

Abstract
The development of therapeutic strategies to inhibit reactive oxygen species (ROS)-mediated damage in blood vessels has been limited by a lack of specific targets for intervention. Targeting ROS-mediated events in the vessel wall is of interest, because ROS play important roles throughout atherogenesis. In early atherosclerosis, ROS stimulate vascular smooth muscle cell (VSMC) growth, whereas in late stages of lesion development, ROS induce VSMC apoptosis, causing atherosclerotic plaque instability. To identify putative protective genes against oxidative stress, mouse aortic VSMC were infected with a retroviral human heart cDNA expression library, and apoptosis was induced in virus-infected cells by 2,3-dimethoxy-1,4-naphthoquinone (DMNQ) treatment. A total of 17 different, complete cDNAs were identified from the DMNQ-resistant VSMC clones by PCR amplification and sequencing. The cDNA encoding PP1cγ1 (catalytic subunit of protein phosphatase 1) was present in several independent DMNQ-resistant VSMC clones. DMNQ increased mitochondrial ROS production, caspase-3/7 activity, DNA fragmentation, and decreased mitochondrial transmembrane potential in VSMC while decreasing PP1cγ1 activity and expression. Depletion of PP1cγ1 expression by short hairpin RNA significantly enhanced basal as well as DMNQ-induced VSMC apoptosis. PP1cγ1 overexpression abrogated DMNQ-induced JNK1 activity, p53 Ser15 phosphorylation, and Bax expression and protected VSMC against DMNQ-induced apoptosis. In addition, PP1cγ1 overexpression attenuated DMNQ-induced caspase-3/7 activation and DNA fragmentation. Inhibition of p53 protein expression using small interfering RNA abrogated DMNQ-induced Bax expression and significantly attenuated VSMC apoptosis. Together, these data indicate that PP1cγ1 overexpression promotes VSMC survival by interfering with JNK1 and p53 phosphorylation cascades involved in apoptosis.
Enhanced reactive oxygen species (ROS)3 generation plays an important role in the proliferation, migration, or apoptosis of vascular smooth muscle cells (VSMC), all of which have been implicated in the pathophysiology of vascular diseases, including atherosclerosis. VSMC proliferation and migration are important in the development of atherosclerotic lesions, and VSMC apoptosis is a histologic hallmark of advanced atherosclerosis (1). Additionally, human VSMC isolated from coronary plaques are more susceptible to apoptosis than VSMC isolated from normal arteries (2). Apoptosis of VSMC in atherosclerotic plaques is accompanied by numerous other events that increase the likelihood of plaque rupture. These include decreases in collagen and extracellular matrix protein production, decreased fibrous cap thickness, accumulation of macrophages along the shoulder of the plaque, and increases in the size of the necrotic core and amount of cellular debris within the plaque (3). A better understanding of the mechanisms that regulate VSMC apoptosis could lead to the development of strategies to stabilize atherosclerotic plaques.
Identifying genes that could protect cells against oxidative stress
requires selection of an oxidant and a screening strategy. A number of
different approaches have been used to induce oxidative stress in cultured
cells and in vivo. The quinones are of particular interest. Quinones
are ubiquitous in nature and are also formed as metabolites from a variety of
drugs, environmental pollutants, and food derivatives by the action of
cytochrome P450 system (4). The
redox-cycling 2,3-dimethoxy-1,4-naphthoquinone (DMNQ) has been proposed as a
model quinone compound to study the role of ROS in cell toxicity and apoptosis
(5). It does not react with
free thiol groups and is nonalkylating and nonadduct-forming. One-electron
reduction of DMNQ by flavoenzymes, such as NADPH-cytochrome P450 reductase or
NADH-cytochrome b5 reductase, yields a semiquinone
radical, which then reacts with oxygen to form superoxide
(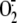 ) and, subsequently, the
dismutation product H2O2. An obligatory two-electron
reducing cytosolic flavoenzyme NADPH oxidoreductase 1, also known as
DT-diaphorase, can compete with one-electron reductases for DMNQ to produce a
hydroquinone, which can undergo autoxidation to yield
H2O2 and the parental quinone.
) and, subsequently, the
dismutation product H2O2. An obligatory two-electron
reducing cytosolic flavoenzyme NADPH oxidoreductase 1, also known as
DT-diaphorase, can compete with one-electron reductases for DMNQ to produce a
hydroquinone, which can undergo autoxidation to yield
H2O2 and the parental quinone.
Retroviral cDNA expression library screening has been used successfully to identify novel oncogenes (6) and modulators of apoptosis (7, 8). The packaging cell lines used in conjunction with retroviral vectors generate high concentrations of infectious virus particles necessary for the efficient infection of large cell populations, including that of primary cells, with a single copy of vector per cell (9). In addition, retroviral delivery systems have the ability to deliver very large libraries to infected cell populations. These properties make retroviral cDNA expression library screening an ideal strategy to identify genes that protect against oxidative stress.
Protein phosphorylation and dephosphorylation play an important regulatory role in apoptosis and other cellular processes (10-12). Protein phosphatase 1 (PP1), 2A (PP2A), 2B (PP2B), and 2C (PP2C) are the four major serine/threonine phosphatases present in eukaryotic cells (13). Of these four, PP1 and PP2A account for the majority of cellular phosphatase activity (13). The catalytic subunit of PP1 exists in four different isoforms (α, γ1, γ2, and δ), which are targeted to various subcellular compartments by association with distinct regulatory proteins (14, 15). The interaction of targeting proteins with the catalytic subunit also modulates substrate specificity, enabling unique and independent roles for the various isoforms in regulating discrete cellular processes (15).
Okadaic acid, a potent inhibitor of PP1 and PP2A, induces apoptosis in several cells, and this was attributed to its preferential inhibition of one or the other isoform depending on the cell type (16-19). Okadaic acid-induced epithelial cell apoptosis and inhibition of PP1 were correlated with increased expression of tumor suppressor gene p53 and proapoptotic gene Bax (19). Serine phosphorylation in both the carboxyl-terminal and N-terminal domains modulates DNA binding ability and transcriptional activity of p53 (20, 21). It has been shown recently that direct dephosphorylation of p53 at Ser15 and Ser37 by PP1 affects its stability and transcriptional and apoptotic activities (22, 23).
Among the kinases that regulate the p53 pathway are the c-Jun NH2-terminal kinase (JNK) group of mitogen-activated protein kinases. JNKs are activated in cells exposed to various environmental stresses (24), and reactive oxygen species (ROS) play an integral part in this activation (25). The 10 members of the JNK family are generated by alternative splicing of transcripts from Jnk1, Jnk2, and Jnk3 genes (24). Jnk1 and Jnk2 double knock-out cells are resistant to apoptosis induced by UV, anisomycin, or DNA damage (26). Proapoptotic proteins Bax and Bak are necessary for JNK-induced apoptosis, and Bax remains inactive in Jnk-deficient fibroblasts exposed to environmental stress (27). JNK is activated by dual phosphorylation at Thr183 and Tyr185 residues (24), which are dephosphorylated by dual specificity mitogen-activated protein kinase phosphatases (28).
In this study, we used retrovirus-mediated insertion of a human heart cDNA library into mouse aortic VSMC to create DMNQ-resistant cell clones. The clones contained 17 full-length cDNAs. We chose to investigate the role of PP1cγ1 (catalytic subunit of protein phosphatase 1) in oxidative stress resistance because the cDNA coding for this protein was present in several independent DMNQ-resistant VSMC clones and because of our continued interest in protein phosphorylation as a regulatory mechanism in atherosclerotic disease process in general and VSMC biology in particular. DMNQ treatment induced mitochondrial ROS production and decreased PP1cγ1 activity in VSMC. Overexpression of PP1cγ1 decreased DMNQ-induced phosphorylation of JNK1 and p53 (Ser15) and protected against DMNQ-induced apoptosis. Further, p53 shRNA transfection protected VSMC against DMNQ-induced apoptosis. Together, these results suggest that PP1cγ1 may regulate molecular mechanisms that mediate VSMC apoptosis in atherosclerosis and restenosis.
EXPERIMENTAL PROCEDURES
Materials—DMNQ and diphenyleneiodonium chloride (DPI) were obtained from Calbiochem. Rotenone, thenoyltrifluoroacetone, carbonyl cyanide m-chlorophenylhydrazone, oxypurinol, proadifen, phenelzine, indomethacin, nordihydroguaiaretic acid, leptin, and antimycin A were purchased from Sigma. MitoTracker Green FM, MitoSOX Red, and 2′,7′-dichlorofluorescein diacetate were purchased from Molecular Probes (Invitrogen). The antibodies used were anti-PP1cγ1, anti-p53, anti-Bax, and anti-JNK1 (Santa Cruz Biotechnology), anti-phospho-p53 (Ser15) (Cell Signaling Technology), and anti-β-actin (Sigma). Glutathione S-transferase-c-Jun-(1-79) recombinant protein was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). [γ-32P]ATP (6,000 Ci/mmol) was obtained from GE Healthcare.
Cell Culture—Aortic VSMC were isolated from 4-month-old male C57BL/6 mice as previously described by us (29). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS) as described previously (30). All experiments were conducted using VSMC between passages 4 and 11 that were growth-arrested by incubation in DMEM containing 0.1% FBS for 72 h.
Quiesced VSMC were treated with 60 μm DMNQ for 16 h. Single cell clones that survived the treatment were isolated by localized trypsinization using cloning cylinders (Sigma) and expanded using the Glasgow modification of Eagle's medium supplemented with 2 mm glutamine, 1 mm sodium pyruvate, 1× nonessential amino acids, 10% (v/v) fetal bovine serum, a 1:1000 dilution of β-mercaptoethanol stock solution (70 μl of β-mercaptoethanol in 20 ml of distilled water), and 500 units/ml of leukocyte-inhibitory factor (Chemicon).
Virus Production—A human heart cDNA expression library was constructed in a replication-defective Moloney murine leukemia virus-based vector, pCFB (Stratagene). The unidirectionally oriented cDNA library contained 1 × 106 primary clones and was amplified only once to ensure the best representation. Retrovirus was produced using the MBS mammalian transfection kit (Stratagene). In brief, the ecotropic virus packaging cell line Phoenix-Eco (Orbigen) was transfected with pCFB-human heart cDNA expression library or retroviral plasmid by the calcium phosphate-DNA co-precipitation method. Retroviruses secreted by transfected cells were collected from cell supernatant 24, 48, and 72 h after transfection and immediately applied to VSMC or 293T cells without any freezing and thawing steps.
The concentration of infectious particles was determined by exposing 293T cells to virus containing the lacZ reporter gene (pFB-Neo-LacZ; Stratagene). In brief, cells were seeded at a density of 2.5 × 105 cells/well in a 6-well plate, 16 h prior to virus infection. Infection/transduction was performed in the presence of 10 μg/ml DEAE-dextran. After 24 h of incubation at 37 °C, cells were stained for β-galactosidase expression using an in situ β-galactosidase staining kit (Stratagene). The number of blue-stained cells was estimated by light microscopy at ×200 magnification.
cDNA Library Screening in VSMC—VSMC (1.0 × 106) were seeded in 15-cm tissue culture dishes. Ten dishes (about 1 × 107 cells) were infected with 2 × 106 infectious particles of human heart cDNA expression library in pCFB retrovirus as described above. 20% infection was achieved as determined by staining for β-galactosidase activity in control cells.
Infected cells were incubated for 48 h at 37 °C to achieve maximal expression of genes (ViraPort XR Plasmid cDNA Library protocol; Stratagene). Then VSMC were treated with 60 μm DMNQ for 16 h. Single cell clones that survived DMNQ treatment were isolated as described above.
Isolation and Sequencing of Transduced cDNAs—Genomic DNA from 132 clones was isolated using the DNeasy tissue kit (Qiagen) and was used for PCR recovery of cDNA inserts. A 5′ Retro primer (5′-GGCTGCCGACCCCGGGGGTGG-3′) and 3′ Retro primer (5′-CGAACCCCAGAGTCCCGCTCA-3′) specific for the virus vector sequence flanking cDNA inserts were used for PCR synthesis of cDNAs. PCR was run for 33 cycles (15 s at 94 °C, 4 min at 68 °C) and a final extension for 10 min at 72 °C. The PCR products were purified using the QIAquick PCR purification kit (Qiagen) and sequenced using the above mentioned primers. The PCR products were analyzed on agarose gels to determine the length of the cDNAs.
Construction of Recombinant Viruses—To make retroviral and adenoviral constructs encoding PP1cγ1 cDNA, PP1cγ1 was subcloned from pEGFP-C1-PP1cγ1 (kindly provided by Dr. Laura Trinkle-Mulcahy (University of Dundee, Scotland)) into pQCXIP (BD Bioscienses) and pShuttle-CMV, respectively. The sequence of PP1cγ1 constructs was confirmed by restriction analysis and DNA sequencing. Retrovirus encoding PP1cγ1 cDNA was generated as described above. Adenovirus encoding PP1cγ1 cDNA was generated after homologous recombination of pShuttle-CMV containing PP1cγ1 cDNA with the adenoviral backbone plasmid pADEasy-1 in BJ5183 Escherichia coli (Stratagene). The recombinant (LE1/E3-deficient) adenoviruses were propagated by the transfection of human embryonic kidney 293 cells using Lipofectamine (Invitrogen). The virus was serially amplified and then purified using the ViraBind adenovirus purification kit (Cell Biolabs, Inc.). A control adenovirus contained an identical adenovirus backbone with green fluorescent protein cDNA (AdGFP).
Two mouse pSM2 retroviral shRNAs for p53 and one retroviral scrambled shRNA construct were purchased from Open Biosystems. The sense sequences of p53 shRNA were 5′-ACCAGTCTACTTCCCGCCATAA-3′ (RHS1764-9208347) and 5′-CCCACTACAAGTACATGTGTAA-3′ (RMM1766-98468519). Constructs were transfected into Phoenix-Eco cells for retrovirus packaging and amplification using the MBS mammalian transfection kit. Retrovirus containing supernatant was used to infect VSMC, and transduced cells were selected using puromycin (2.5 μg/ml)-containing medium. Successful suppression of p53 expression was confirmed by Western blot analysis.
Overexpression of PP1cγ1 Using the Adenoviral Infection System—Adenoviral infection of nearly confluent VSMC was performed at a multiplicity of infection of 100 in DMEM containing 2% FBS. After 16 h of incubation, the cells were quiesced in DMEM containing 0.1% FBS for 72 h.
Retroviral p53 and Pp1cγ1 shRNA Constructs and Infection of Mouse Aortic VSMC—Mouse retroviral shRNAmir against p53 and Pp1cγ1 was purchased from Open Biosystems. The scrambled shRNA encoded a 19-bp scrambled sequence (5'-GCGCGCTTTGTAGGATTCG) with no significant homology to any mouse gene and was cloned into pSUPER.retro.puro vector. Retroviral infection of VSMC was performed by spreading 3 ml of virus supplemented with 10 μg/ml DEAE-dextran on VSMC seeded in 100-mm plates. After 3 h of incubation, an additional 7.0 ml of DMEM containing 10% FBS was added, and the plates were incubated at 37 °C for 21 h. The infection medium was then replaced with fresh DMEM containing 10% FBS. For p53 and Pp1cγ1 shRNA expression, puromycin-resistant clones (selected with 2.5 μg/ml puromycin for 10 days) were expanded prior to their use in experiments.
Cell Viability Assay—VSMC viability after DMNQ treatment was determined by either crystal violet staining or the trypan blue exclusion method. In the former method, DMNQ-treated VSMC in 96-well plates were washed with phosphate-buffered saline and stained with 100 μl of crystal violet solution (0.5% (w/v) crystal violet, 1.5% (v/v) formaldehyde, and 1% (v/v) ethanol) for 30 min. After the wells were washed with water, stained cells were lysed with 1% (w/v) deoxycholate solution, and the absorbance was read at 560 nm using a microplate reader. Trypan blue exclusion was determined microscopically by scoring five fields of 100 cells each.
Intracellular ROS Measurement—VSMC (8 × 105) were seeded in 24-well glass bottom plates, grown for 48 h in DMEM containing 10% FBS, and quiesced for an additional 24 h in culture medium containing 0.1% FBS. Cells were then treated with Hanks' balanced salt solution containing the H2O2-sensitive fluorophore 2′,7′-dichlorofluorescin diacetate (5 μm) for 30 min at 37 °C in darkness. The cells were treated with fresh 2′,7′-dichlorofluorescin diacetate and DMNQ and incubated for another 30 min. For studying the effect of cellular oxidase inhibitors on DMNQ-induced ROS production, VSMC were pretreated with DPI, oxypurinol, proadifen, phenelzine, indomethacin, or nordihydroguaiaretic acid for 30 min before treatment with 2′,7′-dichlorofluorescein diacetate and DMNQ. The fluorescence of DCF was measured using a microplate reader at an excitation wavelength of 485 nm and emission wavelength of 530 nm. Then cells were lysed, and protein content was quantified.
Detection of Mitochondrial ROS—VSMC grown in glass bottom dishes were quiesced and treated with 10 μm DMNQ for 30 min. Cells were washed with Hanks' balanced salt solution and incubated with 5 μm MitoSOX Red and 1 μm MitoTracker Green FM (Molecular Probes) at 37 °C for 10 min. Excess stains were removed, and cells were imaged using Olympus FV500 confocal laser-scanning microscopy. MitoTracker Green FM was visualized at an excitation of 490 nm and an emission of 516 nm, whereas MitoSOX Red was visualized at an excitation of 560 nm and an emission of 600 nm. MitoTracker Green FM preferentially translocates to the mitochondria. MitoSOX Red accumulates in mitochondria and exhibits bright red fluorescence upon oxidation and subsequent binding to mitochondrial DNA.
Detection of Changes in Mitochondrial Transmembrane Potential—Changes in mitochondrial transmembrane potential were detected using the MitoCapture apoptosis detection kit (BioVision Research Products), which provides a fluorescence-based method for distinguishing between healthy and apoptotic cells. VSMC, either untreated or treated with 10 μm DMNQ for 15 h, were incubated with diluted MitoCapture solution at 37 °C in a 5% CO2 incubator for 20 min. The cells were washed with the incubation buffer three times and mounted with Vectashield mounting medium (Vector Laboratories). Cells were photographed by fluorescent microscopy.
Isolation of Mitochondria—Mitochondria were isolated from 80% confluent VSMC using a Pierce mitochondrial isolation kit according to the manufacturer's instructions with the addition of complete EDTA-free protease inhibitor mixture (Roche Applied Science).
Western Blot Analysis—Cells were lysed in radioimmune precipitation buffer (20 mm Tris-HCl, pH 7.6, 150 mm NaCl, 0.05 mm sodium fluoride, 1 mm EDTA, 1% Igepal, 0.05% sodium deoxycholate, 0.1% SDS, and protease inhibitors), and Western analysis was performed as described previously (30). Cell lysates containing 50 μg of protein were analyzed in Western blotting experiments.
Assessment of Apoptosis—For detection of apoptosis, growth-arrested VSMC were harvested after 16 h of treatment with 10 μm DMNQ. Histone-associated DNA fragmentation in cell lysates was determined using the cell death detection ELISAPLUS kit (Roche Applied Science) in accordance with the manufacturer's instructions.
A TUNEL apoptosis detection kit (Upstate Biotechnology, Inc.) was used to measure DNA fragmentation according to the manufacturer's protocol. Briefly, following treatment, the cells were fixed in 4% paraformaldehyde and permeabilized by incubating with 0.5% Tween 20 and 0.2% bovine serum albumin for 15 min. Then cells were incubated with a reaction mix containing biotin-dUTP and terminal deoxynucleotidyltransferase for 60 min. Transfer of biotin-dUTP to the free 3′-OH end of cleaved DNA was visualized by reaction with fluorescein-conjugated avidin (fluorescein isothiocyanate-avidin) for 30 min and photographed by fluorescence microscopy. Cells were counterstained with propidium iodide.
Apoptosis was also assessed by measuring caspase-3/7 activities using an Apo-ONE™ homogeneous caspase-3/7 assay kit (Promega). Briefly, growth-arrested VSMC were treated with 10 μm DMNQ for 6 h. The cells were lysed using bifunctional cell lysis/activity buffer, which contained a profluorescent caspase-3/7 consensus substrate, rhodamine 110 bis-N-benzyloxycarbonyl-l-aspartyl-l-glutamyl-l-valyl-aspartic acid amide (Z-DEVD-R110). After incubation at room temperature for 1 h, aliquots (150 μl) were transferred to a 96-well clear bottom plate. Fluorescence was measured at an excitation wavelength of 485 nm and an emission wavelength of 535 nm using a WALLAC 1420 Multilabel Counter.
Measurement of Pp1cγ1 mRNA Expression—Total RNA from VSMC that were either treated or untreated with DMNQ was extracted using the RNeasy Micro kit (Qiagen). Reverse transcription was performed with 1 μg of total RNA using the TaqMan reverse transcription reagents kit (Applied Biosystems). The Pp1cγ1 gene expression assay (catalog number Mm00849631-s1) was purchased from Applied Biosystems. Real time PCR was performed using the ABI PRISM 7900 HT Sequence Detection System and TaqMan PCR Master Mix according to the manufacturer's recommendations. Sequence Detection System software (version 2.1) (Applied Biosystems) was used for raw data analysis. Pp1cγ1 relative expression was calculated using the Relative Expression Software Tool (31) and by normalization to 18 S ribosomal expression.
Measurement of PP1cγ1 Activity—PP1cγ1 activity was measured using the fluorescence-based RediPlate 96 EnzChek serine/threonine phosphatase assay kit from Molecular Probes. VSMC were lysed in lysis buffer containing 20 mm Tris-Cl, pH 7.4, 132 mm NaCl, 10% glycerol, 1% Triton X-100, and a protease inhibitor mixture. Cell lysates containing 500 μg of protein were immunoprecipitated with anti-PP1cγ1 antibody overnight at 4 °C and then incubated with Protein A-Sepharose beads for another 2 h. The immunoprecipitates were washed three times with lysis buffer and resuspended in 50 μl of assay buffer (50 mm Tris-Cl, pH 7.0, 0.1 mm CaCl2, 2 mm dithiothreitol, 200 μm MnCl2, 125 μg/ml bovine serum albumin, and 0.05% Tween 20) containing 50 μm substrate, 6,8-difluoro-4-methyl-umbelliferyl phosphate. The reaction was performed at 37 °C for 30 min in the dark. 6,8-Difluoro-4-methyl-umbelliferyl fluorescence was measured at an excitation wavelength of 355 nm and an emission wavelength of 460 nm, using a WALLAC 1420 multilabel counter. Phosphatase activity was calculated using a 6,8-difluoro-4-methyl-umbelliferyl standard curve.
Measurement of JNK1 Activity—JNK1 activity was assayed as described elsewhere (32).
Statistical Analysis—All numerical data are expressed as mean ± S.E. Data were analyzed with one-way analysis of variance, and post hoc analysis was performed using the Newman-Keuls test. Statistical significance was accepted at p < 0.05.
RESULTS
Human Heart cDNA Library Screening—Genes that may confer resistance to oxidative stress were identified by screening mouse aortic VSMC infected with a replication-deficient retroviral cDNA library prepared from the human heart. Freshly prepared virus was used for library screening so as to ensure high transduction efficiency. The selection of an intracellular oxidant that is highly apoptotic (close to 100% cell death) is essential for efficient library expression screening for VSMC clones with an oxidative stress-resistant phenotype and for significantly decreasing false positives. In the present investigation, mouse aortic VSMC were incubated with apoptogenic DMNQ (33, 34) for 16 h at increasing concentrations. 10-15 VSMC clones in a 150-mm dish resisted 60 μm DMNQ.
Characterization of VSMC Clones Resistant to DMNQ-induced Apoptosis—To identify cDNAs with an antiapoptotic effect against DMNQ, 1 × 107 VSMC were transduced with 2 × 106 infectious units (2 times the size of the library) of retrovirus containing a human heart cDNA library. This strategy ensured that nearly the entire cDNA library is represented in the screening, and more than 90% of the transduced cells contained one cDNA.
A total of 132 cell clones remained viable after treatment with 60 μm DMNQ and were able to form colonies on enriched Glasgow modification of Eagle's medium. No viable colonies were recovered from the control-transduced cells. In order to identify retrovirally introduced cDNAs responsible for the DMNQ-resistant phenotype of VSMC clones, PCR analysis of genomic DNA was performed using primers specific for the retrovirus vector. We identified a total of 17 different full-length cDNAs in DMNQ-resistant VSMC clones (Table 1).
TABLE 1.
Genes representing full-length cDNAs isolated from DMNQ-resistant VSMC clones
| Gene | Accession number | Function |
|---|---|---|
| N-Acetylglucosamine-1-phosphotransferase, γ subunit, GNPTG | NM_032520 | Metabolism |
| Aldolase A, fructose-bisphosphate, transcript variant 1, ALDOA | NM_000034.2 | Metabolism |
| C9orf112 | DR004665 | Unclassified |
| Cardiac troponin I, type 3, TNNI3 | NM_000363.4 | Muscle and heart contraction |
| CDC42 effector protein 3, CDC42EP3 | NM_006449.3 | Cytoskeleton regulation |
| Coenzyme Q6 homolog monooxygenase, transcript variant 1, COQ6 | NM_182476.1 | Metabolism |
| Eukaryotic translation elongation factor 1 δ, transcript variant 2, EEF1D | NM_001960.2 | Translation |
| The 27-kDa heat shock protein 1, HSPB1 | NM_001540 | Stress resistance, antiapoptosis |
| Malate dehydrogenase, cytosolic, MDH1 | NM_005917.2 | Metabolism |
| MHC class I HLA-B, MHCI | NM_005514.5 | Immune response |
| MHC, class II, DM α, HLA-DMA | NM_006120.2 | Immune response |
| Myoglobin, transcript variant 1, MB | NM_005368.2 | Oxygen transport, antiapoptosis |
| NADH dehydrogenase (ubiquinone) flavoprotein 2, 24 kDa, NDUFV2 | NM_021074.2 | Mitochondrial electron transport |
| Proteasome subunit, β type 1, PSMB1 | NM_002793.2 | Protein catabolism |
| Protein phosphatase I, catalytic subunit, γ isoform, PP1CC | NM_002710.1 | Cell signaling |
| Translationally controlled tumor protein, TPT1 | NM_003295.2 | Antiapoptosis |
| Troponin T2, cardiac, transcript variant 3, TNNT2 | NM_001001431.1 | Muscle and heart contraction |
Four of the 17 genes were previously shown to afford protection against oxidative stress, whereas the remaining genes were not known to modulate either apoptosis or oxidative stress. For example, transcriptional down-regulation of aldolase and other glycolytic enzymes was reported under oxidative stress conditions (35). Further, increased expression of aldolase and enhanced resistance to apoptosis were observed in cancer cell lines with constitutive expression of HIF-1α (36). Although the primary function of Myo (myoglobin) is cellular O2 storage, it also acts as a ROS scavenger (37). Myo-/- mouse hearts had significantly higher levels of ROS than wild-type hearts under ischemia/reperfusion conditions. Translationally controlled tumor protein protects mammalian cells against chemically induced apoptosis and cytotoxicity, and its overexpression inhibited proapoptotic effector caspases (38). Similarly, overexpression of heat shock protein 27 (Hsp27) protects cisplatin-induced apoptosis of mouse fibroblasts (39) and human ovarian tumor cells (40). Interestingly, plasma Hsp27 levels were decreased in atherosclerotic patients compared with healthy subjects (41), and down-regulation of this protein decreases VSMC resistance to proteolytically induced apoptosis (42). We chose to further investigate PP1cγ1, because it was the most common cDNA expressed in VSMC resistant to DMNQ and because inhibitors of PP1cγ1 are proapoptotic (18, 19), indicating a potential role for PP1cγ1 in protecting against apoptosis.
Verification of the Protective Effect of PP1cγ1 Overexpression against DMNQ—DMNQ-resistant VSMC clones were not produced when cells were exposed to 60 μm DMNQ, suggesting that spontaneous development of DMNQ resistance does not occur in VSMC and is an unlikely explanation for DMNQ resistance in PP1cγ1-overexpressing cells. However, it was important to establish that PP1cγ1 overexpression in wild-type VSMC could create DMNQ resistance. Cell viability was reduced by ∼67 and 99% in adenoviral green fluorescent protein-expressing VSMC treated with 10 and 50 μm DMNQ, respectively. Adenoviral overexpression of PP1cγ1 increased cell viability to ∼55 and 28% in VSMC treated with 10 and 50 μm DMNQ (p < 0.001 for each concentration compared with adenoviral green fluorescent protein-expressing cells), respectively (Fig. 1). These data suggest that PP1cγ1 cDNA originally isolated from DMNQ-resistant VSMC clones was involved in interrupting the DMNQ-induced apoptotic signaling pathways in VSMC.
FIGURE 1.

Overexpression of PP1cγ1 increases VSMC viability after DMNQ treatment. VSMC infected with AdGFP or AdPP1cγ1 were quiesced for 3 days and treated with 10 or 50 μm DMNQ or vehicle for 16 h. Cell viability was determined by crystal violet staining. Data presented are mean ± S.E. of three independent experiments (* and **, p < 0.001 compared with respective DMNQ-treated AdGFP VSMC).
DMNQ Induces Mitochondrial ROS Production in VSMC—To characterize the protective effect of PP1cγ1 against DMNQ-induced apoptosis in VSMC, we sought to determine the site of DMNQ-induced intracellular ROS production. First, the effect of DPI, an inhibitor of NADPH oxidase and other flavoprotein enzymes on DMNQ-induced ROS generation was examined by measuring DCF fluorescence. Compared with basal conditions, 10 μm DMNQ increased ROS production to about 1.8-fold (Fig. 2A). DPI (10 μm) had no inhibitory effect on DMNQ-induced ROS production in VSMC. In addition, no difference in DCF fluorescence was observed between wild-type and p47phoxdeficient VSMC treated with DMNQ (data not shown). Next, we examined the effects of other cellular oxidase inhibitors on DMNQ-induced ROS production in VSMC. Oxypurinol (50 μm), an inhibitor of xanthine oxidase; proadifen (30 μm), an inhibitor of cytochrome P450; phenelzine (10 μm), an inhibitor of monoamine oxidase; indomethacin (10 μm), an inhibitor of cyclooxygenase; or nordihydroguaiaretic acid (10 μm), an inhibitor of lipoxygenase, failed to significantly decrease DMNQ-induced ROS production in VSMC.
FIGURE 2.

DMNQ-induced ROS production is not attenuated by inhibitors of endogenous oxidase systems. A, VSMC were pretreated with 10 μm DPI for 30 min and then treated with 10 μm DMNQ for 2 h. ROS generation was determined by measuring DCF fluorescence. Data presented are mean ± S.E. of three independent experiments (*, p < 0.001 compared with control). B, VSMC were pretreated with 50 μm oxypurinol, 30 μm proadifen, 10 μmphenelzine, 10 μm indomethacin, or nordihydroguaiaretic acid for 30 min and then treated with DMNQ for 2 h. Data presented are mean ± S.E. of three independent experiments (*, p < 0.001 compared with control).
To determine whether ROS production is enhanced in mitochondria of VSMC treated with DMNQ, we investigated the colocalization of MitoTracker Green FM, a mitochondria-selective dye, with MitoSOX Red, a superoxide-sensitive fluorescent dye (Fig. 3A). Compared with control cells, DMNQ-treated VSMC showed bright yellow/orange fluorescence in mitochondria, due to colocalization of MitoTracker Green and MitoSOX Red, indicating increased mitochondrial ROS production. To confirm DMNQ-induced increase in mitochondrial ROS production, VSMC were pretreated with 20 μm rotenone, an inhibitor of complex I; 10 μm thenoyltrifluoroacetone, an inhibitor of complex II; or 2 μm carbonyl cyanide m-chlorophenylhydrazone, an uncoupler of oxidative phosphorylation, and then treated with 10 μm DMNQ for 2 h (Fig. 3B). All of the mitochondrial electron transport chain inhibitors abrogated the effect of DMNQ on ROS production, confirming enhanced mitochondrial ROS generation in VSMC treated with DMNQ. Together, these data indicate that increased ROS production by DMNQ occurs predominantly in mitochondria of VSMC.
FIGURE 3.

DMNQ increases mitochondrial ROS production and disrupts mitochondrial transmembrane potential in VSMC. A, confocal laser-scanning microscopy showing colocalization of mitochondria-targeting fluorescent probe MitoSOX Red with the mitochondria-selective dye, MitoTracker Green. Growth-arrested VSMC were treated with 10 μm DMNQ for 30 min. The yellow/orange fluorescence indicates localization of ROS in mitochondria. B, growth-arrested VSMC were pretreated with 20 μm rotenone, 10 μm thenoyltrifluoroacetone, or 2 μm carbonyl cyanide m-chlorophenylhydrazone for 30 min and then treated with 10 μm DMNQ for 2 h. ROS generation was determined by measuring DCF fluorescence. Data presented are mean ± S.E. of three independent experiments (*, p < 0.001 compared with control). C, growth-arrested VSMC were either left untreated (1 and 2) or treated with 10 μm DMNQ for 16 h (3 and 4) and then incubated with diluted MitoCapture solution for 20 min. Cells were washed with incubation buffer and visualized using a fluorescent microscope. Mitochondrial membrane potential is indicated by the red fluorescence of MitoCapture aggregated in the mitochondria, and cells showing green fluorescence are apoptotic.
DMNQ Perturbs VSMC Mitochondrial Transmembrane Potential, Leading to Apoptosis—Enhanced mitochondrial ROS production could result in the induction of potentially deleterious mitochondrial permeability transition, and perturbation of mitochondrial transmembrane potential could lead to apoptosis (43). Therefore, we measured changes in mitochondrial transmembrane potential in VSMC treated with DMNQ, using the MitoCapture apoptosis detection kit. MitoCapture is a cationic dye that aggregates in the mitochondria of healthy cells, giving off a bright red fluorescence, whereas in apoptotic cells, it remains in the cytoplasm in its monomeric form, fluorescing green. The control cells showed only faint green fluorescence but contained foci of red fluorescence in the cells (Fig. 3C). DMNQ treatment for 15 h resulted in significant green fluorescence in the cytoplasm of VSMC and no red fluorescence, indicating an increase in mitochondrial permeability. Together, these data indicate that DMNQ induces VSMC apoptosis through mitochondrial ROS-dependent mechanisms.
PP1cγ1 Activity Is Attenuated by DMNQ, Other Inducers of Mitochondrial ROS, and H2O2—Because our data suggest that overexpression of PP1cγ1 protects VSMC against DMNQ-induced apoptosis, we investigated whether DMNQ treatment inhibits the activity of PP1cγ1. Using immunoprecipitation and a serine/threonine phosphatase assay kit from Molecular Probes, an ∼22% decrease in PP1cγ1 activity (p < 0.05 compared with control) was observed at 2 h after treatment with 10 μm DMNQ (Fig. 4A). PP1cγ1 activity decreased ∼75% at 8 h (p < 0.001 compared with control) after treatment with DMNQ. Next, we investigated whether other agonists that induce mitochondrial ROS production regulate PP1cγ1 activity in VSMC. PP1cγ1 activity was significantly decreased in VSMC at 4 and 0.5 h after treatment with 25 μg/ml antimycin A and 100 ng/ml leptin, respectively (p < 0.001) (Fig. 4, B and C). To confirm that ROS regulate activity, PP1cγ1 immunoprecipitated from VSMC lysates was incubated with 200 μm H2O2. The phosphatase activity was significantly inhibited 10 min after incubation with H2O2 (p < 0.001) and remained inhibited during the 60-min incubation (Fig. 4D). Together, these data indicate that increased mitochondrial ROS production inactivates PP1cγ1 activity.
FIGURE 4.

DMNQ, other inducers of mitochondrial ROS production, and H2O2 decrease PP1cγ1 activity. Growth-arrested VSMC were treated with 10 μm DMNQ (A), 25 μg/ml antimycin A (B), or 100 ng/ml leptin (C) for the indicated times, and cell lysates were immunoprecipitated with anti-PP1cγ1 antibody. Phosphatase activity was measured using 6,8-difluoro-4-methyl-umbelliferyl phosphate as substrate. Data presented are mean ± S.E. (n = 3) and representative of three separate experiments (*, p < 0.05; **, p < 0.001 compared with respective controls). D, PP1cγ1 was immunoprecipitated from VSMC lysates containing equal amounts of protein, immobilized on protein A-agarose beads, and incubated with 200 μm H2O2 for the indicated times. Phosphatase activity was measured as described above, and data presented are mean ± S.E. (n = 3) and representative of three separate experiments (*, p < 0.001 compared with control).
PP1cγ1 Is Abundant in VSMC Mitochondria—The observation that PP1cγ1 activity is regulated by mitochondrial ROS-dependent mechanisms raises the question of whether the phosphatase is present in VSMC mitochondria. Western blot analysis of subcellular fractions of VSMC homogenates demonstrated that PP1cγ1 is present both in cytosol and mitochondria, although it is more abundant in the latter (Fig. 5A).
FIGURE 5.

The subcellular localization and mechanism of expression of PP1cγ1 in DMNQ-treated VSMC. A, VSMC homogenates and mitochondrial and cytosolic fractions were analyzed by Western blotting (WB) with anti-PP1cγ1 antibody, and the blot was reprobed with anti-cytochrome c (Cyt c) antibody. B, growth-arrested VSMC were treated with 10 μm DMNQ for the indicated times, and the cell lysates were analyzed by Western blotting with anti-PP1cγ1 antibody or anti-β-actin antibody. C, densitometric analysis of PP1cγ1 protein levels (mean ± S.E., n = 3; *, p < 0.001 compared with control). D, growth-arrested VSMC were treated with 10 μm DMNQ for the indicated times, and the relative expression of PP1cγ1 mRNA was assayed by real time PCR. PP1cγ1 mRNA expression was normalized to 18 S rRNA (mean ± S.E., n = 3). E, growth-arrested VSMC were pretreated with 5 μm MG132 for 2 h and then treated with DMNQ for the indicated times. Cell lysates were analyzed by Western blotting with anti-PP1cγ1 antibody or anti-β-actin antibody. F, densitometric analysis of PP1cγ1 protein levels (mean ± S.E., n = 3; *, p < 0.001 compared with the respective control).
DMNQ Attenuates VSMC PP1cγ1 Expression by Inducing Proteasome-dependent Degradation—To further define the inhibitory effect of DMNQ on PP1cγ1, we investigated the steady-state protein levels of the phosphatase in VSMC by Western blot analysis of cell lysates. DMNQ treatment for 8 h decreased PP1cγ1 protein levels by ∼59% (p < 0.001) (Fig. 5, B and C). However, Pp1cγ1 mRNA levels in VMSC were unaffected by DMNQ treatment, indicating that decreased protein levels did not result from dysregulation of transcription (Fig. 5D). Next, we examined whether the low levels of PP1cγ1 protein in DMNQ-treated VSMC were caused by enhanced proteasome-mediated degradation. Pretreatment of VSMC with MG132, a proteasome inhibitor, partially but significantly (p < 0.001) attenuated the DMNQ-induced decrease in PP1cγ1 protein levels after 8 h of treatment (Fig. 5, E and F). This was observed in the absence of any change in Pp1cγ1 mRNA levels (data not shown). Together, these data indicate that the DMNQ-induced decrease in PP1cγ1 expression is, in part, mediated by the activation of the ubiquitin-proteasome pathway.
PP1cγ1 Overexpression Abrogates DMNQ-induced Cellular Signaling Pathways—To explore the possible mechanisms by which PP1cγ1 overexpression attenuates DMNQ-induced apoptosis of VSMC, we examined the activity of stress-activated protein kinase JNK1, expression of proapototic gene Bax, and expression and phosphorylation of tumor suppressor gene p53. A significant increase in JNK1 activity (p < 0.001) was observed using an immunocomplex kinase assay in control VSMC at 30 min after treatment with 10 μm DMNQ (Fig. 5, A and B). JNK1 activity in response to DMNQ treatment was not significantly different between control VSMC and cells infected with a control virus expressing enhanced green fluorescent protein (AdGFP). However, adenoviral overexpression of PP1cγ1 (AdPP1cγ1) abrogated DMNQ-induced activation of JNK1 in VSMC (Fig. 6, A and B). To determine whether the observed regulatory effect of PP1cγ1 overexpression on JNK1 activity was specific, we examined stimulation of ERK1/2 in VSMC treated with DMNQ in the presence and absence of PP1cγ1 overexpression. ERK1/2 phosphorylation was significantly enhanced in VSMC after DMNQ treatment (Fig. 6C), and adenoviral overexpression of PP1cγ1 had no inhibitory effect on DMNQ-induced ERK1/2 phosphorylation.
FIGURE 6.

PP1cγ1 overexpression abrogates DMNQ-induced activation of JNK1, phosphorylation and expression of p53, and expression of Bax in VSMC. A, control VSMC and VSMC infected with AdGFP or AdPP1cγ1 were either untreated or treated with 10 μm DMNQ for 30 min, and cell lysates containing equal amounts of protein were immunoprecipitated with anti-JNK1 antibodies, and JNK1 activity was measured by an immunocomplex kinase assay using glutathione S-transferase-c-Jun as a substrate (top). Aliquots of VSMC lysates immunoprecipitated with anti-JNK1 antibodies were analyzed by Western blotting with anti-JNK1 antibody (bottom). B, densitometric analysis of JNK1 activity (glutathione S-transferase-c-Jun phosphorylation). Data presented are mean ± S.E. (n = 3; *, p < 0.001 compared with their respective untreated cells). C, control, AdGFP, and AdPP1cγ1 VSMC were either left untreated or treated with 10 μm DMNQ for 10 min, and cell lysates were analyzed by Western blotting with anti-phosphospecific ERK1/2, anti-ERK1/2, or anti-PP1cγ1 antibodies. D, control, AdGFP, and AdPP1cγ1 VSMC were treated with DMNQ for the indicated times, and cell lysates were analyzed by Western blotting with anti-phosphospecific p53 (Ser15), anti-p53, anti-β-actin, or anti-PP1cγ1 antibodies. E, densitometric analysis of p53 phosphorylation (mean ± S.E., n = 3; * and **, p < 0.001 and p < 0.01, respectively, compared with their controls). F, densitometric analysis of p53 protein levels. G, cell lysates from untreated and DMNQ-treated VSMC were analyzed by Western blotting with anti-Bax, anti-PP1cγ1, or anti-β-actin antibodies. H, densitometric analysis of p53 protein levels (mean ± S.E., n = 3; *, p < 0.001 compared with untreated VSMC). IP, immunoprecipitation; WB, Western blot.
Western blot analysis of cell lysates using phosphospecific antibody showed a significant increase in phosphorylation at Ser15 of p53 in control VSMC (p < 0.001) after a 2-h treatment with DMNQ (Fig. 6, D (top) and E). Similarly, a significant increase in p53 Ser15 phosphorylation (p < 0.01) was observed in AdGFP VSMC treated with DMNQ compared with untreated AdGFP VSMC. Similar to its effect on JNK1, adenoviral overexpression of PP1cγ1 abrogated DMNQ-induced phosphorylation of p53 (Ser15) in VSMC. Interestingly, control and AdGFP VSMC tend to have higher p53 protein expression levels after DMNQ treatment compared with their respective untreated controls (Fig. 6, D (second panel) and F). However, the increase in p53 protein expression in these two groups was not statistically significant. The expression of p53 protein in AdPP1cγ1 VSMC did not change after DMNQ treatment.
Because Bax expression is regulated by both JNK1 (44) and p53 (45), we determined the effect of DMNQ on Bax levels in VSMC. An ∼10-fold increase in Bax protein level was observed in VSMC treated with 10 μm DMNQ for 6 h (Fig. 6, G and H). A similar increase in BAX expression was observed in AdGFP VSMC after DMNQ treatment. However, PP1cγ1 overexpression completely inhibited DMNQ-induced Bax protein level in VSMC. Together, these data indicate that high levels of PP1cγ1 protect VSMC against oxidative stress-induced apoptosis by specifically interfering with JNK1 and p53 activation.
PP1cγ1 Activity and Expression Regulate VSMC Apoptosis—Because suppression of PP1cγ1 activity and expression are correlated with DMNQ-induced VSMC apoptosis and its overexpression increases the viability of VSMC treated with DMNQ, it is necessary to determine the physiological effect of suppression of endogenous PP1cγ1 on VSMC survival. Okadaic acid, a potent protein phosphatase inhibitor, suppresses PP2A activity at 20 nm, whereas it inhibits both PP1 and PP2A activities at 100 nm concentration (19). Treatment of VSMC with 100 nm okadaic acid for 12 h significantly increased histone-associated DNA fragmentation in VSMC (p < 0.001 compared with control), whereas 20 nm okadaic acid had no such effect (Fig. 7A). Further, okadaic acid had an additive effect on DMNQ-induced VSMC apoptosis.
FIGURE 7.

Inhibition of endogenous PP1cγ1 activity/expression induces VSMC apoptosis, whereas its overexpression attenuates DMNQ-induced caspase-3/7 activity and DNA fragmentation. A, VSMC were pretreated with indicated concentrations of okadaic acid (OA) for 12 h and then treated with 10 μm DMNQ for 16 h. Cellular DNA fragmentation was measured using the cell death detection ELISAPLUS kit (mean ± S.E., n = 3 and representative of three separate experiments). B, VSMC stably expressing either retroviral scrambled shRNA construct or retroviral PP1cγ1 shRNA construct were lysed, and cell lysates were analyzed by either anti-PP1cγ1 or β-actin antibody. C, VSMC infected with scrambled shRNA or PP1cγ1 shRNA were either untreated or treated with 5 μm DMNQ for 16 h, and cell lysates were assayed for cellular DNA fragmentation as described above. Data presented are mean ± S.E., n = 12 (*, p < 0.001 compared with respective controls) and representative of three separate experiments. D, VSMC infected with either AdGFP or AdPP1cγ1 were either untreated or treated with 10 μm DMNQ for 8 h and were harvested in bifunctional cell lysis/activity buffer. The cell lysates were incubated with caspase-3/7 consensus substrate, Z-DEVD-R110. The reaction was measured at excitation and emission wavelengths of 485 and 535 nm, respectively. Data presented are mean ± S.E. (*, p < 0.001 compared with control; **, p < 0.001 compared with DMNQ-treated AdGFP VSMC) of three independent experiments. E, VSMC infected with either AdGFP or AdPP1cγ1 were either untreated or treated with DMNQ for 16 h, and cellular DNA fragmentation was measured (mean ± S.E., n = 3; *, p < 0.001 compared with control; **, p < 0.001 compared with DMNQ-treated AdGFP VSMC) and are representative of three separate experiments.
To complement the data indicating that the pharmacological inhibitor of PP1 activity induces apoptosis, we have used retroviral expression of Pp1cγ1 shRNA to specifically silence the expression of endogenous Pp1cγ1 in VSMC. Infection of mouse aortic VSMC with retroviral Pp1cγ1 shRNA construct decreased PP1cγ1 expression by ∼80% compared with cells infected with retroviral scrambled shRNA construct (Fig. 7B). Consistent with induction of apoptosis by okadaic acid, expression of Pp1cγ1 shRNA significantly enhanced histone-associated DNA fragmentation in VSMC compared with cells expressing scrambled shRNA (Fig. 7C). Further, depletion of PP1cγ1 expression by shRNA significantly enhanced DMNQ-induced VSMC apoptosis. In these experiments, we used DMNQ at a 5 μm concentration to better delineate the effect of suppression of endogenous PP1cγ1 on oxidative stress-induced VSMC apoptosis.
PP1cγ1 Overexpression Attenuates DMNQ-induced VSMC Apoptosis—The morphological and cellular changes associated with apoptosis are mediated by the activation of the caspase cascade (46, 47). To determine whether overexpression of PP1cγ1 attenuates apoptotic pathways, we examined DMNQ-induced caspase-3/7 activities in VSMC infected with AdGFP or AdPP1cγ1 constructs by measuring the release of the fluorescent R110 group from the substrate, Z-DEVD-R110. Although other caspases present in the whole cell lysates could cleave this peptide, caspase-3 and-7 preferentially cleave DEVD peptide from Z-DEVD-R110. Caspase-3/7 activity increased ∼5-fold (p < 0.001) in AdGFP VSMC treated with 10 μm DMNQ for 8 h (Fig. 7D). Adenoviral overexpression of PP1cγ1 significantly attenuated (p < 0.001) DMNQ-induced caspase-3/7 activity in VSMC compared with that in DMNQ-treated AdGFP VSMC (Fig. 7D).
Caspases function as effectors of apoptosis (48), and DNA fragmentation as well as nuclear morphological changes occur downstream of caspase activity (49, 50). An ∼8-fold increase in DNA fragmentation (p < 0.001) was observed in AdGFP VSMC after treatment with 10 μm DMNQ (Fig. 7E). Adenoviral overexpression of PP1cγ1 in VSMC significantly attenuated (p < 0.001) DMNQ-induced DNA fragmentation compared with that in AdGFP VSMC treated with DMNQ (Fig. 7E). Together, these data indicate that modulation of PP1cγ1 activity and expression regulates VSMC viability.
Silencing of p53 Expression by shRNA Abrogates DMNQ-induced Bax Protein Levels and Attenuates DMNQ-induced Apoptosis in VSMC—To confirm the role of p53 in mediating apoptosis induced by DMNQ through inhibition of PP1cγ1, we have used retroviral expression of p53 shRNA to silence the expression of the endogenous p53. Infection of mouse aortic VSMC with retroviral p53 shRNA construct decreased p53 expression by ∼85% compared with that in wild-type VSMC or cells infected with retroviral scrambled shRNA construct (Fig. 8A). We next assessed the effect of expression of p53 shRNA on Bax protein levels in VSMC treated with 10 μm DMNQ for 6 h. Expression of p53 shRNA abrogated DMNQ-induced increase in Bax protein levels compared with that in cells expressing scrambled shRNA (Fig. 8, B and C).
FIGURE 8.

p53 shRNA abrogates Bax expression and attenuates apoptosis in VSMC treated with DMNQ. A, VSMC stably expressing either retroviral scrambled shRNA construct or retroviral p53 shRNA construct were lysed, and cell lysates were analyzed by either anti-p53 or β-actin antibody. B, VSMC expressing retroviral scrambled RNA or retroviral p53 shRNA constructs were either left untreated or treated with 10 μm DMNQ for 6 h, and cell lysates were analyzed by Western blotting with either anti-Bax antibody or β-actin antibody. C, densitometric analysis of Bax protein levels. Data presented are mean ± S.E. (*, p < 0.01 compared with control) of three independent experiments. D, VSMC infected with either retroviral scrambled shRNA or retroviral p53 shRNA were either left untreated or treated with DMNQ for 16 h, and cellular DNA fragmentation was measured using the cell death detection ELISAPLUS kit. Data presented are mean ± S.E. (n = 3; *, p < 0.001 compared with control; **, p < 0.001 compared with DMNQ-treated retroviral scrambled RNA expressing VSMC) and are representative of three separate experiments. E, VSMC infected with either retroviral scrambled RNA or retroviral p53 shRNA were either left untreated or treated with 10 μm DMNQ for 16 h, and apoptosis was detected by TUNEL staining (green fluorescence). Cells were counterstained with propidium iodide.
To determine if the observed abrogation of Bax expression in response to DMNQ treatment in retroviral p53 shRNA-expressing cells resulted in decreased apoptosis, detection of nucleosomes in cell lysates was performed. DMNQ-induced DNA fragmentation was significantly decreased (p < 0.001) in VSMC expressing p53 shRNA compared with that in cells expressing scrambled shRNA (Fig. 8D). Consistent with this, VSMC infected with retroviral p53 shRNA showed low levels of TUNEL-positive cells (Fig. 8E). In contrast, VSMC infected with retroviral scrambled shRNA displayed highly elevated levels of TUNEL-positive cells. Taken together, these results indicate that PP1cγ1 expression regulates resistance to oxidantinduced apoptosis in VSMC by modulating p53-dependent cellular apoptotic pathways.
DISCUSSION
The present study was initiated to identify genes that impart resistance to oxidative stress in VSMC. Using retrovirus-mediated cDNA library expression screening, we identified PP1cγ1 and 16 other genes that protected VSMC against DMNQ toxicity. Our data illustrate that PP1cγ1 attenuates VSMC apoptosis by abrogating DMNQ-induced JNK1 activation, p53 phosphorylation, and Bax expression.
Activation of intracellular phosphorylation cascades leading to stimulation of mitogen-activated protein kinases and p53 influences the survival of cells subjected to oxidant injury (51-53). It is well established that inhibitors of PP1 or/and PP2A induce apoptosis in several cell types (19, 54-57). Our results demonstrate a significant decrease in PP1cγ1 activity and expression in VSMC treated with DMNQ. We attribute the decrease in PP1cγ1 activity to increased mitochondrial ROS production upon DMNQ treatment. This observation is supported by our data indicating that PP1cγ1 activity is also decreased in VSMC treated with antimycin and leptin, known inducers of mitochondrial ROS production (58, 59). The redoxsensitive regulation of PP1cγ1 activity is also evident from significant inhibition of its activity in the presence of H2O2 (Fig. 4D). It has been reported that other PP1 isoforms that possess highly homologous catalytic domains to PP1cγ1 undergo reactive site cysteine oxidation and inactivation by ROS (60).
Watanabe and Forman (5) reported that autoxidation of hydroquinone formed from two-electron reduction of DMNQ by NAD(P)H quinone oxidoreductase 1 yields most of the ROS generated in cells treated with low concentrations of DMNQ. In the present study, we did not observe any inhibition of DMNQ-induced ROS production in VSMC by inhibitors of various cellular oxidases (Fig. 2). In contrast, all electron transport chain inhibitors attenuated DMNQ-induced ROS production in VSMC, indicating enhanced mitochondrial ROS generation upon DMNQ treatment. Further support for increased mitochondrial ROS generation is evident from the bright yellow/orange fluorescence resulting from colocalization of MitoTracker Green and MitoSOX Red in the mitochondria of VSMC treated with DMNQ. Therefore, ROS production occurs predominantly in mitochondria of VSMC treated with DMNQ. Similarly, an increase in mitochondrial ROS generation was observed in pancreatic acinar cells treated with menadione, another naphthoquinone (61). However, redox cycling of DMNQ might induce ROS production in the cytosol (5). The presence of PP1cγ1 in both the cytosol and mitochondria (Fig. 5A) makes it a target of increased ROS levels in the cytosol and mitochondria of VSMC treated with DMNQ.
In addition to decreasing activity, DMNQ also decreased PP1cγ1 protein levels in VSMC. However, the decrease in PP1cγ1 protein levels did not result from decreased mRNA levels. Although the decrease in protein levels could result from decreased translation of mRNA, our data indicate that proteasome-mediated degradation partially contributes to lower PP1cγ1 levels observed at 8 h after DMNQ treatment. The ability of DMNQ to induce proteasome-dependent degradation of proteins has been reported recently (62).
The activation of caspases is a central step in the apoptosis signaling cascade, and caspases transduce regulatory upstream signals into the cell death execution machinery (46). Jänicke et al. (49) demonstrated that caspase-3 is required for DNA fragmentation that accompanies apoptosis. Caspase-3 activates an endonuclease, CAD, responsible for fragmentation of the DNA at the linker region between nucleosomes, by inactivating ICAD (DEF45), the inhibitor of CAD (50, 63). Consistent with previous observations (34, 61), DMNQ at a 10 μm concentration caused significant increases in caspase-3/7 activity and DNA fragmentation at 8 and 16 h after treatment. Caspase-3 was shown to disrupt the functions of complex I and II of the electron transport chain, resulting in loss of mitochondrial transmembrane potential and generation of ROS (64). Indeed, we observed a significant decrease in mitochondrial transmembrane potential of VSMC treated with DMNQ.
A major conclusion of our study is that PP1cγ1 has a prosurvival function in the response of VSMC to oxidative stress, and a decrease in its activity and expression results in apoptosis. This notion is supported by our observation that okadaic acid, at a concentration (100 nm) known to inhibit PP1 activity, significantly increased VSMC apoptosis. Further, DMNQ exerted an additive effect on VSMC apoptosis in the presence of okadaic acid. Importantly, suppression of PP1cγ1 expression significantly enhanced basal as well as DMNQ-induced apoptosis in VSMC. Then how could PP1cγ1 overexpression protect VSMC against DMNQ-induced oxidative stress?
One putative mechanism for increased survival of DMNQ-treated VSMC overexpressing PP1cγ1 could be interruption of the JNK1 cascade. JNK1 activation has been implicated in apoptosis (24) and was shown to be necessary for the release of cytochrome c from the mitochondria (26). Further, JNK1 is upstream of caspase-3 activation in apoptosis induced by several stressors (65, 66). Consistent with the observation of Ramachandran et al. (34), a significant increase in JNK1 activity was observed in DMNQ-treated VSMC in the present investigation. However, PP1cγ1 overexpression abrogated DMNQ-induced JNK1 activity. This finding is supported by a recent report that inhibition of PP1 leads to phosphorylation and activation of JNK1 in cancer cells, and PP1 directly inactivates JNK1 in vitro (67). It is noteworthy that suppression of JNK1 provides significant protection against apoptosis (25).
JNK1 is known to phosphorylate p53, resulting in p53 accumulation and activation as a transcriptional regulator (68, 69). p53, a master regulator of apoptosis, regulates cell death by two mechanisms: 1) by transcriptional regulation of expression of genes involved in apoptosis (70, 71); 2) by activation of Bax in mitochondria to antagonize Bcl-2 and Bcl-XL, the antiapoptotic genes (72). The transcriptional activity of p53 is regulated by several post-translational mechanisms, including phosphorylation/dephosphorylation, acetylation/deacetylation, ubiquitylation, sumoylation, and glycosylation (23, 73, 74). Phosphorylation and dephosphorylation impact both stability and function of p53 (73, 74). Consistent with DMNQ-induced apoptosis, we observed increased phosphorylation of p53 at Ser15 in DMNQ-treated VSMC lysates. The increase in p53 Ser15 phosphorylation was abrogated by overexpression of PP1cγ1. These results are supported by an observation that PP1 dephosphorylates p53 at Ser15 and Ser37 in in vitro and in vivo dephosphorylation assays, and PP1 promotes cell survival by negatively regulating the p53-dependent death pathway (23).
Consistent with the report of Sandau et al. (37), we observed increased Bax expression in VSMC treated with DMNQ. p53 was shown to up-regulate Bax expression by binding to the regulatory region of the gene (45). Alternatively, JNK can enhance the expression of Bax by either increasing AP-1 activity (75) or stabilizing the protein (44). Bax is located in the cytosol but inserts into mitochondrial membrane after an apoptotic signal (76). Again, JNK can promote the translocation of Bax to mitochondria through phosphorylation of 14-3-3 protein, a cytoplasmic anchor of Bax (77). It has been suggested that mitochondrial Bax associates with components of the mitochondrial permeability transition pore to cause loss of mitochondrial membrane potential. In fact, Bax translocation from the cytosol to the mitochondria was observed during DMNQ-induced apoptosis (78). Complementary to the reports that inhibition of PP1 induces the expression of p53 and Bax (19), PP1cγ1 overexpression abrogated DMNQ-induced Bax expression in VSMC. Consistent with this, PP1cγ1 overexpression attenuated DMNQ-induced caspase-3/7 activity and DNA fragmentation in VSMC. Further, p53 shRNA abrogated DMNQ-induced Bax expression and attenuated DMNQ-induced apoptosis, confirming that PP1cγ1 negatively regulates VSMC apoptosis cascades by modulating p53 phosphorylation.
In conclusion, our results demonstrate that PP1cγ1 attenuates DMNQ-induced VSMC apoptosis by abrogating JNK1 and p53 Ser15 phosphorylation. It is possible that dephosphorylation of other phosphoproteins by PP1cγ1 might play a role in the survival of VSMC under oxidative stress conditions. This finding supports further investigation of PP1cγ1 expression in restenosis and atherosclerosis, since VSMC apoptosis profoundly affects these arterial diseases.
This work was supported, in whole or in part, by National Institutes of Health Grants HL-57352 and AG 024282. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: ROS, reactive oxygen species; VSMC, vascular smooth muscle cells; DMNQ, 2,3-dimethoxy-1,4-naphthoquinone; JNK, c-Jun N-terminal kinase; DPI, diphenyleneiodonium chloride; DMEM, Dulbecco's modified Eagle's medium; DCF, 2′,7′-dichlorofluorescin; Z-DEVD-R110, bis-N-benzyloxycarbonyl-l-aspartyl-l-glutamyl-l-valyl-aspartic acid amide; FBS, fetal bovine serum; AdGFP, adenovirus backbone with green fluorescent protein cDNA; shRNA, short hairpin RNA; TUNEL, terminal dUTP nick-end labeling.
References
- 1.Delafontaine, P., Song, Y. H., and Li, Y. (2004) Arterioscler. Thromb. Vasc. Biol. 24 435-444 [DOI] [PubMed] [Google Scholar]
- 2.Bennett, M. R., Evan, G. I., and Schwartz, S. M. (1995) J. Clin. Invest. 95 2266-2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clarke, M. C., Figg, N., Maguire, J. J., Davenport, A. P., Goddard, M., Littlewood, T. D., and Bennett, M. R. (2006) Nat Med. 12 1075-1080 [DOI] [PubMed] [Google Scholar]
- 4.Monks, T. J., Hanzlik, R. P., Cohen, G. M., Ross, D., and Graham, D. G. (1992) Toxicol. Appl. Pharmacol. 112 2-16 [DOI] [PubMed] [Google Scholar]
- 5.Watanabe, N., and Forman, H. J. (2003) Arch. Biochem. Biophys. 411 145-157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitehead, I., Kirk, H., and Kay, R. (1995) Mol. Cell. Biol. 15 704-710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hitoshi, Y., Lorens, J., Kitada, S. I., Fisher, J., LaBarge, M., Ring, H. Z., Francke, U., Reed, J. C., Kinoshita, S., and Nolan, G. P. (1998) Immunity 8 461-471 [DOI] [PubMed] [Google Scholar]
- 8.Sandal, T., Ahlgren, R., Lillehaug, J., and Døskeland, S. O. (2001) Cell Death Differ. 8 754-766 [DOI] [PubMed] [Google Scholar]
- 9.Lorens, J. B., Sousa, C., Bennett, M. K., Molineaux, S. M., and Payan, D. G. (2001) Curr. Opin. Biotechnol. 12 613-621 [DOI] [PubMed] [Google Scholar]
- 10.Hunter, T., and Karin, M. (1992) Cell 70 375-387 [DOI] [PubMed] [Google Scholar]
- 11.Elledge, S. J. (1996) Science 274 1664-1672 [DOI] [PubMed] [Google Scholar]
- 12.Gjertsen, B. T., and Doskeland, S. O. (1995) Biochim. Biophys. Acta 1269 187-199 [DOI] [PubMed] [Google Scholar]
- 13.Cohen, P. (1989) Annu. Rev. Biochem. 58 453-508 [DOI] [PubMed] [Google Scholar]
- 14.Shenolikar, S. (1994) Annu. Rev. Cell Biol. 10 55-86 [DOI] [PubMed] [Google Scholar]
- 15.Cohen, P. T. (2002) J. Cell Sci. 115 241-256 [DOI] [PubMed] [Google Scholar]
- 16.Mellgren, G., Vintermyr, O. K., Boe, R., and Doskeland, S. O. (1993) Exp. Cell Res. 205 293-301 [DOI] [PubMed] [Google Scholar]
- 17.Fernandez-Sanchez, M. T., Garcia-Rodriguez, A., Diaz-Trelles, R., and Novelli, A. (1996) FEBS Lett. 398 106-112 [DOI] [PubMed] [Google Scholar]
- 18.Morana, S. J., Wolf, C. M., Li, J., Reynolds, J. E., Brown, M. K., and Eastman, A. (1996) J. Biol. Chem. 271 18263-18271 [DOI] [PubMed] [Google Scholar]
- 19.Li, D. W., Fass, U., Huizar, I., and Spector, A. (1998) Eur. J. Biochem. 257 351-361 [DOI] [PubMed] [Google Scholar]
- 20.Takenaka, I., Morin, F., Seizinger, B. R., and Kley, N. (1995) J. Biol. Chem. 270 5405-5411 [DOI] [PubMed] [Google Scholar]
- 21.Turenne, G. A., Paul, P., Laflair, L., and Price, B. D. (2001) Oncogene 20 5100-5110 [DOI] [PubMed] [Google Scholar]
- 22.Haneda, M., Kojima, E., Nishikimi, A., Hasegawa, T., Nakashima, I., and Isobe, K. (2004) FEBS Lett. 567 171-174 [DOI] [PubMed] [Google Scholar]
- 23.Li, D. W., Liu, J. P., Schmid, P. C., Schlosser, R., Feng, H., Liu, W. B., Yan, Q., Gong, L., Sun, S. M., Deng, M., and Liu, Y. (2006) Oncogene 25 3006-3022 [DOI] [PubMed] [Google Scholar]
- 24.Davis, R. J. (2000) Cell 103 239-252 [DOI] [PubMed] [Google Scholar]
- 25.Shen, H. M., and Liu, Z. G. (2006) Free Radic. Biol. Med. 40 928-939 [DOI] [PubMed] [Google Scholar]
- 26.Tournier, C., Hess, P., Yang, D. D., Xu, J., Turner, T. K., Nimnual, A., Bar-Sagi, D., Jones, S. N., Flavell, R. A., and Davis, R. J. (2000) Science 288 870-874 [DOI] [PubMed] [Google Scholar]
- 27.Lei, K., Nimnual, A., Zong, W. X., Kennedy, N. J., Flavell, R. A., Thompson, C. B., Bar-Sagi, D., and Davis, R. J. (2002) Mol. Cell. Biol. 22 4929-4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camps, M., Nichols, A., and Arkinstall, S. (2000) FASEB J. 14 6-16 [PubMed] [Google Scholar]
- 29.Moon, S. K., Thompson, L. J., Madamanchi, N., Ballinger, S., Papacon-stantinou, J., Horaist, C., Runge, M. S., and Patterson, C. (2001) Am. J. Physiol. Heart Circ. Physiol. 280 H2779-H2788 [DOI] [PubMed] [Google Scholar]
- 30.Madamanchi, N. R., Li, S., Patterson, C., and Runge, M. S. (2001) J. Biol. Chem. 276 18915-18924 [DOI] [PubMed] [Google Scholar]
- 31.Pfaffl, M. W., Horgan, G. W., and Dempfle, L. (2002) Nucleic Acids Res. 30 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao, G. N., Katki, K. A., Madamanchi, N. R., Wu, Y., and Birrer, M. J. (1999) J. Biol. Chem. 274 6003-6010 [DOI] [PubMed] [Google Scholar]
- 33.Sandau, K., Pfeilschifter, J., and Brune, B. (1997) Kidney Int. 52 378-386 [DOI] [PubMed] [Google Scholar]
- 34.Ramachandran, A., Moellering, D., Go, Y. M., Shiva, S., Levonen, A. L., Jo, H., Patel, R. P., Parthasarathy, S., and Darley-Usmar, V. M. (2002) Biol. Chem. 383 693-701 [DOI] [PubMed] [Google Scholar]
- 35.Hamm-Kunzelmann, B., Schafer, D., Weigert, C., and Brand, K. (1997) FEBS Lett. 403 87-90 [DOI] [PubMed] [Google Scholar]
- 36.Akakura, N., Kobayashi, M., Horiuchi, I., Suzuki, A., Wang, J., Chen, J., Niizeki, H., Kawamura, K.-I., Hosokawa, M., and Asaka, M. (2001) Cancer Res. 61 6548-6554 [PubMed] [Google Scholar]
- 37.Flogel, U., Godecke, A., Klotz, L. O., and Schrader, J. (2004) FASEB J. 18 1156-1158 [DOI] [PubMed] [Google Scholar]
- 38.Li, F., Zhang, D., and Fujise, K. (2001) J. Biol. Chem. 276 47542-47549 [DOI] [PubMed] [Google Scholar]
- 39.Zhang, Y., and Shen, X. (2007) Clin. Cancer Res. 13 2855-2864 [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto, K., Okamoto, A., Isonishi, S., Ochiai, K., and Ohtake, Y. (2001) Cancer Lett. 168 173-181 [DOI] [PubMed] [Google Scholar]
- 41.Martin-Ventura, J. L., Duran, M. C., Blanco-Colio, L. M., Meilhac, O., Leclercq, A., Michel, J. B., Jensen, O. N., Hernandez-Merida, S., Tunon, J., Vivanco, F., and Egido, J. (2004) Circulation 110 2216-2219 [DOI] [PubMed] [Google Scholar]
- 42.Martin-Ventura, J. L., Nicolas, V., Houard, X., Blanco-Colio, L. M., Leclercq, A., Egido, J., Vranckx, R., Michel, J. B., and Meilhac, O. (2006) Arterioscler. Thromb. Vasc. Biol. 26 1337-1343 [DOI] [PubMed] [Google Scholar]
- 43.Orrenius, S., Gogvadze, V., and Zhivotovsky, B. (2007) Annu. Rev. Pharmacol. Toxicol. 47 143-183 [DOI] [PubMed] [Google Scholar]
- 44.Papadakis, E. S., Finegan, K. G., Wang, X., Robinson, A. C., Guo, C., Kayahara, M., and Tournier, C. (2006) FEBS Lett. 580 1320-1326 [DOI] [PubMed] [Google Scholar]
- 45.Miyashita, T., and Reed, J. C. (1995) Cell 80 293-299 [DOI] [PubMed] [Google Scholar]
- 46.Green, D. R., and Reed, J. C. (1998) Science 281 1309-1312 [DOI] [PubMed] [Google Scholar]
- 47.Woo, M., Hakem, R., Soengas, M. S., Duncan, G. S., Shahinian, A., Kagi, D., Hakem, A., McCurrach, M., Khoo, W., Kaufman, S. A., Senaldi, G., Howard, T., Lowe, S. W., and Mak, T. W. (1998) Genes Dev. 12 806-819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardone, M. H., Roy, N., Stennicke, H. R., Salvesen, G. S., Franke, T. F., Stanbridge, E., Frisch, S., and Reed, J. C. (1998) Science 282 1318-1321 [DOI] [PubMed] [Google Scholar]
- 49.Janicke, R. U., Sprengart, M. L., Wati, M. R., and Porter, A. G. (1998) J. Biol. Chem. 273 9357-9360 [DOI] [PubMed] [Google Scholar]
- 50.Nagata, S., Nagase, H., Kawane, K., Mukae, N., and Fukuyama, H. (2003) Cell Death Differ. 10 108-116 [DOI] [PubMed] [Google Scholar]
- 51.Kyriakis, J. M., and Avruch, J. (1996) J. Biol. Chem. 271 24313-24316 [DOI] [PubMed] [Google Scholar]
- 52.Wang, X., Martindale, J. L., Liu, Y., and Holbrook, N. J. (1998) Biochem. J. 333 291-300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng, W. H., Zheng, X., Quimby, F. R., Roneker, C. A., and Lei, X. G. (2003) Biochem. J. 370 927-934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bøe, R., Gjertsen, B. T., Vintermyr, O. K., Houge, G., Lanotte, M., and Døskeland, S. O. (1991) Exp. Cell Res. 195 237-246 [DOI] [PubMed] [Google Scholar]
- 55.Kiguchi, K., Glesne, D., Chubb, C. H., Fujiki, H., and Huberman, E. (1994) Cell Growth & Differ. 5 995-1004 [PubMed] [Google Scholar]
- 56.Yan, Y., Shay, J. W., Wright, W. E., and Mumby, M. C. (1997) J. Biol. Chem. 272 15220-15226 [DOI] [PubMed] [Google Scholar]
- 57.Garcia, A., Cayla, X., Guergnon, J., Dessauge, F., Hospital, V., Rebollo, M. P., Fleischer, A., and Rebollo, A. (2003) Biochimie (Paris) 85 721-726 [DOI] [PubMed] [Google Scholar]
- 58.Yamagishi, S. I., Edelstein, D., Du, X. L., Kaneda, Y., Guzmán, M., and Brownlee, M. (2001) J. Biol. Chem. 276 25096-25100 [DOI] [PubMed] [Google Scholar]
- 59.Park, W. H., Han, Y. W., Kim, S. H., and Kim, S. Z. (2007) J. Cell. Biochem. 102 98-109 [DOI] [PubMed] [Google Scholar]
- 60.Kim, H. S., Song, M. C., Kwak, I. H., Park, T. J., and Lim, I. K. (2003) J. Biol. Chem. 278 37497-37510 [DOI] [PubMed] [Google Scholar]
- 61.Criddle, D. N., Gillies, S., Baumgartner-Wilson, H. K., Jaffar, M., Chinje, E. C., Passmore, S., Chvanov, M., Barrow, S., Gerasimenko, O. V., Tepikin, A. V., Sutton, R., and Petersen, O. H. (2003) J. Biol. Chem. 281 40485-40492 [DOI] [PubMed] [Google Scholar]
- 62.Callapina, M., Zhou, J., Schmid, T., Köhl, R., and Brüne, B. (2005) Free Radic. Biol. Med. 39 925-936 [DOI] [PubMed] [Google Scholar]
- 63.Sakahira, H., Enari, M., and Nagata, S. (1998) Nature 391 96-99 [DOI] [PubMed] [Google Scholar]
- 64.Ricci, J. E., Gottlieb, R. A., and Green, D. R. (2003) J. Cell Biol. 160 65-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen, Y. R., Wang, X., Templeton, D., Davis, R. J., and Tan, T. H. (1996) J. Biol. Chem. 271 31929-31936 [DOI] [PubMed] [Google Scholar]
- 66.Ho, F. M., Liu, S. H., Liau, C. S., Huang, P. J., and Lin-Shiau, S. Y. (2000) Circulation 101 2618-2624 [DOI] [PubMed] [Google Scholar]
- 67.Chu, S., and Ferro, T. J. (2006) Am. J. Physiol. 291 L983-L992 [DOI] [PubMed] [Google Scholar]
- 68.Fuchs, S. Y., Adler, V., Pincus, M. R., and Ronai, Z. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 10541-10546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oleinik, N. V., Krupenko, N. I., and Krupenko, S. A. (2007) Oncogene 26 7222-7230 [DOI] [PubMed] [Google Scholar]
- 70.Miyashita, T., Krajewski, S., Krajewska, M., Wang, H. G., Lin, H. K., Liebermann, D. A., Hoffman, B., and Reed, J. C. (1994) Oncogene 9 1799-1805 [PubMed] [Google Scholar]
- 71.Polyak, K., Xia, Y., Zweier, J. L., Kinzler, K. W., and Vogelstein, B. (1997) Nature 389 300-305 [DOI] [PubMed] [Google Scholar]
- 72.Mihara, M., Erster, S., Zaika, A., Petrenko, O., Chittenden, T., Pancoska, P., and Moll, U. M. (2003) Mol Cell. 11 577-590 [DOI] [PubMed] [Google Scholar]
- 73.Giaccia, A. J., and Kastan, M. B. (1998) Genes Dev. 12 2973-2983 [DOI] [PubMed] [Google Scholar]
- 74.Bode, A. M., and Dong, Z. (2004) Nat. Rev. Cancer. 4 793-805 [DOI] [PubMed] [Google Scholar]
- 75.Mandal, M., Olson, D. J., Sharma, T., Vadlamudi, R. K., and Kumar, R. (2001) Gastroenterology 120 71-78 [DOI] [PubMed] [Google Scholar]
- 76.Goping, I. S., Gross, A., Lavoie, J. N., Nguyen, M., Jemmerson, R., Roth, K., Korsmeyer, S. J., and Shore, G. C. (1998) J. Cell. Biol. 143 207-215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tsuruta, F., Sunayama, J., Mori, Y., Hattori, S., Shimizu, S., Tsujimoto, Y., Yoshioka, K., Masuyama, N., and Gotoh, Y. (2004) EMBO J. 23 1889-1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tamm, C., Robertson, J. D., Sleeper, E., Enoksson, M., Emgard, M., Orrenius, S., and Ceccatelli, S. (2004) Eur. J. Neurosci. 19 2613-2621 [DOI] [PubMed] [Google Scholar]