

Abstract
A subset of methanogenic archaea synthesize the cysteinyl-tRNACys (Cys-tRNACys) needed for protein synthesis using both a canonical cysteinyl-tRNA synthetase (CysRS) as well as a set of two enzymes that operate via a separate indirect pathway. In the indirect route, phosphoseryl-tRNACys (Sep-tRNACys) is first synthesized by phosphoseryl-tRNA synthetase (SepRS), and this misacylated intermediate is then converted to Cys-tRNACys by Sep-tRNA:Cys-tRNA synthase (SepCysS) via a pyridoxal phosphate-dependent mechanism. Here, we explore the function of all three enzymes in the mesophilic methanogen Methanosarcina mazei. The genome of M. mazei also features three distinct tRNACys isoacceptors, further indicating the unusual and complex nature of Cys-tRNACys synthesis in this organism. Comparative aminoacylation kinetics by M. mazei CysRS and SepRS reveals that each enzyme prefers a distinct tRNACys isoacceptor or pair of isoacceptors. Recognition determinants distinguishing the tRNAs are shown to reside in the globular core of the molecule. Both enzymes also require the S-adenosylmethione-dependent formation of m1G37 in the anticodon loop for efficient aminoacylation. We further report a new, highly sensitive assay to measure the activity of SepCysS under anaerobic conditions. With this approach, we demonstrate that SepCysS functions as a multiple-turnover catalyst with kinetic behavior similar to bacterial selenocysteine synthase and the archaeal/eukaryotic SepSecS enzyme. Together, these data suggest that both metabolic routes and all three tRNACys species in M. mazei play important roles in the cellular physiology of the organism.
Methanosarcina mazei strain Gö1 is a methanogenic archaeon capable of utilizing a broad spectrum of growth substrates, including H2/CO2, acetate, methanol, and methylamines (1). The capacity of M. mazei to adapt to changing environmental conditions is a hallmark of the Methanosarcina. Organisms belonging to this genus coexist with fermenting bacteria in freshwater and marine sediments, where their capacity to metabolically transform bacterially produced acetate into methane forms a closed carbon cycle that helps to determine atmospheric levels of this greenhouse gas (2). In some marine environments, the Methanosarcina also flourish together with methanotrophs and sulfate-reducing bacteria, where they participate in a microbial community that generates very high sulfate reduction rates via anaerobic methane oxidation (3). Comparative genomic studies of M. mazei, Methanosarcina barkeri, and Methanosarcina acetivorans C2A, the three Methanosarcina for which complete sequences are available, show that the 4.1-megabase M. mazei genome is the smallest so far known in the genus and may approximate the ancestral state (1, 4, 5). Of the 3371 M. mazei open reading frames identified, 1043 find their closest homolog in the bacterial domain (1), suggesting that horizontal gene transfer has played an important evolutionary role in the development of the organism.
The distinguishing methanogenesis pathway, present in all methanogens, produces methane as a catabolic product while also generating an electrochemical gradient that is harnessed to synthesize ATP (5, 6). Methanogens also possess many subsidiary metabolic activities that function to directly support methane production, including those involved in cofactor biosynthesis and in energy transduction through proton and sodium gradients (6). Interestingly, genomic data reveal a further correlation; all but two methanogens for which full sequence information is available also possess a unique two-step pathway by which cysteine is incorporated into proteins (7). In this pathway, tRNACys is first aminoacylated with the nonstandard amino acid phosphoserine by phosphoseryl-tRNA synthetase (SepRS).2 The misacylated Sep-tRNACys is then converted to Cys-tRNACys through the action of the PLP-dependent enzyme SepCysS. For some methanogens, which lack a canonical cysteinyl-tRNA synthetase (CysRS), this indirect route is the sole pathway to synthesis of Cys-tRNACys. However, other methanogens, including all three Methanosarcina for which genome sequences are available, possess both pathways. Comparative phylogenetic studies have shown that both the direct CysRS route and the indirect two-step pathway were in existence at the time of the last universal common ancestor (8). The coincident phylogenetic distributions of SepRS/SepCysS and the enzymes of methanogenesis also suggest the existence of a linkage between primary energy production and protein synthesis in contemporary methanogens, although the nature of and biological rationale for such a linkage have so far been obscure (8). The only nonmethanogen known to possess the two-step Cys-tRNACys biosynthetic pathway is the hyperthermophilic sulfate-reducing archaeon Archaeoglobus fulgidus, a closely related archaeon that retains nearly all of the methanogenesis enzymes and cofactors needed for the hydrogenotrophic pathway, with the exception of the essential methyl-CoM reductase (9).
Recently, it was reported that SepRS and CysRS from Methanococcus maripaludis share major identity elements for heterologous aminoacylation of an unmodified, in vitro transcript corresponding to the single tRNACys species present in the M. jannaschii genome (10). Consistent with other studies in which a common tRNA is recognized by distinct tRNA synthetases, these data were interpreted to support a general hypothesis that aminoacyl-tRNA synthetases evolved in the context of preestablished tRNA identity. Other work on the M. jannaschii pathway has shown that SepRS and SepCysS form a stable binary complex that assists in conversion of the intermediate Sep-tRNACys to Cys-tRNACys, a complex reminiscent of the transamidosome particle that facilitates prokaryotic tRNA-dependent asparagine biosynthesis (11, 12).
Here, we sought to address distinct questions pertaining to the contemporary function of apparently redundant Cys-tRNACys synthesis pathways in M. mazei. Both M. mazei and M. barkeri possess genes encoding three distinct isoacceptors of tRNACys, a feature not known in other bacteria or archaea and suggestive that the machinery for incorporation of cysteine into proteins in these organisms may interact with other cellular components (1, 4, 8). By examining aminoacylation of the three wild-type as well as a set of hybrid tRNACys species as substrates for either CysRS or the combined SepRS/SepCysS pathway, we show that each enzyme prefers a distinct tRNACys isoacceptor or pair of isoacceptors and that the nucleotides determining CysRS versus SepRS selectivity among tRNACys species are located in the globular core of the tRNA. Through the development and application of a new anaerobic assay for the quantitative analysis of SepCysS kinetics, we also provide evidence that the tRNACys isoacceptor preferences of SepRS and SepCysS are identical. This supports the notion that tRNACys acceptors functioning in the two-step pathway could be efficiently shuttled between these two enzymes. Together, these findings suggest that both pathways may play an important role in the cellular physiology. We also show that the aminoacylation efficiency of M. mazei CysRS is very significantly enhanced by the addition of a methyl group to the N1 position of G37 by the S-adenosylmethione-dependent Trm5 enzyme, consistent with a similar dependence found for SepRS (11, 13). Since S-adenosylmethione is synthesized from methionine (14), the tRNACys methylation requirement for aminoacylation by both CysRS and SepRS links protein synthesis with sulfur metabolism in this organism.
EXPERIMENTAL PROCEDURES
Expression, Purification, and Quaternary Structure Determination of M. mazei CysRS and SepCysS—M. mazei genomic DNA was purchased from the American Type Culture Collection and used as template for PCR amplification of the CysRS and SepCysS open reading frames. For SepCysS, the resulting DNA fragments were digested with NdeI and XhoI and inserted into pet22b+ for expression of His-tagged proteins in Escherichia coli Rosetta2(DE3) pLysS cells. Cultures were grown aerobically at 37 °C in LB medium supplemented with 100 μg/ml ampicillin, 34 μg/ml chloramphenicol, and 0.01% pyridoxine. When the cultures reached an A600 of 0.5, expression of His-tagged protein was induced by adding isopropyl-β-d-thiogalactoside to a final concentration of 0.6 mm. Cultures were then grown for a further 5 h at 37 °C prior to harvesting.
Cells expressing SepCysS were resuspended in a buffer containing 50 mm NaH2PO4 (pH 8.0), 20 mm NaCl, 3 mm DTT, 20 μm PLP, and 10% glycerol and were disrupted by a French press. Under aerobic conditions, the clarified lysate was applied to a 30-ml DEAE column preequilibrated with 50 mm NaH2PO4 (pH 8.0), 20 mm NaCl, 3 mm DTT, and 20 μm PLP. SepCysS was eluted with a gradient from 20–500 mm NaCl, and the pooled fractions were dialyzed into a solution containing 50 mm NaH2PO4 (pH 8.0), 150 mm NaCl, 5 mm βME, and 20 μm PLP. The enzyme was then applied to a Superdex 200 gel filtration column preequilibrated with 50 mm NaH2PO4 (pH 8.0), 150 mm NaCl, 5 mm βME, and 20 μm PLP. Fractions containing SepCysS were then directly applied to a Ni2+-nitrilotriacetic acid column equilibrated with 50 mm NaH2PO4 (pH 8.0), 300 mm NaCl, 5 mm βME, and 20 μm PLP. After washing of the column in this buffer, the enzyme was then eluted by the addition of 150 mm imidazole. The enzyme was then dialyzed into a solution containing 10 mm NaH2PO4 (pH 7.4), 20 mm NaCl, 2 mm MgCl2, 1 mm βME, 100 μm PLP, and 50% glycerol and stored at -20 °C. His-tagged SepCysS was recovered at better than 95% purity as judged by SDS-polyacrylamide gel electrophoresis, at a yield of 2 mg of protein/liter of culture. The PLP content of SepCysS was verified using a colorimetric assay in which the titration of PLP released upon boiling is monitored (15).
Construction of the recombinant DNA vector, induction of enzyme expression, and purification of M. mazei CysRS were accomplished by procedures identical to those described for M. mazei SepRS (13). The yield of highly purified CysRS was ∼5 mg of protein/liter of culture.
The oligomeric structures of CysRS and SepCysS were determined by analytic size exclusion chromatography, using conditions and molecular mass markers identical to those reported in the accompanying paper (13). The 473-amino acid M. mazei CysRS enzyme (calculated Mr = 53,861) exhibits 40.2% sequence identity with the monomeric E. coli CysRS and migrates as a monomer, as expected. M. mazei SepCysS belongs to the PLP-dependent superfamily of enzymes, which typically function as dimers with one molecule of PLP bound in each active site (16). As expected, the 386-amino acid enzyme (predicted Mr = 42,324 Da) elutes at a position corresponding to a molecular mass of 84 kDa, consistent with dimer formation (13).
Preparation and Methylation of tRNAs—To prepare tRNA for
kinetic analysis, the three M. mazei tRNACys genes were
each transcribed from a synthetic duplex DNA template, methylated at the N1
position of G37, purified, and refolded using procedures identical to those
described in the accompanying paper
(13). The oligonucleotides
used for the  and
and
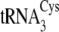 genes were as follows:
genes were as follows:
 ,
5′-AATTCCTGCCGTAATACGACTCACTATAGCCAAGGTGGCGGAGCGGTCACGCAATCGC-3′
and
5′-mUmGGAGCCAAGGTCCGGATTTGAACCAGGACTGAATCGCTCTGCTGGCGATTGCGT-3′;
,
5′-AATTCCTGCCGTAATACGACTCACTATAGCCAAGGTGGCGGAGCGGTCACGCAATCGC-3′
and
5′-mUmGGAGCCAAGGTCCGGATTTGAACCAGGACTGAATCGCTCTGCTGGCGATTGCGT-3′;
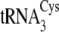 ,
5′-AATTCCTGCAGTAATACGACTCACTATAGCCAAGGTGGCGGAGCGGCTACGCAATCGCCT-3′
and
5′-mUmGGAGCCAAGGTCCGGATTCGAACCGGAATGGTATCGCTCTGCAGGCGATTGC-3′,
where 2′-O-methyl nucleotides are represented by mU and mG, the
underlined portions represent the overlap region, and boldface indicates the
T7 RNA polymerase promoter. Oligonucleotides used to construct the
,
5′-AATTCCTGCAGTAATACGACTCACTATAGCCAAGGTGGCGGAGCGGCTACGCAATCGCCT-3′
and
5′-mUmGGAGCCAAGGTCCGGATTCGAACCGGAATGGTATCGCTCTGCAGGCGATTGC-3′,
where 2′-O-methyl nucleotides are represented by mU and mG, the
underlined portions represent the overlap region, and boldface indicates the
T7 RNA polymerase promoter. Oligonucleotides used to construct the
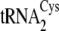 template are listed in
the accompanying paper (13).
To remove unmethylated transcripts, the 16-mer oligodeoxynucleotides
complementary to the tRNA sequence flanking nucleotide G37 were as follows:
template are listed in
the accompanying paper (13).
To remove unmethylated transcripts, the 16-mer oligodeoxynucleotides
complementary to the tRNA sequence flanking nucleotide G37 were as follows:
 ,
5′-mUmGmAmAmUmCmGmCTCTGmCmUmGmG-3′;
,
5′-mUmGmAmAmUmCmGmCTCTGmCmUmGmG-3′;
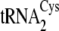 and
and
 ,
5′-mGmGmUmAmUmCmGmCTCTGmCmAmGmG-3′ (where “m”
indicates a 2′-O-methyl ribose modification). Individual
tRNACys isoacceptors are depicted in
Fig. 1B.
,
5′-mGmGmUmAmUmCmGmCTCTGmCmAmGmG-3′ (where “m”
indicates a 2′-O-methyl ribose modification). Individual
tRNACys isoacceptors are depicted in
Fig. 1B.
FIGURE 1.

A, dependence of reaction rate on tRNA concentration for
aminoacylation by M. mazei SepRS. Replot of
 concentration
versus initial velocities determined from time courses. The
inset shows the imaged TLC plate of a single time course for
concentration
versus initial velocities determined from time courses. The
inset shows the imaged TLC plate of a single time course for
 . B, the
secondary structures of M. mazei tRNACys isoacceptors
. B, the
secondary structures of M. mazei tRNACys isoacceptors
 ,
,
 , and
, and
 , are depicted from
left to right. The 7-66 base pair at the bottom of the
acceptor stem, which distinguishes
, are depicted from
left to right. The 7-66 base pair at the bottom of the
acceptor stem, which distinguishes
 from
from
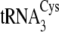 , is shown in
boldface type on the
, is shown in
boldface type on the
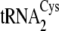 cloverleaf at the
center. Nucleotides distinguishing
cloverleaf at the
center. Nucleotides distinguishing
 (left) from
both
(left) from
both 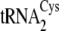 and
and
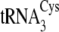 are shown in
boldface type on the
are shown in
boldface type on the
 cloverleaf. Mutations
of
cloverleaf. Mutations
of 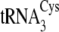 (right)
made in this study are circled. The correspondence between the
nucleotides and the mutations described under “Results and
Discussion” and in Table
1 is as follows: tRNACysΔ2, introduction of A33
from
(right)
made in this study are circled. The correspondence between the
nucleotides and the mutations described under “Results and
Discussion” and in Table
1 is as follows: tRNACysΔ2, introduction of A33
from  into
into
 ;
tRNACysΔ3, introduction of A57 from
;
tRNACysΔ3, introduction of A57 from
 into
into
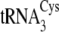 ;
tRNACysΔ5, introduction of both C49 and U51 from
;
tRNACysΔ5, introduction of both C49 and U51 from
 into
into
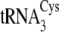 ;
tRNACysΔ4, introduction of the five D-loop and variable loop
nucleotides U20, C21, U44, A46, and G47 from
;
tRNACysΔ4, introduction of the five D-loop and variable loop
nucleotides U20, C21, U44, A46, and G47 from
 into
into
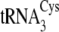 .
.
Steady-state Aminoacylation Kinetics—SepRS plateau aminoacylation levels and assays to determine steady-state kinetic parameters were carried out using tRNA 32P-labeled at the 3′-internucleotide linkage, using the methodology reported in the accompanying paper (13, 17). Measurements of aminoacylation by CysRS with all tRNACys species were performed with a conventional assay that tracks incorporation of 35S-labeled cysteine into tRNA, as described (18). Saturation for tRNA was not observed in any CysRS reaction, limiting the analysis to determination of kcat/Km. kcat/Km was derived by fitting initial velocities to the following linear relation,
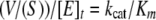 |
(Eq. 1) |
where (v/S) is the slope of the linear fit, and [E]t is the enzyme concentration. Errors are reported in Tables 1, 2, 3 as the mean and S.D. values of at least two measurements.
TABLE 1.
Determinants of tRNA specificity in CysRS and SepRS
|
SepRS
|
CysRS
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| kcat | Km | kcat/Km | pmol/OD | kcat | Km | kcat/Kma | pmol/OD | Specificityb | |
| s–1 | μm | s–1m–1 | s–1 | μm | s–1m–1 | ||||
 c c
|
0.12 ± 0.01 | 6.4 ± 0.2 | (2.0 ± 0.3) × 104 | 1250 ± 60 | >0.17 | >80 | 2070 ± 33 | 780 ± 50 | 9.5 |
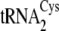 c c
|
0.51 ± 0.11 | 6.0 ± 0.8 | (8.5 ± 0.9) × 104 | 1360 ± 80 | >0.06 | >160 | 380 ± 60 | 830 ± 70 | 223 |
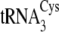 c c
|
0.58 ± 0.09 | 7.6 ± 0.3 | (8.0 ± 0.1) × 104 | 1300 ± 80 | >0.03 | >140 | 229 ± 21 | 530 ± 40 | 143 |
| tRNACysΔ1d | 0.23 ± 0.02 | 13.1 ± 2.8 | (1.6 ± 0.3) × 104 | 840 ± 40 | >0.16 | >90 | 1700 ± 57 | 720 ± 40 | 9.4 |
| tRNACysΔ2e | 0.52 ± 0.10 | 10.2 ± 1.2 | (5.1 ± 0.4) × 104 | 990 ± 70 | >0.05 | >130 | 409 ± 83 | 580 ± 30 | 125 |
| tRNACysΔ3f | 0.30 ± 0.04 | 9.9 ± 0.7 | (3.0 ± 0.1) × 104 | 660 ± 40 | >0.09 | >120 | 752 ± 95 | 600 ± 40 | 39.5 |
| tRNACysΔ4g | 0.25 ± 0.03 | 10.1 ± 1.5 | (2.5 ± 0.3) × 104 | 890 ± 50 | >0.14 | >110 | 1250 ± 43 | 480 ± 20 | 20 |
| tRNACysΔ5h | NDi | ND | ND | 140 ± 20 | ND | ND | ND | 125 ± 15 | ND |
| tRNACysΔ6j | 0.18 ± 0.02 | 8.6 ± 0.6 | (2.1 ± 0.3) × 104 | 900 ± 30 | >0.18 | >90 | 1950 ± 44 | 740 ± 50 | 10.8 |
kcat/Km values were determined from initial velocities using the relation, ν = kcat/Km[E][S]
Specificity for each tRNA is determined as the ratio, kcat/Km(SEPRS)/kcat/Km(CYSRS)
Structures of the M. mazei isoacceptors
 ,
,
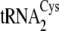 , and
, and
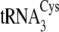 are shown in
Fig. 1B
are shown in
Fig. 1B
The A33U mutation in

The U33A mutation in
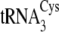
The G57A mutation in
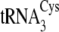
The C20U/U21C/A44U/C46A/A47G mutations in
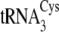
The U49C/C51U mutations in
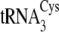
ND, not determined
The C20U/U21C/A44U/C46A/A47G/G57A mutations in
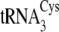
TABLE 2.
Alternative sulfur donors used by SepCysS
| Conversion to Cys-tRNACys | |
|---|---|
| % | |
| HS– | 72.1 ± 9.5 |
| Thiophosphate | 52.3 ± 4.5 |
| Cysteine | 55.4 ± 5.0 |
TABLE 3.
tRNA discrimination by SepCysS
| Percentage conversion to Cys-tRNACys | Kd | kcat/Kma | |
|---|---|---|---|
| % | μm | min–1 μm–1 | |
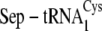 |
52.2 ± 9.5 | 1.1 ± 0.2 | 0.009 ± 0.002 |
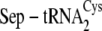 |
75.3 ± 4.5 | 0.4 ± 0.15 | 0.014 ± 0.003 |
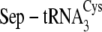 |
72.2 ± 5.0 | 0.55 ± 0.18 | 0.012 ± 0.002 |
| tRNACysΔ2 | 72.0 ± 8.0 | NDb | ND |
| tRNACysΔ4 | 50.0 ± 6.0 | ND | ND |
kcat/Km values were determined from initial velocities using the relation ν = kcat/Km [E][S]
ND, not determined
SepCysS Activity Assay—SepCysS was assayed anaerobically under an atmosphere of 95% N2, 5% H2/CO2 in an anaerobic chamber (Coy Laboratories). All enzymes, buffers, and substrates entering the chamber were first deoxygenated by five cycles of vacuuming followed by purging with argon (19). Anoxic solutions were verified by testing larger volumes with a BBL anaerobic test strip. Anoxic solutions were kept in sealed glass vials and stored at -20 °C until needed.
The Sep-tRNACys substrate needed for the assay of SepCysS was generated in one of two ways. A 2-fold excess of SepRS, usually 30–40 μm, was incubated with 10–20 μm tRNACys in a buffer containing 50 mm Tris (pH 7.5), 20 mm KCl, 10 mm MgCl2, 5 mm ATP, 3 mm phosphoserine, and 5 mm DTT for 8 min. 32P-labeled tRNA sample mixes were prepared as described above for aminoacylation assays. Aliquots were removed for evaluation of plateau aminoacylation levels. After phosphoserylation, the reactions were diluted 2-fold in water, and concentrated sodium acetate (pH 5.0) was then added to a final concentration of 200 mm to acidify the reaction and prevent tRNA deacylation. The reaction was then extracted with phenol/chloroform/isoamyl alcohol, and the phosphoserylated tRNA was recovered by ethanol precipitation. tRNA was then resuspended in the anaerobic chamber in an anoxic solution consisting of 20 mm sodium cacodylate (pH 6.0), 5 mm MgCl2. Aminoacylation levels were requantified by removing a small aliquot and digesting with P1 nuclease followed by TLC separation as described above. The aminoacylated tRNA was then stored in a sealed glass vial at -20 °C until needed.
Alternatively, Sep-tRNACys was generated in the same reaction as used for the SepCysS assay, by first performing the aminoacylation reaction with SepRS under anaerobic conditions. Excess SepCysS and sulfur donor are then added to initiate the SepCysS activity assay. In this approach, 20 μm 32P-labeled tRNACys was first heated to 85 °C and slowly cooled under anaerobic conditions in the presence of 2 mm MgCl2. Sep-tRNACys was then synthesized in a buffer consisting of 50 mm Tris (pH 7.5), 20 mm KCl, 10 mm MgCl2, 5 mm ATP, 3 mm phosphoserine, and 5 mm DTT. The phosphoserylation reaction was initiated by adding SepRS to 20–30 μm and incubating for 8 min at 37 °C. SepCysS was preincubated at 37 °C in a buffer containing 10 mm NaH2PO4 (pH 7.4), 20 mm NaCl, 2 mm MgCl2, 1 mm βME, 100 μm PLP, and 50% glycerol and was then added to concentrations of 0.05–40 μm, followed immediately by sodium sulfide to a concentration of 5 mm. Typical concentrations of Sep-tRNACys used in the second reaction, after dilution with reaction components, were 5–10 μm. In reactions where Sep-tRNACys was generated outside of the anaerobic chamber, it was first preincubated in the chamber at 37 °C with 50 mm Tris (pH 7.5), 20 mm KCl, 10 mm MgCl2, and 5 mm DTT for several minutes before the addition of SepCysS and sodium sulfide. Anhydrous sodium sulfide was purchased from Aldrich (product 407410) and was opened and stored in an anaerobic environment. The pH of cysteine and sodium sulfide stock solutions was adjusted to neutrality.
Sep-tRNACys, Cys-tRNACys, an intermediate-tRNACys (each as 3′-aminoacylated A76), and nonreacted substrate (as unmodified AMP) were separated by TLC on polyethyleneimine-cellulose plates, which were run in 1 m acetic acid with adjustment of the pH to 3.5 using concentrated ammonium hydroxide. These conditions for TLC separation were reoptimized for visualization of the Cys-AMP product and differ from those employed to characterize SepRS reactions alone (13). Quantitation was done by phosphorimaging analysis, as described previously (17). Time courses from 0.5 to 60 min were fit to linear or sigmoidal equations. Results using either method of Sep-tRNACys preparation were identical.
tRNA Binding to SepCysS—Equilibrium fluorescence titrations were performed at ambient temperature with 0.25 μm M. mazei SepCysS in 20 mm Tris-HCl (pH 7.5), 50 mm NaCl, and 5 mm βME. Tryptophan fluorescence was excited at 295 nm, and the emission was monitored from 300 to 400 nm. An emission wavelength of 350 nm was used to quantitate binding after correction for dilution and for intrinsic quenching due to RNA absorbance at 295 nm, according to the formula, Fc = Fobs × antilog((A295 + A350)/2) (20). Control solutions of bovine serum albumin or of tryptophan showed no fluorescence response to tRNA. tRNA was titrated in the concentration range of 0.05–10 μm. The Kd values were derived by fitting the data to a quadratic binding equation using Scientist software.
RESULTS AND DISCUSSION
Differential Utilization of tRNACys Isoacceptors by SepRS and CysRS—We chose to study Cys-tRNACys formation in M. mazei because genetic techniques have been developed in this organism (21) and because its biosynthetic apparatus is expanded to its known limit in M. mazei and M. barkeri, suggesting the possibility that the macromolecules involved may be regulated in unusual ways or may perform physiological roles unrelated to protein synthesis. Both of these Methanosarcina possess all three enzymes corresponding to the two distinct pathways, CysRS, SepRS, and SepCysS, as well as three tRNACys isoacceptors (Fig. 1). In contrast, M. acetivorans, the other Methanosarcina for which the genome sequence is available, possesses all three enzymes but only one tRNACys acceptor that reads both the UGU and UGC codons. Among the other sequenced methanogens, M. maripaludis and Methanospirillum hungatei possess biosynthetic apparati with identical components to M. acetivorans, whereas Methanothermobacter thermautotrophicus, M. jannaschii, Methanopyrus kandleri, and Methanococcoides burtonii possess only the two-step indirect pathway with a single tRNACys isoacceptor. Only the human intestinal archaea Methanosphaera stadtmanae and Methanobrevibacter smithii provide examples of methanogens possessing just the canonical CysRS with one tRNACys acceptor and lacking the SepRS/SepCysS pathway (22, 23).
Among all sequenced bacteria and archaea, the presence of three
tRNACys isoacceptors is found only in M. mazei and M.
barkeri. Indeed, analysis of genomic data shows that only 10 bacterial or
archaeal species possess even two tRNACys isoacceptors. In addition
to the GCA anticodon, all three M. mazei isoacceptors possess the U73
discriminator base typical of tRNACys species; these four
nucleotides form a key part of the “identity set” for recognition
by both CysRS and SepRS (10,
24).
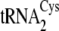 and
and
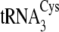 are of canonical
structure and are highly similar to each other, being distinguished only by
the base pair at position 7-66 at the bottom of the acceptor stem
(Fig. 1). However,
are of canonical
structure and are highly similar to each other, being distinguished only by
the base pair at position 7-66 at the bottom of the acceptor stem
(Fig. 1). However,
 possesses several
unusual features: the presence of a rare purine at position 33 (found in only
four other known tRNAs, two of which are also tRNACys acceptors
from methanogens), a GU wobble pair at position 51-63 of the T-stem, and an
A65-C49 mismatch at the inside of the T-stem (found in only one other known
tRNA, the homologous
possesses several
unusual features: the presence of a rare purine at position 33 (found in only
four other known tRNAs, two of which are also tRNACys acceptors
from methanogens), a GU wobble pair at position 51-63 of the T-stem, and an
A65-C49 mismatch at the inside of the T-stem (found in only one other known
tRNA, the homologous  isoacceptor from M. barkeri).
isoacceptor from M. barkeri).
 also differs from the
other two M. mazei tRNACys isoacceptors in featuring A57
in the T-loop as well as sequence differences at positions 20, 21, 44, 46, and
47 in the D-loop and variable loops. The last five differences probably
produce distinct base triples and/or stacking interactions in the augmented
D-stem region of the globular core. Despite the unusual structure, analysis of
the genomic DNA surrounding
also differs from the
other two M. mazei tRNACys isoacceptors in featuring A57
in the T-loop as well as sequence differences at positions 20, 21, 44, 46, and
47 in the D-loop and variable loops. The last five differences probably
produce distinct base triples and/or stacking interactions in the augmented
D-stem region of the globular core. Despite the unusual structure, analysis of
the genomic DNA surrounding
 reveals the presence
of both the A-box and B-box transcriptional initiation motifs
(25). Therefore, this unusual
tRNA may well be expressed in vivo. Resequencing of the genomic DNA
in this region also revealed no errors in the deposited sequence (data not
shown).
reveals the presence
of both the A-box and B-box transcriptional initiation motifs
(25). Therefore, this unusual
tRNA may well be expressed in vivo. Resequencing of the genomic DNA
in this region also revealed no errors in the deposited sequence (data not
shown).
We sought to explore the biological rationale for the existence of redundancy in the machinery for Cys-tRNACys synthesis by determining the efficiency by which each of the three tRNAs is processed by the two pathways. The three tRNAs were synthesized by in vitro transcription, were each methylated at position m1G37 using recombinant M. jannaschii Trm5, and were purified from unmethylated transcripts (see “Experimental Procedures” and Refs. 11 and 13). SepRS aminoacylation kinetics was measured as described (13) (Fig. 1A), whereas CysRS aminoacylation kinetics was measured using a filter binding assay in which [35S]cysteine is attached to tRNA (18). Preliminary plateau aminoacylation assays showed that SepRS aminoacylates each transcript to ∼1300 pmol/OD (78–85% aminoacylation), whereas CysRS aminoacylates the transcripts to lower levels (33–52% aminoacylation) (Table 1). As observed for SepRS (11, 13), plateau levels for aminoacylation of all three tRNACys species by M. mazei CysRS were very low in the absence of methylation by Trm5 (Fig. 2).
FIGURE 2.

Time course for plateau cysteinylation by M. mazei CysRS in the
absence or presence of m1G37 in
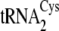 . The effect of
methylating
. The effect of
methylating  and
and
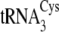 is nearly identical
(not shown).
is nearly identical
(not shown).
Steady-state parameters for phosphoserylation and cysteinylation of the
three isoacceptors revealed that each enzyme possesses distinct preferences.
SepRS preferentially aminoacylates
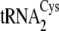 and
and
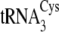 by a factor of
4–5-fold as compared with
by a factor of
4–5-fold as compared with
 , with the selectivity
manifested almost entirely in kcat
(Table 1). In contrast, CysRS
aminoacylates
, with the selectivity
manifested almost entirely in kcat
(Table 1). In contrast, CysRS
aminoacylates  5–9-fold more efficiently than the other isoacceptors. We also find that
SepRS is the better functioning enzyme for all isoacceptors; phosphoserylation
is favored over cysteinylation by 140–220-fold for
5–9-fold more efficiently than the other isoacceptors. We also find that
SepRS is the better functioning enzyme for all isoacceptors; phosphoserylation
is favored over cysteinylation by 140–220-fold for
 and
and
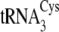 and by 9.5-fold for
and by 9.5-fold for
 . The
kcat of 0.5–0.6 s-1 toward preferred
tRNAs by SepRS is comparable with many other tRNA synthetases, including the
M. jannaschii SepRS
(11), and is ∼5-fold
greater than reported for phosphoserylation of fully unmodified M.
jannaschii tRNACys by M. maripaludis SepRS
(10). The primary defect in
aminoacylation by M. mazei CysRS is the inability to observe tRNA
saturation at concentrations up to 100 μm, limiting analyses to
the determination of kcat/Km. However,
it is also notable that the unusual structure of
. The
kcat of 0.5–0.6 s-1 toward preferred
tRNAs by SepRS is comparable with many other tRNA synthetases, including the
M. jannaschii SepRS
(11), and is ∼5-fold
greater than reported for phosphoserylation of fully unmodified M.
jannaschii tRNACys by M. maripaludis SepRS
(10). The primary defect in
aminoacylation by M. mazei CysRS is the inability to observe tRNA
saturation at concentrations up to 100 μm, limiting analyses to
the determination of kcat/Km. However,
it is also notable that the unusual structure of
 that apparently
produces antideterminants for phosphoserylation nonetheless operates to
provide positive recognition features for cysteinylation that are not present
in
that apparently
produces antideterminants for phosphoserylation nonetheless operates to
provide positive recognition features for cysteinylation that are not present
in 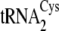 or
or
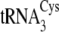 . This suggests the
operation of an evolutionary selection by which the parallel direct pathway is
retained by the organism.
. This suggests the
operation of an evolutionary selection by which the parallel direct pathway is
retained by the organism.
To examine the structural basis for the tRNACys isoacceptor
preferences of SepRS and CysRS, we constructed a series of tRNA mutants in
which structural elements of
 were systematically
transplanted into
were systematically
transplanted into 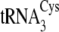 and
evaluated the effectiveness of the mutants as CysRS and SepRS substrates by
steady-state aminoacylation (Fig.
1 and Table 1). As
observed for the three wild-type isoacceptors, all of the tRNA mutants are
aminoacylated by SepRS more efficiently than by CysRS. A33 is not a
distinguishing determinant, because introduction of this nucleotide into
and
evaluated the effectiveness of the mutants as CysRS and SepRS substrates by
steady-state aminoacylation (Fig.
1 and Table 1). As
observed for the three wild-type isoacceptors, all of the tRNA mutants are
aminoacylated by SepRS more efficiently than by CysRS. A33 is not a
distinguishing determinant, because introduction of this nucleotide into
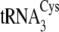 does not significantly
alter the relative preferences of SepRS and CysRS for that substrate
(tRNACysΔ2; Table
1). (In a separate experiment, mutation of A33 to U33 in the
does not significantly
alter the relative preferences of SepRS and CysRS for that substrate
(tRNACysΔ2; Table
1). (In a separate experiment, mutation of A33 to U33 in the
 framework also does
not shift enzyme preferences (tRNACysΔ1;
Table 1)). Incorporation of A57
into
framework also does
not shift enzyme preferences (tRNACysΔ1;
Table 1)). Incorporation of A57
into 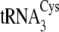 (tRNACysΔ3), however, both decreases the efficiency of
phosphoserylation and increases the efficiency of cysteinylation; the
SepRS/CysRS preference of this mutant is decreased from about 140- to 40-fold.
Incorporation into
(tRNACysΔ3), however, both decreases the efficiency of
phosphoserylation and increases the efficiency of cysteinylation; the
SepRS/CysRS preference of this mutant is decreased from about 140- to 40-fold.
Incorporation into 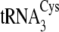 of
the
of
the  nucleotides in the
augmented D-stem/variable loop region (C20U/U21C/A44U/C46A/A47G) has
qualitatively similar but more marked effects, so that the SepRS/CysRS
preference is decreased to 20-fold (tRNACysΔ4;
Table 1). Finally, combining
the tRNACysΔ3 and tRNACysΔ4 substitutions
generates a mutant that fully recapitulates the SepRS/CysRS preferences of
nucleotides in the
augmented D-stem/variable loop region (C20U/U21C/A44U/C46A/A47G) has
qualitatively similar but more marked effects, so that the SepRS/CysRS
preference is decreased to 20-fold (tRNACysΔ4;
Table 1). Finally, combining
the tRNACysΔ3 and tRNACysΔ4 substitutions
generates a mutant that fully recapitulates the SepRS/CysRS preferences of
 (tRNACysΔ6; Table
1). Therefore, A57 together with the five nucleotides
U20/C21/U44/A46/G47 represents the distinguishing identity set
(Fig. 1B).
Interestingly, the unusual T-stem base pairs in
(tRNACysΔ6; Table
1). Therefore, A57 together with the five nucleotides
U20/C21/U44/A46/G47 represents the distinguishing identity set
(Fig. 1B).
Interestingly, the unusual T-stem base pairs in
 appear not to play a
role in modulating the SepRS/CysRS preference. Independent introduction of
these two base pairs into
appear not to play a
role in modulating the SepRS/CysRS preference. Independent introduction of
these two base pairs into
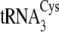 produced a mutant that
could not be efficiently aminoacylated by either enzyme
(tRNACysΔ5; Table
1). This suggests that other structural elements in
produced a mutant that
could not be efficiently aminoacylated by either enzyme
(tRNACysΔ5; Table
1). This suggests that other structural elements in
 are required to
maintain the structural stability of its unusual core domain.
are required to
maintain the structural stability of its unusual core domain.
These data are consistent with studies of other SepRS enzymes. Work in the
M. maripaludis system indicates that the G6:C67 base pair, C20, G37,
A47, and A59 are putative tRNACys identity elements for SepRS
(10).
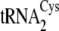 and
and
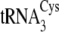 of M. mazei
each possess all six of these nucleotides, whereas
of M. mazei
each possess all six of these nucleotides, whereas
 is missing C20 and
A47. Further, the crystal structure of the A. fulgidus
SepRS·tRNACys complex shows that U33 does not contact the
enzyme (26), consistent with
the ineffectiveness of this nucleotide in modulating the SepRS/CysRS
preferences. The importance of T-loop, D-loop, and variable loop nucleotides
shown in this work, together with similar findings from studies of the M.
maripaludis enzyme, suggest that SepRS makes important interactions with
the globular tRNA core. Because the A. fulgidus SepRS crystallized as
an unproductive complex in which interactions are made only with the tRNA
anticodon (26), further
structural analyses will be required to provide a reliable basis for these
observations.
is missing C20 and
A47. Further, the crystal structure of the A. fulgidus
SepRS·tRNACys complex shows that U33 does not contact the
enzyme (26), consistent with
the ineffectiveness of this nucleotide in modulating the SepRS/CysRS
preferences. The importance of T-loop, D-loop, and variable loop nucleotides
shown in this work, together with similar findings from studies of the M.
maripaludis enzyme, suggest that SepRS makes important interactions with
the globular tRNA core. Because the A. fulgidus SepRS crystallized as
an unproductive complex in which interactions are made only with the tRNA
anticodon (26), further
structural analyses will be required to provide a reliable basis for these
observations.
The tRNACys Km values of 6–8 μm measured toward SepRS are some 10-fold higher than typically observed for highly efficient cognate aminoacylation by tRNA synthetases, and the equivalent values toward CysRS are more than 100-fold higher. It is notable that the addition of the m1G37 modification, which substantially improves both initial velocities of aminoacylation and aminoacylation plateau values for both enzymes (11, 13) (Fig. 2), still produces such weak apparent complementarity in the enzyme-tRNA interfaces. It appears quite possible, therefore, that further base or ribose modification may be required for optimal Cys-tRNACys formation by SepRS, CysRS, or both enzymes. A positive tRNA modification requirement for efficient aminoacylation has been observed previously in only a small number of cases, primarily involving the wobble nucleotide at the 5′-position of the anticodon in the isoleucyl, glutamyl, and glutaminyl systems (27–31). The only previous observation of a positive tRNA methylation requirement for aminoacylation is the need for 1-methyladenosine at position 9 of nematode mitochondrial tRNAs lacking the T-arm (32). In these cases, however, the methyl group clearly plays a compensating structural role in repairing the tRNA core and may not be directly important for interaction with the tRNA synthetase. It is also of interest to note that the m1G37 modification functions as a negative determinant to ensure aminoacylation specificity of yeast tRNAAsp; when G37 is unmodified in that tRNA, it is significantly misacylated by arginyl-tRNA synthetase (33). We know of no tRNA-synthetase system, however, in which modification at two separate nucleotides has been shown to be required for full aminoacylation efficiency in vitro.
Embedded metabolic linkages among macromolecules responsible for synthesis of Cys-tRNACys, for methanogenesis, and for other aspects of sulfur metabolism could help to explain why the presence of the canonical CysRS has not resulted in elimination of the SepRS/SepCysS pathway in methanogens and the related A. fulgidus, as it has in all other archaea (8). The unusually high number of Fe-S cluster proteins in methanogens may provide one rationale for the existence of unique linkages in these organisms (34). We suggest that the S-adenosylmethione-dependent methylation requirement for Cys-tRNACys synthesis shown here also points to such linkages, since S-adenosylmethione is synthesized intracellularly from methionine (14). Further, free cysteine is known to be a source of metabolic sulfur for biosynthesis in bacteria (35). Hence, the finding that deletion of the SepRS gene in M. maripaludis leads to cysteine auxotrophy suggested a clear connection between the SepRS/SepCysS pathway and cysteine biosynthesis (7). Finally, the finding that SepCysS remains essential in the context of this SepRS deletion (where Cys-tRNACys is provided by CysRS), suggests a further independent role for that enzyme (36).
No correlation is evident between the pathway of cysteine biosynthesis and
the number of tRNACys isoacceptors among the known
Methanosarcina. M. acetivorans, which retains only duplicate copies
of a single tRNACys gene that is highly similar to
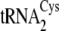 and
and
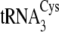 of M. mazei,
possesses both the bacterial and mammalian enzymes needed for cysteine
biosynthesis. However, M. barkeri, which similarly possesses
redundant cysteine biosynthesis pathways, contains all three tRNAs. By
contrast, M. mazei lacks clear homologs for either the
O-acetylserine sulfhydrylase or the cystathionine β-synthase
enzymes needed for the bacterial and mammalian pathways, respectively
(37). Cysteine may be
synthesized in M. mazei entirely by means of deacylating
tRNACys; alternatively, the organism might use a weakly related
O-phosphoserine sulfhydrylase to synthesize cysteine from
phosphoserine and sulfide ions
(38,
39); this enzyme exists in
some archaea and provides an alternative to the bacterial pathway from
O-acetylserine.
of M. mazei,
possesses both the bacterial and mammalian enzymes needed for cysteine
biosynthesis. However, M. barkeri, which similarly possesses
redundant cysteine biosynthesis pathways, contains all three tRNAs. By
contrast, M. mazei lacks clear homologs for either the
O-acetylserine sulfhydrylase or the cystathionine β-synthase
enzymes needed for the bacterial and mammalian pathways, respectively
(37). Cysteine may be
synthesized in M. mazei entirely by means of deacylating
tRNACys; alternatively, the organism might use a weakly related
O-phosphoserine sulfhydrylase to synthesize cysteine from
phosphoserine and sulfide ions
(38,
39); this enzyme exists in
some archaea and provides an alternative to the bacterial pathway from
O-acetylserine.
Despite the inconclusive results from these genome comparisons, it is of
interest to note that the presence of A33 in the unusual
 species may render it
a less efficient substrate for the ribosome, because U33 is required for the
anticodon loop to adopt the conserved U-turn structure. Several studies have
indicated that tRNAs lacking the highly conserved U33 bind the ribosome with
lower efficiency (40,
41), although it has also been
shown that U33 is not essential for tRNAs to perform in eukaryotic in
vitro suppression assays
(42). The dedication of
species may render it
a less efficient substrate for the ribosome, because U33 is required for the
anticodon loop to adopt the conserved U-turn structure. Several studies have
indicated that tRNAs lacking the highly conserved U33 bind the ribosome with
lower efficiency (40,
41), although it has also been
shown that U33 is not essential for tRNAs to perform in eukaryotic in
vitro suppression assays
(42). The dedication of
 to a function other
than protein synthesis would suggest a further linkage between
Cys-tRNACys synthesis and other aspects of metabolism. Although
to a function other
than protein synthesis would suggest a further linkage between
Cys-tRNACys synthesis and other aspects of metabolism. Although
 is clearly an unusual
tRNA, its selective aminoacylation by CysRS, capacity to be methylated by
Trm5, conservation in M. barkeri, and retention of transcription
activation signals all suggest a true functional role.
is clearly an unusual
tRNA, its selective aminoacylation by CysRS, capacity to be methylated by
Trm5, conservation in M. barkeri, and retention of transcription
activation signals all suggest a true functional role.
Sulfur Donor and tRNACys Isoacceptor Substrate Preference of SepCysS—Rigorous characterization of the enzymatic activity of SepCysS, which catalyzes the conversion of Sep-tRNACys to Cys-tRNACys, is challenging because of the need to prepare the misacylated substrate and because the assay requires anaerobic conditions. To first characterize the ligand-bound state of the M. mazei enzyme purified from recombinant E. coli cells, we monitored the UV-visible absorbance of an extensively dialyzed sample. Distinct peaks were observed at 230, 280, and 415 nm; the latter peak corresponds to the lysine-bound ketoenamine form of the cofactor (Fig. 3A) (43). Colorimetric analysis of SepCysS reveals that the molar ratio of PLP to enzyme subunits is 1.2:1, indicating that both active sites bind the cofactor (data not shown) (13). Consistent with this analysis, the crystal structure of A. fulgidus SepCysS showed PLP bound in a covalent aldimine linkage to Lys209 in both subunits (44).
FIGURE 3.

A, UV-visible absorbance spectrum of SepCysS revealing the
presence of bound PLP in the overexpressed recombinant enzyme. B,
imaged TLC plate showing time-dependent formation of Cys-tRNACys
catalyzed by SepCysS. Conditions were as follows: 50 mm Tris (pH
7.5), 20 mm KCl, 10 mm MgCl2, 5 mm
ATP, 3 mm phosphoserine, 5 mm DTT, 10 μm
tRNACys, and 20 μm SepRS. After 5 min,
Cys-tRNACys formation was initiated by the addition of SepCysS to
20 μm and sodium sulfide to 5 mm. The percentage
conversion to Cys-tRNACys is determined by the ratio of the
intensities for Sep-AMP and Cys-AMP and converted to molarity by multiplying
by the concentration of Sep-tRNACys determined from the plateau
aminoacylation value. C, plot of the velocity of
Cys-tRNACys synthesis catalyzed by SepCysS versus enzyme
concentration under steady-state conditions. No activity is observed at enzyme
concentrations below 50 nm. A constant
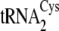 concentration of 5
μm was used.
concentration of 5
μm was used.
We next considered how best to quantitate SepCysS activity. The only reported functional assay monitors the PLP-dependent transformation of 14C-labeled phosphoserine attached to tRNACys into 14C-labeled cysteine (7). Because this assay is insensitive and requires the use of commercially obtained radiolabel at prohibitive cost, we sought to develop an alternative that would be cost-effective, highly sensitive, and hence more suitable for detailed investigation of enzymatic properties. We found that the assay reported here and elsewhere for SepRS function, which generates 32P-labeled Sep-tRNACys with the radiolabel positioned at the 3′-internucleotide linkage of the tRNA, can be extended to monitor the conversion of Sep-tRNACys to Cys-tRNACys (11, 13, 45). After the SepCysS reaction using 32P-labeled Sep-tRNACys as substrate, the tRNA is hydrolyzed to its constituent nucleotides, and Sep-Ap* (substrate) is separated from Cys-Ap* (product) by thin layer chromatography (Fig. 3B). Separate SepRS and CysRS reactions are used as controls to identify the observed spots. The assay allows for straightforward determination of the fraction of substrate tRNA that is phosphoserylated, to control for the possibility that the input Sep-tRNACys may become deacylated in the processing steps between reactions.
We generated the Sep-tRNACys substrate in two ways. First, tRNACys was phosphoserylated to 30–85% plateau levels in an aerobic environment at 37 °C and then isolated by acidic phenol/chloroform extraction and ethanol precipitation. The Sep-tRNACys was then reconstituted in an anaerobic environment, in a buffer consisting of 10 mm sodium cacodylate (pH 6.0) and 2 mm MgCl2. Alternatively, tRNACys transcripts were lyophilized, transported into the anaerobic environment, and subjected to consecutive SepRS and SepCysS reactions without recovering the Sep-tRNACys intermediate. SepCysS converted identical proportions of substrate to product with either method of substrate preparation. Consequently, most experiments were performed using the latter approach, since it obviated the need to isolate Sep-tRNACys and provided continuous assay capability requiring only supplementation of the SepRS reaction with PLP-bound enzyme and sulfur donor. We also verified that both the proportion of overall substrate conversion to product by SepCysS and the rates of conversion were independent of the initial plateau level of phosphoserylation, in the range of 30–85%. Many SepCysS reactions were performed using tRNACys at 40–50% phosphoserylation levels, solely for reasons of economy and efficiency in generating the Sep-tRNACys. All SepCysS reactions were performed under strictly anoxic conditions, using reagents made anoxic through cycles of vacuum followed by purging with oxygen-free argon gas.
All reactions also utilized tRNACys transcripts modified to carry the m1G37 modification. Whether the added methyl group influences the activity of SepCysS is not known, because in the absence of methylation, the levels of Sep-tRNACys that can be synthesized by SepRS are too low to permit measurement of sulfide transfer.
Initial characterization of SepCysS was performed under conditions of molar
enzyme excess over tRNA. Previously, sodium sulfide was used as the sulfur
donor in SepCysS reactions (7).
To explore the sulfur donor requirement more thoroughly, we carried out
reactions using Sep-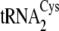 as
substrate, in the presence of 5 mm sodium sulfide, 5 mm
thiophosphate, or 5 mm cysteine. Higher concentrations of any of
these reagents did not increase the overall conversion to product. Each
compound functioned as a sulfur donor, with sodium sulfide (72% end point
conversion of Sep-tRNACys to Cys-tRNACys) providing
higher activity than either thiophosphate or cysteine (52–55%
conversion; Table 2). Only
sodium sulfide yields the correct Cys-tRNACys product; the capacity
of the enzyme to utilize the other donors demonstrates the presence of some
structural adaptability in the active site.
as
substrate, in the presence of 5 mm sodium sulfide, 5 mm
thiophosphate, or 5 mm cysteine. Higher concentrations of any of
these reagents did not increase the overall conversion to product. Each
compound functioned as a sulfur donor, with sodium sulfide (72% end point
conversion of Sep-tRNACys to Cys-tRNACys) providing
higher activity than either thiophosphate or cysteine (52–55%
conversion; Table 2). Only
sodium sulfide yields the correct Cys-tRNACys product; the capacity
of the enzyme to utilize the other donors demonstrates the presence of some
structural adaptability in the active site.
To evaluate whether SepCysS exhibits substrate preference among the three
M. mazei tRNACys isoacceptors, a 5 μm
concentration of each Sep-tRNACys species was incubated with 30
μm SepCysS and 5 mm sodium sulfide, and the reactions
were allowed to proceed to completion over 1 h. The percentage conversion to
Cys-tRNACys was reproducibly higher for
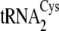 and
and
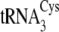 (72–75%) as
compared with
(72–75%) as
compared with  (53%)
(Table 3). By this measure,
then, SepCysS exhibits the same tRNA substrate preference observed for SepRS
(Table 1). Measurement of the
SepCysS substrate conversion efficiencies for tRNA mutants
tRNACysΔ2 and tRNACysΔ4 further confirmed
the SepCysS preference. tRNACysΔ2, which is strongly
preferred by SepRS, is also a good substrate for SepCysS, whereas
tRNACysΔ4, which is shifted substantially toward CysRS
preference, is a poorer SepCysS substrate (Tables
1 and
3). Part of the SepCysS
preference appears to arise from differences in binding affinity to the tRNA
body; measurements of equilibrium dissociation constants for SepCysS
interaction with each native unacylated isoacceptor, by fluorescence quenching
(20), showed that
Kd for
(53%)
(Table 3). By this measure,
then, SepCysS exhibits the same tRNA substrate preference observed for SepRS
(Table 1). Measurement of the
SepCysS substrate conversion efficiencies for tRNA mutants
tRNACysΔ2 and tRNACysΔ4 further confirmed
the SepCysS preference. tRNACysΔ2, which is strongly
preferred by SepRS, is also a good substrate for SepCysS, whereas
tRNACysΔ4, which is shifted substantially toward CysRS
preference, is a poorer SepCysS substrate (Tables
1 and
3). Part of the SepCysS
preference appears to arise from differences in binding affinity to the tRNA
body; measurements of equilibrium dissociation constants for SepCysS
interaction with each native unacylated isoacceptor, by fluorescence quenching
(20), showed that
Kd for
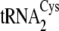 and
and
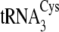 are 2–3-fold
lower than Kd for
are 2–3-fold
lower than Kd for
 (Table 3).
(Table 3).
To further characterize the properties of M. mazei SepCysS, we investigated the kinetics of converting Sep-tRNACys to Cys-tRNACys. Under steady-state conditions at 1–50 nm Sep-CysS, we observed no reaction when Sep-tRNACys substrate levels were maintained below 1 μm. However, product formation was detected at 50 nm SepCysS and 5 μm Sep-tRNACys. At this substrate concentration, reaction velocities then were found to increase with enzyme concentration (Fig. 3C). It is likely that this behavior arises from dissociation of the SepCysS dimer into monomers, with Kd for subunit association in the range of 10-7 m. This interpretation is consistent with the crystal structure of A. fulgidus SepCysS, which shows that each active site is composed of amino acids from both subunits, suggesting that monomers do not retain activity (44). No Sep-tRNACys saturation was observed under steady-state conditions of substrate excess ([S]/[E] > 20) and with Sep-tRNACys concentrations up to 15 μm. However, the initial velocities allow estimation of kcat/Km at 0.009–0.014 min-1 μm-1, with no significant differences measured among the three isoacceptors (Table 3). Levels of product formation substantially exceed the amount of enzyme present, demonstrating unambiguously that SepCysS is able to function as a multiple-turnover catalyst.
The higher substrate conversion efficiency and binding affinities for
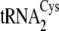 and
and
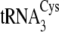 by SepCysS matches the
preference of SepRS and suggests that these two species may be dedicated to
the indirect pathway. This may represent an adaptation that serves to maximize
the efficiency of Cys-tRNACys synthesis in vivo. However,
the SepCysS selectivity is not large and is also not detected at the level of
kcat/Km. Further, our work in assay
development showed that the presence of SepRS in the SepCysS reactions does
not affect either the rate or plateau level of Sep-tRNACys
conversion under these experimental conditions. Nevertheless, work on the
homologous M. jannaschii pathway showed that SepRS and SepCysS from
that organism do form a binary complex that binds tRNACys and that
catalyzes the SepRS reaction with 2-fold higher kcat than
SepRS alone (11). Thus, a
modest enhancement in substrate throughput may be a common characteristic of
the two-step indirect pathway in methanogens. In addition, a further important
role for the formation of the SepRS-SepCysS-tRNACys ternary complex
is to sequester the misacylated Sep-tRNACys from EF-1α, thus
preventing misincorporation of phosphoserine into cellular proteins
(11).
by SepCysS matches the
preference of SepRS and suggests that these two species may be dedicated to
the indirect pathway. This may represent an adaptation that serves to maximize
the efficiency of Cys-tRNACys synthesis in vivo. However,
the SepCysS selectivity is not large and is also not detected at the level of
kcat/Km. Further, our work in assay
development showed that the presence of SepRS in the SepCysS reactions does
not affect either the rate or plateau level of Sep-tRNACys
conversion under these experimental conditions. Nevertheless, work on the
homologous M. jannaschii pathway showed that SepRS and SepCysS from
that organism do form a binary complex that binds tRNACys and that
catalyzes the SepRS reaction with 2-fold higher kcat than
SepRS alone (11). Thus, a
modest enhancement in substrate throughput may be a common characteristic of
the two-step indirect pathway in methanogens. In addition, a further important
role for the formation of the SepRS-SepCysS-tRNACys ternary complex
is to sequester the misacylated Sep-tRNACys from EF-1α, thus
preventing misincorporation of phosphoserine into cellular proteins
(11).
Mechanism of SepCysS—The kinetic experiments performed in the presence of a molar excess of enzyme showed that the required period of incubation to achieve maximal conversion of Sep-tRNACys to Cys-tRNACys by SepCysS is quite long (Fig. 3B). However, 10–40-min time courses were required for maximal substrate conversion by eukarytic selenocysteine synthase, another PLP-dependent enzyme that acts on aminoacylated tRNA (46). The kcat/Km for the M. mazei SepCysS reaction with bisulfide as sulfur donor is also very similar to that reported for the analogous conversion of Ser-tRNASec to Sec-tRNASec by bacterial selenocysteine synthase when thiophosphate is employed as sulfur donor in that enzyme (47).
To further examine the kinetics of SepCysS, we measured substrate conversion while systematically varying the concentration of enzyme. At 5 μm Sep-tRNACys and at SepCysS concentrations above 200 nm, product formation exhibits an upward slope after reactions proceed for ∼30 min (Fig. 4A). Further, at equimolar E/S ratios and under conditions of enzyme molar excess, the reaction progress curve is sigmoidal. For example, at 10 μm Sep-tRNACys and 15–30 μm SepCysS, a distinct lag phase is followed by a rapid increase in product formation, with maximal levels reached at 50–60 min. During the slow lag phase, an intermediate that migrates farthest on the TLC plate is observed to first accumulate and then to disappear as Cys-tRNACys formation begins to rapidly increase (Figs. 3B and 4B). It appears that formation of this intermediate is rate-limiting under single-turnover conditions.
FIGURE 4.

A, time courses of Cys-tRNACys synthesis. Sigmoidal behavior is observed under conditions of enzyme excess. The conditions used were 0.10–30 μm SepRS as indicated by the different symbols, 5 mm sodium sulfide, and 5 μm Sep-tRNACys. Error bars are omitted for clarity but are similar to those depicted in Fig. 3C. B, plot of the decrease in Sep-tRNACys concentration together with Cys-tRNACys formation. Quantitation of spots on the imaged TLC plate resulting in concentration of each species present was plotted against time. The conditions were as follows: 50 mm Tris (pH 7.5), 20 mm KCl, 10 mm MgCl2, 5 mm ATP, 3 mm phosphoserine, 5 mm DTT, 6 μm Sep-tRNACys, and 20 μm SepCysS. A small amount of an intermediate species is observed that forms quickly and then disappears as Cys-tRNACys formation increases.
The distinct lag phase suggests a rate-limiting step associated with the
mechanistic step in which the exogenous sulfur reacts
(Fig. 5). Further, the measured
kcat/Km for conversion of
Sep-tRNACys to Cys-tRNACys is ∼500-fold lower than
kcat/Km for phosphoserylation of
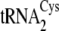 and
and
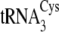 by SepRS (Tables
1 and
3). Thus, the rates by which
the substrate is processed by each enzyme appear to be greatly mismatched;
this would produce a highly inefficient pathway if present in vivo.
Both of these observations suggest that the sulfur donors employed in these
in vitro experiments are not likely to be used in vivo, so
that a major experimental challenge associated with elucidating the true
pathway of sulfur delivery to the active site remains.
by SepRS (Tables
1 and
3). Thus, the rates by which
the substrate is processed by each enzyme appear to be greatly mismatched;
this would produce a highly inefficient pathway if present in vivo.
Both of these observations suggest that the sulfur donors employed in these
in vitro experiments are not likely to be used in vivo, so
that a major experimental challenge associated with elucidating the true
pathway of sulfur delivery to the active site remains.
FIGURE 5.

Proposed mechanism for the synthesis of Cys-tRNACys by SepCysS. The mechanism is based upon a proposed sulfur relay system and on similarity to Sec-tRNA[Ser]Sec biosynthesis. The movement of electrons into PLP has been omitted. The inclusion of bisulfide as the sulfur donor indicates how the enzyme functions in this in vitro study (bottom right; the PLP generated would reform the internal aldimine with Lys222). The identification of Lys222 as the likely side chain that ligates PLP is based on a structure-based sequence alignment with A. fulgidus SepCysS.
By analogy with the mechanism proposed for the PLP-dependent conversion of seryl-tRNASec to selenocysteyl-tRNASec by bacterial selenocysteine synthase (48), we suggest that binding of Sep-tRNACys to SepCysS is followed by formation of a Schiff base between the α-amino group of the phosphoserine residue and the formyl group of pyridoxal 5-phosphate, resulting in elimination of the phosphate group and formation of an aminoacrylyl-tRNACys, which would correspond to the intermediate observed on TLC as aminoacrylyl-A76 (Figs. 3B, 4B, and 5). In our in vitro reactions, HS- or another donor then adds slowly to the double bond, in the rate-limiting step, to generate product (Fig. 5).
Use of the natural donor selenophosphate provides a 330-fold rate enhancement to selenocysteine synthase as compared with thiophosphate (47); such an improvement, if conferred to SepCysS via use of the proper sulfur donor, would suffice to nearly match the catalytic efficiency of SepRS (Tables 1 and 3). It is most likely that the true sulfur donor to tRNA is a persulfide group formed from one or more of the three conserved cysteines in the active site (Cys51, Cys54, and Cys260 in the M. mazei enzyme) (44). The persulfide group provides sulfur for both cofactor and nucleotide biosynthesis in a wide variety of biological systems (49) and could presumably function to increase the reaction rate here by properly positioning the sulfur for transfer in relation to the bound Sep-tRNACys. Interestingly, SepCysS itself is strongly homologous to the cysteine desulfurases CsdB, NifS, and IscS, suggesting that it may function as both a sulfur recipient to form Cys-tRNACys and a sulfur donor in other, as yet unknown, reactions (8, 49). In this regard, the essentiality of the enzyme in the context of a cell deleted for SepRS may be relevant (36). The identity of the enzyme that may convert one of the SepCysS active site cysteines to a persulfide is also unknown at this time; by analogy to pathways such as the sulfuration of tRNA, it is possible that the route of sulfur delivery to SepCysS involves transfer through multiple proteins (50). After the sulfur transfer, the resulting enzyme-bound disulfide would then be reduced by reaction with another cysteine residue, releasing PLP and Cys-tRNACys (Fig. 5). This mechanism predicts essentiality for at least two of the three conserved active site cysteines. The sulfur relay system would then regenerate persulfided SepCysS for the next catalytic cycle.
Formation of Cys-tRNACys by SepCysS probably also has many elements in common with the formation of selenocysteyl-tRNASec in archaea and eukaryotes. In these domains, serylation of tRNA[Ser]Sec by SerRS is followed by a phosphorylation reaction leading to formation of a phosphoserylated tRNA: Sep-tRNA[Ser]Sec. This intermediate is then converted to Sec-tRNASec by another PLP-dependent enzyme designated alternatively as selenocysteine synthase (46, 51) or SepSecS (52). This enzyme uses selenophosphate as a selenide donor and is weakly homologous to SepCysS (46). Kinetic analysis of the selenocysteine synthase reaction has shown that, as proposed for SepCysS and demonstrated for bacterial selenocysteine synthase (48), generation of Sec-tRNA[Ser]Sec from Sep-tRNA[Ser]Sec also occurs in conjunction with an intermediate that is likely to be aminoacrylyl-tRNA[Ser]Sec (46). Interestingly, selenocysteine synthase can also utilize phosphoseryl-tRNA[Ser]Sec as a substrate (46), suggesting that comparative studies among all three enzymes may be a productive approach to acquiring detailed insight into the mechanisms of these remarkable RNA-dependent amino acid modification reactions.
Perspectives—The selectivities of CysRS and SepRS for distinct tRNACys isoacceptors clearly suggest that both synthetic routes and all three tRNAs have physiological significance. The detailed nature of how the tRNAs are partitioned between pathways in vivo, however, and the extent to which one or more of them may play other metabolic roles remain as open questions for future research. In many simpler tRNA-synthetase systems, in vitro reconstitution by expression of a single enzyme, together with use of unmodified tRNA transcripts, suffices to recapitulate most biochemical properties of interest. By contrast, the Cys-tRNACys biosynthetic system in M. mazei illustrates how the full reconstitution of activities is likely to require multiple tRNA modifications as well as essential cofactor proteins. Identification of additional tRNACys-modifying enzymes and of the enzyme that delivers sulfur to SepCysS may suffice for a definitive understanding of tRNA selectivities in CysRS, SepRS, and SepCysS. Such identifications may provide clues that, together with genetic, biochemical, and bioinformatic studies, could elucidate novel metabolic relationships connecting the protein synthesis machinery with sulfur and energy metabolism in methanogens. The crucial role of these archaebacteria in the biogeochemical processes determining global climate change offers strong impetus for these experiments.
Acknowledgments
We thank Ya-Ming Hou for helpful discussions and Dave Valentine for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM63713 (to J. J. P.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: SepRS, phosphoseryl-tRNA synthetase; Sep-CysS, Sep-tRNA:Cys-tRNA synthase; Sep-tRNACys, phosphoseryl-tRNACys; CysRS, cysteinyl-tRNA synthetase; DTT, dithiothreitol; PLP, pyridoxal phosphate; βME, β-mercaptoethanol.
References
- 1.Deppenmeier, U., Johann, A., Hartsch, T., Merkl, R., Schmitz, R. A., Martinez-Arias, R., Henne, A., Wiezer, A., Bäumer, S., Jacobi, C., Brüggemann, H., Lienard, T., Christmann, A., Bömeke, M., Steckel, S., Bhattacharyya, A., Lykidis, A., Overbeek, R., Klenk, H.-P., Gunsalus, R. P., Fritz, H.-J., and Gottschalk, G. (2002) J. Mol. Microbiol. Biotechnol. 4 453-461 [PubMed] [Google Scholar]
- 2.Hippe, H., Caspari, C., Fiebig, K., and Gottschalk, G. (1979) Proc. Natl. Acad. Sci. U. S. A. 76 494-498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boetius, A., Ravenschlag, K., Schubert, C. J., Rickert, D., Widdel, F., Gleseke, A., Amann, R., Jergensen, B. B., Witte, U., and Pfannkuche, O. (2000) Nature 407 623-626 [DOI] [PubMed] [Google Scholar]
- 4.Maeder, D. L., Anderson, I., Brettin, T., Bruce, D., Gilna, P, Han, C. S., Lapidus, A., Metcalf, W. W., Saunders, E., Tapia, R., and Sowers, K. R. (2006) J. Bacteriol. 188 7922-7931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galagan, J. E., Nusbaum, C., Roy, A., Endrizzi, M. G., Macdonald, P., FitzHugh, W., Calvo, S., Engels, R., Smirnov, S., Atnoor, D., Brown, A., Allen, N., Naylor, J., Stange-Thomann, N., and DeArellano, K., et al. (2002) Genome Res. 12 532-542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deppenmeier, U. (2002) Prog. Nucleic Acids Res. Mol. Biol. 71 223-383 [DOI] [PubMed] [Google Scholar]
- 7.Sauerwald, A., Zhu, W., Major, T., Roy, H., Palioura, S., Jahn, D., Whitman, W. B., Yates, J. R., III, Ibba, M., and Soll, D. (2005) Science 307 1969-1972 [DOI] [PubMed] [Google Scholar]
- 8.O'Donoghue, P., Sethi, A., Woese, C. R., and Luthey-Schulten, Z. A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 19003-19008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klenk, H.-P., Clayton, R. A., Tomb, J. F., White, O., Nelson, K. E., Ketchum, K. A., Dodson, R. J., Gwinn, M., Hickey, E. K., Peterson, J. D., Richardson, D. L., Kerlavage, A. R., Graham, D. E., Kyrpides, N. C., Fleischmann, R. D., et al. (1997) Nature 390 364-370 [DOI] [PubMed] [Google Scholar]
- 10.Hohn, M. J., Park, H. S., O'Donoghue, P., Schnitzbauer, M., and Soll, D. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 18095-18100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang, C.-M., Liu, C., Slater, S., and Hou, Y.-M. (2008) Nat. Struct. Mol. Biol. 15 507-514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailly, M., Blaise, M., Lorber, B., Becker, H. D., and Kern, D. (2007) Mol. Cell 28 228-239 [DOI] [PubMed] [Google Scholar]
- 13.Hauenstein, S., Hou, Y.-M., and Perona, J. J. (2008) J. Biol. Chem. 283 21997-22006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Markham, G. D., Hafner, E. W., Tabor, C. W., and Tabor, H. (1980) J. Biol. Chem. 255 9082-9092 [PubMed] [Google Scholar]
- 15.Wada, H., and Snell, E. E. (1961) J. Biol. Chem. 236 2089-2095 [PubMed] [Google Scholar]
- 16.Eliot, A. C., and Kirsch, J. F. (2004) Ann. Rev. Biochem. 73 383-415 [DOI] [PubMed] [Google Scholar]
- 17.Bullock, T. L., Uter, N., Nissan, T. A., and Perona, J. J. (2003) J. Mol. Biol. 328 395-408 [DOI] [PubMed] [Google Scholar]
- 18.Hou, Y.-M., Westof, E., and Giege, R. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 6776-6780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sowers, K. R. (1995) in Archaea: A Laboratory Manual (Sowers, K. R., and Schreier, H. J., eds) pp. 38-39, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- 20.Lakowicz, J. R. (1999) Principles of Fluorescence Spectroscopy, pp. 53-55, Kluwer Academic/Plenum Publishers, New York
- 21.Ehlers, C., Weidenbach, K., Deppenmeier, U., Metcalf, W. W., and Schmitz, R. A. (2005) Mol. Genet. Genomics 273 290-298 [DOI] [PubMed] [Google Scholar]
- 22.Fricke, W. F., Seedorf, H., Henne, A., Kruer, M., Liesegang, H., Hedderich, R., Gottschalk, G., and Thauer, R. K. (2006) J. Bacteriol. 188 642-658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samuel, B. S., Hansen, E. E., Manchester, J. K., Coutinho, P. M., Henrissat, B., Fulton, R., Latreille, P., Kim, K., Wilson, R. K., and Gordon, J. I. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 10643-10648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Komatsoulis, G. A., and Abelson, J. (1993) Biochemistry 32 7435-7444 [DOI] [PubMed] [Google Scholar]
- 25.Wich, G., Hummel, H., Jarsch, M., Bar, U., and Bock, A. (1986) Nucleic Acids Res. 14 2459-2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukunaga, R., and Yokoyoma, S. (2007) Nat. Struct. Mol. Biol. 14 272-279 [DOI] [PubMed] [Google Scholar]
- 27.Muramatsu, T., Nishikawa, K., Nemoto, F., Kuchino, Y., Nishimura, S., Miyazawa, T., and Yokoyama, S. (1988) Nature 336 179-181 [DOI] [PubMed] [Google Scholar]
- 28.Senger, B., Auxilien, S., Englisch, U., Cramer, F., and Fasiolo, F. (1997) Biochemistry 36 8269-8275 [DOI] [PubMed] [Google Scholar]
- 29.Rogers, K. C., and Soll, D. (1993) Biochemistry 32 14210-14219 [DOI] [PubMed] [Google Scholar]
- 30.Kruger, M. K., and Sorensen, M. A. (1998) J. Mol. Biol. 284 609-620 [DOI] [PubMed] [Google Scholar]
- 31.Madore, E., Florentz, C., Giege, R., Sekine, S., Yokoyama, S., and Lapointe, J. (1999) Eur. J. Biochem. 266 1128-1135 [DOI] [PubMed] [Google Scholar]
- 32.Sakurai, M., Ohtsuki, T., and Watanabe, K. (2005) Nucleic Acids Res. 33 1653-1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Putz, J., Florentz, C., Benseler, F., and Giege, R. (1994) Nat. Struct. Biol. 1 580-582 [DOI] [PubMed] [Google Scholar]
- 34.Major, T. A., Burd, H., and Whitman, W. R. (2004) FEMS Microbiol. Lett. 239 117-123 [DOI] [PubMed] [Google Scholar]
- 35.Milhara, H., and Esaki, N. (2002) Appl. Microbiol. Biotechnol. 60 12-23 [DOI] [PubMed] [Google Scholar]
- 36.Sheppard, K., Yuan, J., Hohn, M. J., Jester, B., Devine, K. M., and Soll, D. (2008) Nucleic Acids Res. 36 1813-1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ambrogelly, A., Kamtekar, S., Sauerwald, A., Ruan, B., Tumbula-Hansen, D., Kennedy, D., Ahel, I., and Soll, D. (2004) Cell. Mol. Life Sci. 61 2437-2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mino, K., and Ishikawa, K. (2003) J. Bacteriol. 185 2277-2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oda, Y., Mino, K., Ishikawa, K., and Ataka, M. (2005) J. Mol. Biol. 351 334-344 [DOI] [PubMed] [Google Scholar]
- 40.Dix, D. B., Wittenberg, W. L., Uhlenbeck, O. C., and Thompson, R. C. (1986) J. Biol. Chem. 261 10112-10118 [PubMed] [Google Scholar]
- 41.Ashraf, S. S., Ansari, G., Guenther, R., Sochacka, E., Malkiewicz, A., and Agris, P. F. (1999) RNA 5 503-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bare, L., Bruce, A. G., Gesteland, R., and Uhlenbeck, O. C. (1983) Nature 305 554-556 [DOI] [PubMed] [Google Scholar]
- 43.Zhou, X., and Toney, M. D. (1999) Biochemistry 38 311-320 [DOI] [PubMed] [Google Scholar]
- 44.Fukunaga, R., and Yokoyoma, S. (2007) J. Mol. Biol. 370 128-141 [DOI] [PubMed] [Google Scholar]
- 45.Wolfson, A. D., and Uhlenbeck, O. C. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 5965-5970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu, X.-M., Carlson, B. A., Mix, H., Zhang, Y., Saira, K., Glass, R. S., Berry, M. J., Gladyshev, V. N., and Hatfield, D. L. (2007) PLoS Biol. 5 96-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tormay, P., Wilting, R., Lottspeich, F., Mehta, P. K., Christen, P., and Bock, A. (1998) Eur. J. Biochem. 254 655-661 [DOI] [PubMed] [Google Scholar]
- 48.Forchhammer, K., and Bock, A. (1991) J. Biol. Chem. 266 6324-6328 [PubMed] [Google Scholar]
- 49.Mueller, E. G. (2006) Nat. Chem. Biol. 2 185-195 [DOI] [PubMed] [Google Scholar]
- 50.Ikeuchi, Y., Shigi, N., Kato, J., Nishimura, A., and Suzuki, T. (2006) Mol. Cell 21 97-108 [DOI] [PubMed] [Google Scholar]
- 51.Ganichkin, O. M., Xu, X.-M., Carlson, B. A., Mix, H., Hatfield, D. L., Gladyshev, V. N., and Wahl, M. C. (2008) J. Biol. Chem. 283 5849-5865 [DOI] [PubMed] [Google Scholar]
- 52.Yuan, J., Palioura, S., Salazar, J. C., Su, D., O'Donoghue, P., Hohn, M. J., Cardoso, A. M., Whitman, W. B., and Soll, D. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 18923-18927 [DOI] [PMC free article] [PubMed] [Google Scholar]