetramer binds two tRNA molecules. Although increasing the stoichiometry of tRNA·SepRS monomers above 1:2 does reveal additional quenching, the very sharp break at this point in the titration strongly suggests that the additional interactions are nonnative and most likely represent formation of nonspecific higher order aggregates (Fig. 2C). Binding of two tRNAs by SepRS appears consistent with half-of-the-sites reactivity in formation of phosphoseryl adenylate, assuming, of course, that the tRNAs are particularly oriented such that their reactive 3′-acceptor ends are positioned in the same two active sites that also adopt catalytically active conformations in the first half-reaction. The essential enigmas, however, are the same in SepRS as were recognized previously with TyrRS. By what mechanism are half of the active sites rendered inactive despite their chemical identity with those that do function, and what, if any, is the biological rationale for this phenomenon? Given the chemical identity of the four subunits in SepRS, it is not surprising that, although the crystal structure of the A. fulgidus enzyme reveals only two bound tRNAs, docking of two additional tRNAs to produce a symmetric particle with four copies of each macromolecule is easily achieved (8).
The data presented in Fig. 2 provide an essential foundation to examine the catalytic cycle of SepRS in detail. With respect to the phenomenon of half-of-the-sites activity, several observations made in the studies of the class I homodimeric TyrRS provide a framework that may be helpful in considering how the mechanism may function in the new homotetramer context. Detailed study of Bacillus stearothermophilus TyrRS showed that the enzyme also exhibits half-of-the-sites activity with respect to tRNATyr aminoacylation (40, 54). However, it was further demonstrated that a second molecule of tyrosyl adenylate can be formed on the enzyme but that accumulation is very slow, with a half-life some 6000-fold longer than for formation of the first molecule (55). Crystal structures of B. stearothermophilus TyrRS bound to tyrosine and of T. thermophilus TyrRS bound to tRNATyr and tyrosinol reflect the inherent capacity of the enzyme to bind substrates stoichiometrically over the long periods required for crystallization, since these studies revealed symmetrical binding of both the small ligands and tRNA to both subunits (56, 57). In contrast, the A. fulgidus SepRS crystal structure bound to phosphoserine and tRNACys shows that amino acid and tRNA are each bound to only two of the four subunits (8). Thus, in this case, the x-ray structure appears to reflect the half-of-the-sites activity for phosphoseryl adenylate formation shown here. Further detailed enzymological studies will be required to establish whether SepRS is capable of binding or catalyzing the formation of stoichiometric quantities of substrate and product under different conditions and time scales, whether the apparent half-of-the-sites activity might arise in part from distinct subpopulations of fully active and inactive enzyme, and whether the phenomenon extends as well to the aminoacylation of tRNACys, as predicted by the structure. Of course, as mentioned above, it is important to recall that the crystal structure that is available is of a nonproductive complex; further efforts in determining the structure of functional complexes are also required. It is instructive also to note that the cocrystal structure of TyrRS, although symmetrical in tRNA binding, does not reveal ordered docking of the 3′-CCA termini into the two active sites (57). Indeed, Cusack and co-workers (57) have suggested that a crystal of an asymmetric form of the TyrRS dimer with only one bound tRNA may be required to observe the 3′-end of the tRNA functionally bound in the active site.
The half-of-the-sites activity in TyrRS has been characterized as an example of extreme negative cooperativity; binding of tyrosine to one subunit induces a conformational change that destroys the symmetry of the homodimer (58). Although this proposed mechanism suggests that the enzyme is symmetric in the unliganded state, later studies using heterodimers, in which one subunit is truncated and unable to bind tRNATyr, indicated instead that the wild-type enzyme is inherently asymmetric even in the absence of any bound substrate (39). An inherent structural asymmetry may also be a property of SepRS enzymes. The crystal structure of M. maripaludis SepRS, which crystallized with the full tetramer in the asymmetric unit, showed that the enzyme possesses only approximate 222 point group symmetry (9). The core regions of each monomer, which include the central canonical class II catalytic domain, obey the symmetry much more strictly than do several peripheral domains comprising the N terminus, C terminus, and an inserted domain not present in PheRS. Overall, the structure is highly interdigitated, with numerous points of contact among the four subunits. This gives rise to substantially more potential for structural asymmetry than may be present in homodimers such as TyrRS, ProRS, and HisRS. The asymmetry at the level of intersubunit associations then provides a basis for divergence in the structures of the active sites, even in the unliganded state. Because the homodimeric and homotetrameric tRNA synthetases bind the same classes of substrates and catalyze stereochemically equivalent reactions, the discovery of half-of-the-sites activity in SepRS opens a further opportunity for detailed comparative investigation of the structural origins of the phenomenon. Half-of-the-sites activity provides an essential basis for a proposed detailed catalytic model for HisRS, in which the two subunits alternately bind tRNA, and may be similarly crucial to the catalytic cycles of tetrameric enzymes (53).
Amino Acid Discrimination by SepRS—The observation that SepRS discriminates against noncognate amino acids to a greater extent in the overall aminoacylation reaction, as compared with adenylate synthesis alone, demonstrates that tRNA binding enhances the capacity of the enzyme to select the specific substrate. This phenomenon has been observed in other tRNA synthetases as well; e.g. E. coli glutaminyl-tRNA synthetase discriminates against glutamate by 107-fold in tRNA aminoacylation but only by 104-fold in synthesis of glutaminyl adenylate (45). A likely explanation for these observations is that tRNA binding alters the conformation of the amino acid binding pocket not only to render it more complementary toward the cognate substrate but also less hospitable for the noncognate ligand.
The level of discrimination observed in both the adenylate synthesis and aminoacylation reactions is high enough to allow us to predict with confidence that SepRS does not require an editing activity to hydrolyze misactivated adenylate or misacylated tRNACys. The crystal structure of A. fulgidus SepRS bound to phosphoserine provides a structural basis for the observed specificity against serine and glutamate (Fig. 4C). All three nonesterified oxygens of the side-chain phosphate group accept hydrogen bonds from enzyme moieties, with a total of four hydrogen bonds made in all (8). Serine would not be capable of making any of these contacts, whereas glutamate would be able to form at most two or three of the four. In addition to possessing only two oxygen atoms, the shape and charge distribution of the Glu carboxylate also substantially differ from phosphate. Because neither Ser nor Glu can be easily excluded from the active site on steric grounds, the most likely basis for the observed selectivity is that the additional contacts possible with phosphate serve to fix the orientation of the attacking α-carboxylate oxygen nucleophile with respect to the α-phosphate of ATP. The absence of some or all of these contacts might lead to increased mobility or to an alternative binding mode not conducive to proper juxtaposition of the reactive substrate groups.
Finally, it is of interest to note that, although phosphothreonine is not a cellular metabolite against which discrimination is required and possesses only an additional methyl group as compared with phosphoserine, it is nonetheless selected against by a factor of over 104-fold in aminoacylation (Table 2). Further, the modest 3-fold improvement in synthesis of phosphothreonyl adenylate afforded by the space-creating T307S mutation is obviated in the second step of the reaction, providing another example of how tRNA binding enhances amino acid selectivity in this system. There are no other positions in the amino acid pocket that suggest themselves as evident targets of rational mutagenesis to confer phosphothreonylation activity or indeed to alter the specificity in favor of any other amino acid. Although it has been proposed that the tRNA synthetase subclass IIc members should be particularly favorable for protein engineering experiments to alter amino acid specificity (11), we would suggest instead that analysis of the SepRS cocrystal structure bound to phosphoserine indicates otherwise. In general, the alteration of substrate specificity in complex oligomeric induced fit enzymes that modify nucleic acids with high sequence selectivity is not likely to be achieved by mutation solely in the local region of the substrate binding cleft.
Supplementary Material
Acknowledgments
We are grateful to Nick Dupuis and Professor Michael T. Bowers for collection and analysis of mass spectrometry data. M. T. B. thanks Waters Corp. for donation of the Synapt prototype instrument to the Bowers research group and the National Science Foundation (Grant CHE-0503728) for funding of the mass spectrometry laboratory.
This work was supported, in whole or in part, by National Institutes of Health Grants GM63713 (to J. P.) and GM066267 (to Y. M. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
Footnotes
The abbreviations used are: SepRS, phosphoseryl-tRNA synthetase; PheRS, phenylalanyl-tRNA synthetase; GlyRS, glycyl-tRNA synthetase; AlaRS, alanyl-tRNA synthetase; βME, β-mercaptoethanol; DTT, dithiothreitol.
References
- 1.Ibba, M., and Söll, D. (2004) Genes Dev. 18 731–738 [DOI] [PubMed] [Google Scholar]
- 2.Sauerwald, A., Zhu, W., Major, T., Roy, H., Palioura, S., Jahn, D., Whitman, W. B., Yates, J. R., III, Ibba, M., and Söll, D. (2005) Science 307 1969–1972 [DOI] [PubMed] [Google Scholar]
- 3.Tumbula, D. L., Becker, H. D., Chang, W. Z., and Söll, D. (2000) Nature 407 106–110 [DOI] [PubMed] [Google Scholar]
- 4.O'Donoghue, P., Sethi, A., Woese, C. R., and Luthey-Schulten, Z. A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 19003–19008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanke, T., Bartmann, P., Hennecke, H., Kosakowski, H. M., Jaenicke, R., Holler, E., and Bock, A. (1974) Eur. J. Biochem. 43 601–607 [DOI] [PubMed] [Google Scholar]
- 6.Ostrem, D. L., and Berg, P. (1974) Biochemistry 13 1338–1348 [DOI] [PubMed] [Google Scholar]
- 7.Putney, S. D., Sauer, R. T., and Schimmel, P. (1981) J. Biol. Chem. 256 198–204 [PubMed] [Google Scholar]
- 8.Fukunaga, R., and Yokoyama, S. (2007) Nat. Struct. Mol. Biol. 14 272–279 [DOI] [PubMed] [Google Scholar]
- 9.Kamtekar, S., Hohn, M. J., Park, H-S., Schnitzbauer, M., Sauerwald, A., Soll, D., and Steitz, T. A. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 2620–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srinivasan, G., James, C. M., and Krzycki, J. (2002) Science 296 1459–1462 [DOI] [PubMed] [Google Scholar]
- 11.Kavran, J. M., Gundllapalli, S., O'Donoghue, P., Englert, M., Soll, D., and Steitz, T. A. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 11268–11273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arnez, J. G., Dock-Bregeon, A. C., and Moras, D. (1999) J. Mol. Biol. 286 1449–1459 [DOI] [PubMed] [Google Scholar]
- 13.Xie, W., Nangle, L. A., Zhang, W., Schimmel, P., and Yang, X. L. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 9976–9981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swairjo, M. A., Otero, F. J., Yang, X. L., Lovato, M. A., Skene, R. J., McRee, D. E., Ribas de Pouplana, L., and Schimmel, P. R. (2004) Mol. Cell 13 829–841 [DOI] [PubMed] [Google Scholar]
- 15.Goldgur, Y., Mosyak, L., Reshetnikova, L., Ankilova, V., Khodyreva, S., Lavrik, O., and Safro, M. (1997) Structure 5 59–68 [DOI] [PubMed] [Google Scholar]
- 16.Mosyak, L., Reshetnikova, L., Goldgur, Y., Delarue, M., and Safro, M. (1995) Nat. Struct. Biol. 2 537–547 [DOI] [PubMed] [Google Scholar]
- 17.Shiba, K., Schimmel, P., Motegi, H., and Noda, T. (1994) J. Biol. Chem. 269 30049–30055 [PubMed] [Google Scholar]
- 18.Mosyak, L., and Safro, M. (1993) Biochimie (Paris) 75 1091–1098 [DOI] [PubMed] [Google Scholar]
- 19.Fersht, A. R., Ashford, J. S., Bruton, C. J., Jakes, R., Koch, G. L., and Hartley, B. S. (1975) Biochemistry 14 1–4 [DOI] [PubMed] [Google Scholar]
- 20.Jasin, M., Regan, L., and Schimmel, P. (1983) Nature 306 441–447 [DOI] [PubMed] [Google Scholar]
- 21.Ribas de Pouplana, L., and Schimmel, P. (1997) Biochemistry 36 15041–15048 [DOI] [PubMed] [Google Scholar]
- 22.Hohn, M. J., Park, H.-S., O'Donoghue, P., Schnitzbauer, M., and Soll, D. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 18095–18100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komatsoulis, G. A., and Abelson, J. (1993) Biochemistry 32 7435–7444 [DOI] [PubMed] [Google Scholar]
- 24.Ehlers, C., Weidenbach, K., Deppenmeier, U., Metcalf, W. W., and Schmitz, R. A. (2005) Mol. Genet. Genomics 273 290–298 [DOI] [PubMed] [Google Scholar]
- 25.Deppenmeier, U., Johann, A., Hartsch, T., Merkl, R., Schmitz, R. A., Martinez-Arias, R., Henne, A., Wiezer, A., Bäumer, S., Jacobi, C., Brüggemann, H., Lienard, T., Christmann, A., Bömeke, M., Steckel, S., Bhattacharyya, A., Lykidis, A., Overbeek, R., Klenk, H.-P., Gunsalus, R. P., Fritz, H.-J., and Gottschalk, G. (2002) J. Mol. Microbiol. Biotechnol. 4 453–461 [PubMed] [Google Scholar]
- 26.Maeder, D. L., Anderson, I., Brettin, T., Bruce, D., Gilna, P., Han, C. S., Lapidus, A., Metcalf, W. W., Saunders, E., Tapia, R., and Sowers, K. R. (2006) J. Bacteriol. 188 7922–7931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sherlin, L. D., Bullock, T. L., Nissan, T. A., Perona, J. J., Lariviere, F. J., Uhlenbeck, O. C., and Scaringe, S. A. (2001) RNA 7 1671–1678 [PMC free article] [PubMed] [Google Scholar]
- 28.Hou, Y. M., Li, Z., and Gamper, H. (2006) Nucleic Acids Res. 34 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christian, T., Evilia, C., Williams, S., and Hou, Y.-M. (2004) J. Mol. Biol. 339 707–719 [DOI] [PubMed] [Google Scholar]
- 30.Chen, Z., and Ruffner, D. E. (1996) BioTechniques 21 820–822 [DOI] [PubMed] [Google Scholar]
- 31.Pringle, S. D., Giles, K., Wildgoose, J. L., Williams, J. P., Slase, S. E., Thalassinos, K., Bateman, R. H., Bowers, M. T., and Scrivens, J. H. (2007) Int. J. Mass Spectrom. 261 1–12 [Google Scholar]
- 32.Giles, K., Pringle, S. D., Worthington, K. R., Little, D., Wildgoose, J. L., and Bateman, R. H. (2004) Rapid Commun. Mass Spectrom. 18 2401–2414 [DOI] [PubMed] [Google Scholar]
- 33.Hernandez, H., and Robinson, C. V. (2007) Nat. Prot. 2 715–726 [DOI] [PubMed] [Google Scholar]
- 34.Lakowicz, J. R. (1999) Principles of Fluorescence Spectroscopy, pp. 53–55, Kluwer Academic/Plenum Publishers, New York
- 35.Uter, N. T., Gruic-Sovulj, I., and Perona, J. J. (2005) J. Biol. Chem. 280 23966–23977 [DOI] [PubMed] [Google Scholar]
- 36.Bullock, T. L., Uter, N., Nissan, T. A., and Perona, J. J. (2003) J. Mol. Biol. 328 395–408 [DOI] [PubMed] [Google Scholar]
- 37.Wolfson, A. D., and Uhlenbeck, O. C. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 5965–5970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fasiolo, F., Ebel, J. P., and Lazdunski, M. (1977) Eur. J. Biochem. 73 7–15 [DOI] [PubMed] [Google Scholar]
- 39.Ward, W. H., and Fersht, A. R. (1988) Biochemistry 27 1041–1049 [DOI] [PubMed] [Google Scholar]
- 40.Ward, W. H., and Fersht, A. R. (1988) Biochemistry 27 5525–5530 [DOI] [PubMed] [Google Scholar]
- 41.Liu, C., Gamper, H., Shtivelband, S., Hauenstein, S., Perona, J. J., and Hou, Y. M. (2007) J. Mol. Biol. 367 1063–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lipman, R. S., Chen, J., Evilia, C., Vitseva, O., and Hou, Y.-M. (2003) Biochemistry 42 7487–7496 [DOI] [PubMed] [Google Scholar]
- 43.Roy, H., Ling, J., Irnov, M., and Ibba, M. (2004) EMBO J. 23 4639–4648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uter, N. T., and Perona, J. J. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 14396–14401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gruic-Sovulj, I., Uter, N. T., Bullock, T. L., and Perona, J. J. (2005) J. Biol. Chem. 280 23978–23986 [DOI] [PubMed] [Google Scholar]
- 46.Hou, Y.-M., Westof, E., and Giege, R. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 6776–6780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sankaranarayanan, R., and Moras, D. (2001) Acta Biochim. Pol. 48 323–335 [PubMed] [Google Scholar]
- 48.Jakubowski, H., and Goldman, E. (1992) Microbiol. Rev. 56 412–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beebe, K., Ribas De Pouplana, L., and Schimmel, P. (2003) EMBO J. 22 668–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roy, H., Ling, J., Alfonzo, J., and Ibba, M. (2005) J. Biol. Chem. 280 38186–38192 [DOI] [PubMed] [Google Scholar]
- 51.Ambrogelly, A., Kamtekar, S., Stathopoulos, C., Kennedy, D., and Soll, D. (2005) FEBS Lett. 579 6017–6022 [DOI] [PubMed] [Google Scholar]
- 52.Guth, E., Connolly, S. H., Bovee, M., and Francklyn, C. S. (2005) Biochemistry 44 3785–3794 [DOI] [PubMed] [Google Scholar]
- 53.Guth, E., and Francklyn, C. S. (2007) Mol. Cell 25 531–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bedouelle, H. (1990) Biochimie (Paris) 72 589–598 [DOI] [PubMed] [Google Scholar]
- 55.Mulvey, R. S., and Fersht, A. R. (1977) Biochemistry 16 4005–4013 [DOI] [PubMed] [Google Scholar]
- 56.Brick, P., and Blow, D. M. (1987) J. Mol. Biol. 194 287–297 [DOI] [PubMed] [Google Scholar]
- 57.Yaremchuk, A., Kriklivyi, I., Tukalo, M., and Cusack, S. (2002) EMBO J. 21 3829–3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fersht, A. R., Mulvey, R. S., and Koch, G. L. E. (1975) Biochemistry 14 13–18 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.