

Abstract
Arf GAPs are a family of enzymes that catalyze the hydrolysis of GTP bound to Arf. Arf GAP1 is one member of the family that has a critical role in membrane traffic at the Golgi apparatus. Two distinct models for the regulation of Arf GAP1 in membrane traffic have been proposed. In one model, Arf GAP1 functions in a ternary complex with coat proteins and is inhibited by cargo proteins. In another model, Arf GAP1 is recruited to a membrane surface that has defects created by the increased membrane curvature that accompanies transport vesicle formation. Here we have used kinetic and mutational analysis to test predictions of models of regulation of Arf GAP1. We found that Arf GAP1 has a similar affinity for Arf1·GTP as another Arf GAP, ASAP1, but the catalytic rate is ≈0.5% that of ASAP1. Coatomer stimulated Arf GAP1 activity; however, different from that predicted from the current model, coatomer affected the Km and not the kcat values. Effects of most mutations in Arf GAP1 paralleled those in ASAP1. Mutation of an arginine that aligned with an arginine presumed to be catalytic in ASAP1 abrogated activity. Peptide from the cytoplasmic tail of cargo proteins inhibited Arf GAP1; however, the unrelated Arf GAP ASAP1 was also inhibited. The curvature of the lipid bilayer had a small effect on activity of Arf GAP1 under the conditions of our experiments. We conclude that coatomer is an allosteric regulator of Arf GAP1. The relevance of the results to the two models of Arf GAP1-mediated regulation of Arf1 is discussed.
Arf family GTP-binding proteins are critical regulators of membrane traffic and the actin cytoskeleton (1, 2). Many of the protein components of Arf-regulated membrane traffic have been identified, and several models for the function of Arf have been proposed. In the prevailing models, vesicle coat proteins bind to membranes and drive the formation of transport intermediates (3–6). For the transport intermediates to interact with a target membrane, the coat proteins dissociate. Arf·GTP, which binds to membranes and to coat proteins simultaneously, recruits the coat proteins to membranes. Arf·GDP does not tightly associate with membranes and does not bind coat proteins. The cycle of coat protein binding and dissociation is linked to the cycle of Arf binding to and hydrolysis of GTP. Arf proteins, however, do not have detectable intrinsic GTPase activity and depend on accessory proteins called GTPase-activating proteins (GAPs)2 for the hydrolysis of GTP (7–10).
Arf GAPs are a family of proteins that induce the hydrolysis of GTP bound to Arf (2, 8–10). Humans have 31 genes that encode proteins with Arf GAP domains. Of these proteins, 18 have demonstrated GAP activity, and 8 with GAP activity have been found to affect membrane traffic. Arf GAP1, the first Arf GAP to be purified and cloned (11), is the most extensively studied. It regulates membrane traffic at the Golgi by inducing hydrolysis of GTP bound to Arf1 (11–14). Two distinct regulatory mechanisms that ensure that GTP on Arf1 is hydrolyzed after the polymerization of vesicle coat and formation of a transport intermediate have been proposed. One mechanism involves vesicle coat proteins and cargo (15, 16). Coatomer was found to increase the reaction velocity when an Arf1 mutant, [Δ17]Arf1, was used as a substrate. The stimulation is thought to be due to coatomer introducing a catalytic residue into the Arf1·GTP·Arf GAP1 complex. Another group reported that coatomer has a limited effect if wild type myristoylated Arf1 (myrArf1), the naturally occurring form of Arf1, was used as a substrate, which has been taken to refute the idea that coatomer binding to Arf GAP1 regulates activity in normal physiology (17). Nevertheless, coatomer binds to cargo, and a peptide from the cytoplasmic tail of a cargo protein, p24, was found to inhibit GAP activity. Based on the inhibition, a proofreading mechanism for cargo sorting was proposed (15). The slow GTPase rate in the presence of cargo was thought to be sufficient to explain GTP hydrolysis following transport vesicle formation.
Another proposed regulatory mechanism invokes the ability of Arf GAP1 to sense defects in membrane surfaces that accompany increases in membrane curvature (18–20). Antonny and co-workers (19, 21) have defined the mechanism by which Arf GAP1 detects curvature. Arf GAP1 has two motifs called Arf GAP lipid packing sensors (ALPS). The motifs are amphipathic helices induced when inserted into the hydrophobic center of a lipid bilayer. The hydrophobic center becomes available when the packing density of the lipid head groups is decreased, which occurs when the curvature increases. In the Antonny model, Arf GAP1 is recruited to membranes containing its substrate Arf1·GTP when curvature increases consequent to forming a transport intermediate. Recruitment is the primary means of regulating the GAP.
The models for Arf GAP1 regulation of membrane traffic invoke changes in enzymatic rate in response to either protein binding or the physical state of the lipid bilayer; however, enzymatic parameters for Arf GAP1 have not been determined. To date, ASAP1 has been the only Arf GAP for which catalytic rates and affinities have been determined (22). Different from one model for Arf GAP1, ASAP1 is able to catalyze GTP hydrolysis in a bi-protein complex with Arf. The catalytic rate is 5–20-fold greater than the rates reported for Ras and Rho GAPs (23–26). The affinity for Arf is ∼2 μm. Both the affinity and the catalytic rate are affected by the phospholipid composition of the bilayer on which the reaction occurs. The geometry of the bilayer surface does not affect the catalytic rate, which is different from that reported for Arf GAP1.
Here we report the examination of the enzymology of Arf GAP1. We found that Arf GAP1 has less enzymatic power than ASAP1 and that coatomer allosterically modifies Arf GAP1 activity, affecting the affinity of Arf GAP1 for Arf1·GTP but not the catalytic constant. Our results support the idea that coatomer has a regulatory role in Arf GAP1 function.
EXPERIMENTAL PROCEDURES
Plasmids—Plasmids for the bacterial expression of [Δ17]Arf1, Arf1, and N-myristoyltransferase have been described previously (27–29). The plasmid for expression of [1–257]Arf GAP1-His6 was obtained from Victor Hsu and was described by Cassel and co-workers (30). The plasmids used to prepare baculovirus for expression of [1–415]Arf GAP1-His6, called [1–415]Arf GAP1-His throughout the text, and [1–415]Arf GAP1-His-GFP, called [1–415]Arf GAP1-GFP, were prepared by amplifying the open reading frame for Arf GAP1-GFP from a plasmid, generously provided by J. Lippincott-Schwartz, by PCR and subcloning it into a modified pDONr201 donor vector (Invitrogen) forming the entry clone. The reading frame was then subcloned into the baculovirus expression vector pDEST8 with the Gateway LR recombination reaction. The plasmid containing the reading frame for His10 [325–724]ASAP1 in pET19b has been described (31). Point mutations of [1–415]Arf GAP1-His and [1–415]Arf GAP1-GFP were generated using the QuickChange site-directed mutagenesis kit according to the manufacturer's protocols (Stratagene). The mutations were confirmed by sequencing.
Protein Purification—Human myrArf1 protein was expressed in and purified from bacteria as described (22, 28, 29, 32). [Δ17]Arf1 was expressed and purified as described (27, 33). His10 [325–724]ASAP1 was expressed and purified as described (31). [1–257]Arf GAP-His6 protein was purified from inclusion bodies (31). [1–415]Arf GAP1-His and [1–415]Arf GAP1-GFP were expressed in H5 insect cells with the BAC-to-BAC® baculovirus expression system following the manufacturer's protocol (Invitrogen). Pellets from 200 ml of H5 insect cells containing the expressed proteins were lysed in 20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 10% glycerol, 0.1% Triton X-100 with a protease inhibitor mixture (Complete™, Roche Applied Science). The lysate was fractionated using a 5-ml HiTrap Q column developed in a linear gradient of 30–400 mm NaCl in 100 mm Tris-HCl, pH 8.0, 10% glycerol. Fractions containing Arf GAP1 were applied to a 1-ml of HisTrap HP column (GE Healthcare) and eluted using an imidazole gradient from 20 to 600 mm in 20 mm Tris-HCl, pH 8.0, 500 mm NaCl, and 10% glycerol. Imidazole was removed by dialysis against 20 mm Tris HCl, pH 8.0, 400 mm NaCl, and 10% glycerol. In the indicated experiments, [1–415]Arf GAP1-His was treated with 6 m urea for 10 min. The protein was refolded by diluting the urea 200-fold when adding the protein to the GAP reaction. Arf GAP1 treated with urea is referred to as Arf GAP1dr (dr indicates denaturation-renaturation). Coatomer was purified from frozen rat liver according to Pavel et al. (50). The coatomer fractions were identified by Western blotting using anti-β-COP antibody.
Large Unilamellar Vesicles (LUVs)—LUVs for most experiments were formed by extrusion as described (34) and consisted of 50% phosphatidylcholine (PC), 19% phosphatidylethanolamine (PE), 5% phosphatidylserine (PS), 10% phosphatidylinositol (PI), and 16% cholesterol. In LUVs containing phosphatidylinositol 4-phosphate (PI4P), phosphatidylinositol 4,5-bisphosphate (PIP2), dioleoylglycerol, or phosphatidic acid, PC concentration was reduced to maintain a total lipid concentration of 500 μm. Extrusion through filters with pore sizes of 1.0, 0.4, 0.1, 0.05, and 0.03 μm yielded vesicles with approximate mean radii of 176, 129, 70, 54, and 38 nm, as estimated by dynamic light scattering (34).
GAP Assays—GAP activity was measured at 30 °C and was assayed as described previously (31). Briefly, the hydrolysis of GTP bound to Arf1 was determined in one of two ways. First, [α-32P]GTP was used to follow the reaction. Arf1 was incubated with [α-32P]GTP to form the complex Arf1·[α-32P]GTP (31), which was used as a substrate for single turnover experiments, GAP titrations used to determine C50 values (see below), and in some time courses. Second, tryptophan fluorescence, determined by exciting with 297 nm light and measuring emission at 340 nm, was used to follow Arf1·GTP to Arf1·GDP conversion (22, 31). Arf1·GTP has a greater signal than does Arf1·GDP. Tryptophan fluorescence could be continuously monitored and was used to determine initial rates for saturation kinetics. To determine the C50 values, Arf GAPs were titrated into a reaction mixture containing 0.5 μm myrArf1·[α-32P]GTP and LUVs prepared as indicated. The reaction was stopped at 3 min by dilution and temperature shift, and the relative amounts of GTP and GDP bound to Arf were determined (31, 35). Time courses of GTP hydrolysis under conditions in which the GAP was limiting were performed by removing samples from a reaction after variable times, stopping the reaction as for the C50 determination, and measuring relative levels of GDP and GTP bound to Arf1. Single turnover experiments, in which GAP was in excess and Arf1·GTP was limiting, were performed using a quench flow instrument (KinTek, Inc., Austin, TX) (22).
Tryptophan fluorescence was determined using a FluorMax 3 spectrophotometer from Jobin Yvon Horiba (Edison, NJ) (22). MyrArf1 or [Δ17]Arf1 were loaded with GTP in a buffer containing 20 mm Hepes, pH 7.5, 100 mm NaCl, 1 mm dithiothreitol, 0.5 mm MgCl2, 1 mm EDTA, 100 μm GTP, and LUVs for 30 min. The concentration of MgCl2 was adjusted to a final concentration of 1.5 mm before Arf GAPs were added to initiate the reaction.
KES Peptide Association with LUVs—A peptide composed of the ALPS domain from the yeast protein KES fused to two lysines at the amino terminus and conjugated to nitrobenzoxadiazole (NBD) at the COOH terminus (KKSSSWTSFLKSIASFNGDLSSLSAK- NBD) was incubated at a concentration of 100 nm in phosphate-buffered saline with either no LUVs or LUVs prepared by extrusion through membranes with the indicated diameter pores. Fluorescence spectra were determined exciting at 460 nm and scanning for emission from 490 to 610 nm. Signal due to light scatter from the vesicles was subtracted.
Lipid Binding Assay—Sucrose-loaded LUVs were prepared as described (34, 36). 0.5 μm of [1–257]Arf GAP1 or [1–415]Arf GAP1-His were incubated with 0.2 μm of coatomer and sucrose-loaded LUVs in 25 mm Hepes, pH 7.5, 100 mm NaCl, 2 mm MgCl2 and 1 mm EDTA for 5 min at 30 °C. The LUVs were pelleted by centrifugation at 100,000 × g for 15 min at 4 °C. The Arf GAP1 bound to the vesicles was separated by SDS-PAGE and visualized using Coomassie Blue dye. The signal was quantified by densitometry using Scion image.
Miscellaneous—All lipids were obtained from Avanti Polar Lipids (Alabaster, AL), including dioleoylglycerol, phosphatidic acid (from chicken egg PC), PC (chicken egg), PE (bovine liver), PS (porcine brain), PI (bovine liver), PI4P (porcine brain), PIP2 (porcine brain), and cholesterol. KES-NBD peptide and p24β cargo peptide (YYLKRFFEVRRVV) (15) were synthesized by Anaspec (San Jose, CA). Frozen rat livers for purification of coatomer were purchased from Pelfreez Biological (Rogers, AR). Rabbit polyclonal anti-β-COP antibody to detect β-COP for coatomer purification was from ABR Affinity Bioreagents (Golden, CO). Protein concentrations were estimated using the Bio-Rad assay. All data presented are either the summary of the indicated number of experiments or representative data of at least two experiments with similar results.
RESULTS
We set out to test two hypotheses about the regulation of Arf GAP1. The first hypothesis is that Arf GAP1 is regulated by curvature of the lipid bilayer. The second is that coatomer provides the catalytic residue to the Arf1·GTP·Arf GAP1 complex to initiate GTP hydrolysis. We have used preparations of recombinant full-length Arf GAP1 fused to either a hexahistidine tag ([1–415]Arf GAP1-His) or green fluorescent protein ([1–415]Arf GAP1-GFP) at the COOH terminus that were expressed and purified from insect cells.
To optimize lipid conditions prior to characterizing baseline kinetics of Arf GAP1, we determined the amount of enzyme required for 50% hydrolysis in a fixed time (referred to as the C50) using LUVs of different sizes as a reaction surface. The LUVs were prepared by extrusion through filters with the indicated diameter pores to control mean vesicle diameter (see “Experimental Procedures”). The reaction was followed by the conversion of [α-32P]GTP bound to Arf1 to [α-32P]GDP (Fig. 1A and Table 1). Using [1–415]Arf GAP1-His, the C50 was independent of LUV size. The report in which vesicle size dependence was described used a different assay for GAP activity. The conversion of Arf1·GTP to Arf1·GDP was followed by a change in tryptophan fluorescence (20). To ensure that the difference in results was not consequent to the assay, the activity of Arf GAP1 was determined following changes in tryptophan fluorescence. Vesicle size affected the apparent catalytic rate by about 50% (Fig. 1B).
FIGURE 1.

Dependence of Arf GAP1 activity on the size of vesicles. The hydrolysis of GTP bound to myrArf1 was induced by [1–415]Arf GAP1-His in the presence of LUVs formed by extrusion through membranes with the indicated pore sizes. Total phospholipid concentration was 500 μm. A, determination of C50. [1–415]Arf GAP1-His was titrated into a reaction containing [α-32P]GTP·myrArf1. The fraction of bound GTP converted to GDP in 3 min was determined as described under “Experimental Procedures.” B, time course of Arf1·GTP to Arf1·GDP conversion determined by change in tryptophan fluorescence. The reaction contained 2 μm myrArf1·GTP and 40 nm [1–415]Arf GAP1-His.
TABLE 1.
Effect of vesicle size on Arf GAP1 activity
C50 values for [1–415]Arf GAP1-His, [1–257]Arf GAP1, and [1–415]Arf GAP1dr were determined using myrArf1·[α-32P]GTP as a substrate in reactions containing LUVs formed by extrusion through membranes with pores of the indicated diameters at a total phospholipid concentration of 500 μm. The data presented are the average and standard deviation of three experiments.
|
C50 (nm)
|
|||
|---|---|---|---|
| 0.4 μm | 0.1 μm | 0.03 μm | |
| [1-415]Arf GAP1-His | 33.7 ± 6.0 | 30.4 ± 3.0 | 35.2 ± 14.8 |
| [1-257]Arf GAP1 | 41.0 ± 10.8 | 123.0 ± 25.9 | 24.1 ± 2.8 |
| [1-415]Arf GAP1dr | 51.1 ± 9.9 | 38.0 ± 4.2 | 50.5 ± 4.2 |
We considered reasons for the lack of vesicle curvature dependence in our experiments. There were three differences between the experimental conditions we used and those used in the reports of curvature dependence. The first difference was the assay. As described above, it alone could not account for the difference in results. The recombinant proteins were also prepared differently. In our experiments, we used Arf GAP1 expressed in insect cells. The protein was purified from a soluble fraction of the cell lysate. Most work previously reported has used protein expressed in bacteria and purified from inclusion bodies using a denaturant to solubilize the proteins. The third difference in experimental conditions was lipid concentration. Although we typically used total phospholipid concentrations of 500–1000 μm, the reports of curvature sensitivity used 200 μm. Using 200 μm lipid, and examining tryptophan fluorescence to measure the conversion of Arf1·GTP to Arf1·GDP, bacterially expressed [1–257]Arf GAP1 activity had a greater dependence on curvature than we had observed with [1–415]Arf GAP1-His, with about 10-fold more activity observed with the smaller vesicles (Fig. 2A). The effect, however, was less than that reported previously (19, 20). If lipid concentration was raised to 500 μm, the effect of vesicle size on apparent activity was smaller, with a difference in rate of about 2-fold (Fig. 2B). Also, [1–415]Arf GAP1-His expressed and purified from insect cells was less sensitive, with a 40% difference in reaction rate under conditions in which a 10-fold difference was observed for [1–257]Arf GAP1 (compare Fig. 2, C with A). [1–415]Arf GAP1-His expressed and purified from insect cells, denatured with urea (designated [1–415]Arf GAP1dr) and then diluted into the reaction, had a greater sensitivity to curvature than protein that had not been treated with urea, with a large decrease in tryptophan fluorescence observed in the first 10–30 s following addition of GAP and an apparent 12-fold greater rate with the smaller vesicles (compare Fig. 2, D with C). We also determined whether using an assay that relied on the conversion of myrArf1·[α-32P]GTP to myrArf1·[α-32P]GDP influenced the ability to detect an effect on vesicle size on GAP activity. When examining either enzymatic power by determination of C50 or time dependence of [α-32P]GTP hydrolysis, effects of vesicle size were observed, but they were of a smaller magnitude and were less consistent than those observed with the assay based on tryptophan fluorescence of Arf1 (Fig. 3 and Table 1).
FIGURE 2.

Factors influencing the dependence of the GAP reaction on the size of vesicles. A, protein preparation and lipid concentration used in the original report of curvature dependence. Tryptophan fluorescence was used to monitor the conversion of myrArf1·GTP to myrArf1·GDP in an assay containing 2 μm myrArf1·GTP, 40 nm [1–257]Arf GAP1, and LUVs formed by extrusion through membranes with either 0.4- or 0.03-μm pores. Total phospholipid concentration was 200 μm. B, effect of lipid concentration on vesicle size dependence of GAP reaction. The conditions were identical to those described in A, except total phospholipid concentration was 500 μm. C, effect of vesicle size on activity of [1–415]Arf GAP1-His under lipid conditions used in first report of curvature dependence. Conditions were identical to those in A, but 40 nm [1–415]Arf GAP1-His was used as the enzyme. D, effect of denaturing and renaturing [1–415]Arf GAP1-His on curvature dependence. Conditions for the assay were identical to those described in A, but [1–415]Arf GAP1-His was treated with 6 m urea and then diluted into the assay to a final concentration of 40 nm. The protein that had been treated with urea is indicated as [1–415]Arf GAP1dr.
FIGURE 3.

Effect of vesicle size on GAP-induced hydrolysis of [α32P]GTP. A and B, effect on C50. [1–257]Arf GAP1 (A) or [1–415]Arf GAP1dr (B) was titrated into a reaction containing [α-32P]GTP·Arf1 as a substrate, and LUVs were extruded through membranes with pores of the indicated size. Total phospholipid concentration was 500 μm. The extent of hydrolysis of GTP bound to myrArf1 in 3 min was determined. C, time dependence of hydrolysis of [α-32P]GTP induced by [1–257]Arf GAP1. 1.6 nm [1–257]Arf GAP1 was incubated with 0.2 μm myrArf1·[α-32P]GTP, and LUVs were extruded through membranes with pores of the indicated sizes. Total phospholipid concentration was 200 μm. Samples of the reaction were taken at the indicated times and stopped, and extent of GTP hydrolysis was determined.
Vesicle size has also been reported to affect the affinity of vesicles for Arf GAP1 and other proteins or peptides containing ALPS domains. We first tested this idea with a peptide comprising the ALPS domain of a yeast protein called KES. The assay used a peptide conjugated to a fluorescent molecule, NBD. Binding to lipids was detected as an increase in fluorescent emission. As reported previously (19, 21), we found that binding efficiency of the peptide was inversely proportional to the diameter of the vesicles (Fig. 4). To determine protein binding to vesicles of different curvatures, we prepared sucrose-filled LUVs by extrusion through membranes with the pore sizes indicated in Fig. 5. The vesicles were incubated with Arf GAP1 and rapidly precipitated by centrifugation. Protein in the pellet was fractionated by gel electrophoresis and visualized with Coomassie Blue Dye. [1–257]Arf GAP1, prepared from bacteria, bound to the vesicles dependent on the vesicle size. However, the effect was less than 2-fold (Fig. 5A), and an effect of vesicle size was not observed if total phospholipid concentration was increased to 1 mm (Fig. 5B). Binding of [1–415]Arf GAP1-His was not dependent on vesicle size at either phospholipid concentration (Fig. 5, C and D). Although these results do not exclude curvature sensitivity of Arf GAP1 (see “Discussion”), we did not observe it and, therefore, did not consider the effects of curvature in kinetic analyses.
FIGURE 4.

Binding of an isolated ALPs domain to vesicles. The emission spectra of a peptide, comprised of the ALPS domain of the yeast protein KES conjugated to NBD, incubated with no vesicles, and LUVs formed by extrusion through membranes with 1-μm pores or LUVs extruded through membranes with 0.05-μm pores were determined as described under “Experimental Procedures.”
FIGURE 5.

Association of Arf GAP1 with LUVs, dependence on vesicle size, lipid concentration, and coatomer. The indicated preparations of Arf GAP1 were incubated with sucrose-filled LUVs formed by extrusion through membranes of the indicated pore sizes and at the indicated total phospholipid concentration. Where indicated, 0.48 μm coatomer was included in the incubation. The vesicles were separated from the bulk solution by centrifugation, and the proteins associated with the vesicles were measured. A, [1–257]Arf GAP1 and 200 μm phospholipids. B, [1–257]Arf GAP1 and 1 mm phospholipids. C, [1–415]Arf GAP1-His and 200 μm phospholipids. D, [1–415]Arf GAP1-His and 1 mm phospholipids.
In our kinetic analysis, Arf1·GTP was treated as a substrate and Arf GAP1 as the enzyme. Km and Vmax values were determined by saturation kinetics using [1–415]Arf GAP1-His and LUVs extruded through membranes with 1-μm pores (Fig. 6A and Table 2). MyrArf1 is the naturally occurring form of Arf1 and was used as a substrate in these experiments. The Km value for myrArf1 as a substrate was similar to that reported for ASAP1, but the kcat was less than 1/250th that of ASAP1. MyrArf1 bound to GTP associates tightly with hydrophobic surfaces. [Δ17]Arf1, which has the α-helical amino-terminal extension deleted, is soluble when GTP-bound and is technically less challenging to prepare in large quantities. It has been used for studying Arf GAP1 in reports describing the proofreading model. To be able to make comparisons with work using [Δ17]Arf1, we also determined Km and Vmax values using [Δ17]Arf1 as a substrate. We had previously reported that [Δ17]Arf1 is not used as efficiently as myrArf1. Consistent with this observation, the Km value was greater and the kcat value, calculated from the Vmax, was less than the corresponding parameters for myrArf1 (Fig. 6B and Table 2).
FIGURE 6.

Kinetic analysis of Arf GAP1 activity. A, saturation kinetics with myrArf1 as a substrate. The conversion of Arf1·GTP to Arf·GDP was followed using tryptophan fluorescence in a reaction containing 40 nm [1–415]Arf GAP1-His, LUVs extruded through a 1-μm pore filter, and the indicated concentrations of myrArf1·GTP. Total phospholipid concentration was 500 μm. Initial rates were estimated, and the plot of initial rate versus myrArf1·GTP was fit to the Michaelis-Menten equation to estimate the Km and Vmax. B, saturation kinetics using [Δ17]Arf1·GTP as a substrate. The reaction conditions were the same as described in A, except 3.6 μm [1–415]Arf GAP1-His was used, and the substrate was [Δ17]Arf1. Reactions contained 0.48 μm coatomer where indicated. C, single turnover kinetics. The rate of GTP hydrolysis using 0.5 μm myrArf1·[α-32P]GTP as a substrate and the indicated concentrations of [1–415]Arf GAP1-His was determined as described under “Experimental Procedures.” The hydrolysis rate was determined from the plots of %GDP versus time. The rate versus [1–415]Arf GAP1-His concentration was plotted and fit to a hyperbola to determine kcat and Km values, as described under “Experimental Procedures” and in Ref. 22.
TABLE 2.
Kinetic parameters for [1–415]Arf GAP1-His
Saturation kinetics to determine Km and Vmax were performed by titrating myrArf1 or [Δ17]Arf1 into a reaction containing 40 nm [1–415]Arf GAP1-His when using myrArf1 as a substrate or 3.6 μm [1–415]Arf GAP1-His when using [Δ17]Arf1 as a substrate. The reaction mixture also contained LUVs formed by extrusion through membranes with 1-μm diameter pores. Total phospholipid concentration was 500 μm. Where indicated the reaction also contained 0.48 μm coatomer. The data were analyzed under the steady state assumption. The kcat value was calculated as kcat = Vmax/total enzyme. For single turnover kinetics, 0.5 μm myrArf·[α-32P]GTP was used as substrate, and [1–415]Arf GAP1-His was titrated into the reaction.
|
Substrate
|
Steady state
|
Single turnover
|
||
|---|---|---|---|---|
| Km | kcat | Km | kcat | |
| μm | s | μm | s | |
| myrArf1 | 1.4 ± 0.6 | 0.23 ± 0.017 | 0.96 ± 0.2 | 0.18 ± 0.04 |
| [Δ17]Arf1 | 68.4 ± 12.6 | 0.046 ± 0.0006 | ||
| [Δ17]Arf1 + coatomer | 25.3 ± 5.9 | 0.05 ± 0.0.0003 | ||
Single turnover kinetic analysis was used to address the concern that the Arf GAP1 preparation used was not properly folded resulting in a low apparent kcat. In these experiments, Arf GAP1 was titrated into a reaction containing a limiting concentration of Arf1·GTP, and the reaction was stopped using a rapid mixing system (quench flow). At saturating concentrations of enzyme, the rate of hydrolysis was the kcat. The value was similar to that determined by standard kinetic analysis in which the substrate was titrated into the reaction, and the data were analyzed using the steady state or rapid equilibrium assumption (Fig. 6C and Table 2). The Km value was also similar, indicating that the substrate, Arf1, was properly folded.
We analyzed the effect of mutations in Arf GAP1 in residues analogous to those in ASAP1 known to contribute to GAP activity (Fig. 7). The amount of [1–415]Arf GAP1-His required to achieve 50% hydrolysis of GTP in a fixed time point assay (the C50) was determined (Table 3). Similar to ASAP1, changing arginine 50, analogous to arginine 497 of ASAP1, to lysine resulted in a protein with little detectable GAP activity. Also similar to ASAP1, changing aspartate 65, analogous to aspartate 512 of ASAP1, to alanine resulted in a protein with ∼1/300th the activity of wild type protein. However, there were a number of differences between the proteins. Notably, changing tryptophan 32 in Arf GAP1 resulted in a protein with ¼ the activity of wild type; changing the analogous residue in ASAP1 resulted in a protein with 1/4000th the activity of wild type protein. We also prepared Arf GAP1 recombinant proteins with changes in the ALPS domain (Table 3). Recombinant Arf GAP1-His with leucine 197 or phenylalanine 204 changed to alanine had ¼ the activity of wild type protein. Recombinant [W201A]Arf GAP1 had ¼ the activity of wild type protein. These results were obtained with freshly prepared proteins. One freeze-thaw cycle resulted in a significant but variable loss of activity.
FIGURE 7.

Sequence alignment of Arf GAP domain of Arf GAP1 and ASAP1. The GenBank™ accession number for Arf GAP1 is U35776 and for ASAP1 is AF075461. [1–257]Arf GAP1 was aligned with [441–724]ASAP1 using ClustalW2.
TABLE 3.
Mutational Analysis
The C50 values for the indicated point mutants of [1–415]Arf GAP1-His and [325–724]ASAP1 were determined using myrArf·[α-32P]GTP as a substrate in a reaction containing LUVs formed by extrusion through a 1-μm diameter pore filter. Total phospholipid concentration was 500 μm. [W32A]Arf GAP1 was expressed as a fusion protein with GFP instead of His6 because the His-tagged protein was not stable. The number in parentheses indicates the number of experiments.
|
His10[325-724]
ASAP1a
|
[1-415]Arf GAP1
|
|||
|---|---|---|---|---|
| Protein | C50 | Protein | C50 | kcat |
| nm | nm | s-1 | ||
| Wild type | 0.08 ± 0.009 (4) | Wild type | 31.5 ± 3.4 (4) | 0.23 ± 0.017 |
| W479A | 313 ± 53 (3) | W32A-GFP | 145 ± 27 (3) | |
| I490A | 14 ± 1.4 (3) | L43A | 88 ± 6.0 (3) | |
| R497K | >10,000 (3) | R50K | >17500 (3)b | <0.0002 |
| R505A | 34 ± 3 (3) | F58A | 1410 ± 210.1 (3)c | 0.003 ± 0.0002 |
| L511A | 9.8 ± 1.8 (3) | M64A | 1230 ± 33 (3) | |
| D512A | 35 ± 2.8 (3) | D65A | >10,000d | 0.0007 ± 0.00004 |
| L197A | 60.0 ± 7.2 (3) | |||
| W201A | 121 ± 19 (3) | |||
| F204A | 52.2 ± 6.0 (3) | |||
Data were taken from Luo et al. (22).
Data show less than 2% GTP hydrolysis in 3 min with 1.75 μm [R50K]Arf GAP1.
Data show less than 60% GTP hydrolysis in 3 min with 5.9 μm [F58A]Arf GAP1.
Data show less than 15% GTP hydrolysis in 3 min with 3.7 μm [D56A]Arf GAP1.
Coatomer and cargo are important components in one model for regulation of Arf GAP1. We examined the contribution of coatomer to enzymatic activity. In the proposed model, Arf GAP1, Arf1·GTP, and coatomer form a complex, with Arf1·GTP binding to both coatomer and Arf GAP1. Coatomer contributes a catalytic residue that induces hydrolysis of GTP. We developed kinetic models to test this idea. We considered two cases. In one, coatomer functions as a conventional modifier, forming a complex with the enzyme-substrate complex and increasing enzymatic power (Fig. 8, Scheme 1). Using this scheme, we derived equations under rapid equilibrium assumptions (37). A plot of initial reaction velocity versus coatomer concentration is a hyperbola. If coatomer changes the affinity for substrate (i.e. Km), the proportional effect of coatomer on velocity would decrease with increasing substrate (i.e. Arf1·GTP) concentration (see “Appendix”). If the effect were the result of a change in Vmax, the proportional effect would be independent of substrate concentration. We also considered the possibility that coatomer is able to bind both the GAP and Arf1. In this model, coatomer binding to Arf1 is independent of GAP. Therefore, coatomer is able to sequester Arf1·GTP (Fig. 8, Schemes 2 and 3). We derived equations under rapid equilibrium assumptions based on this scheme (see “Appendix”). First, we considered the case in which Arf1·GTP concentration was small compared with coatomer. Under this condition and with these assumptions, initial velocity would be a biphasic function of coatomer concentration. We also derived the equation assuming substrate concentration was significantly greater than coatomer (see “Appendix”). The relationship between initial rate and coatomer concentration would be a hyperbola. In either case, if the effect of coatomer were because of a change in Km values for the enzyme (Arf GAP1), the proportional effect of coatomer on activity would decrease with increasing Arf1·GTP concentrations. If the effect were because of a change in Vmax, the proportional effect would be independent of Arf1·GTP concentration.
FIGURE 8.

Kinetic schemes. Abbreviations used are as follows: E, Arf GAP1; S, Arf1·GTP; C, coatomer.
With these considerations, we first examined coatomer activation of Arf GAP1 using myrArf1 as a substrate (Fig. 9, A and B). Using 0.4 μm Arf1·GTP, the effect of coatomer was biphasic, with peak activity observed at 0.2 μm coatomer. At 4 μm myrArf1·GTP, the activity dependence on coatomer was hyperbolic, but the effect observed was proportionally less than observed with 0.4 μm Arf1·GTP. In previous reports of the effect of coatomer on Arf GAP activity, [Δ17]Arf1 was used as a substrate. We repeated the experiments using this truncated form of Arf1 at two concentrations (Fig. 9, C and D). Coatomer increased activity with a hyperbolic dependence. The proportional effect was less at the higher concentration of [Δ17]Arf1. Coatomer did not affect ASAP1 activity (not shown). AP-1 did not affect Arf GAP1 activity at the concentrations we tested (not shown). In summary, coatomer stimulated activity of Arf GAP1 using either myrArf1 or [Δ17]Arf1 as a substrate. The biphasic curve observed with myrArf1 is consistent with substrate sequestration by coatomer, which does not appear to be significant when using [Δ17]Arf1 as a substrate. The dependence of the effect on Arf1·GTP concentration is consistent with coatomer affecting the Km value, rather than the kcat value, of the reaction.
FIGURE 9.

Effect of coatomer on Arf GAP1 activity. Coatomer was titrated into reactions containing LUVs extruded through 1-μm pore filters, [1–415]Arf GAP1-His, and either myrArf1·[α-32P]GTP or [Δ17]Arf1·[α-32P]GTP. Total phospholipid concentration was 500 μm. The reaction was followed by the conversion of [α-32P]GTP to [α-32P]GDP. A, 3.5 nm [1–415]Arf GAP1-His and 0.4 μm myrArf1·GTP. B, 3.5 nm [1–415]Arf GAP1-His and 4.7 μm myrArf1·GTP. C, 660 nm
[1–415]Arf GAP1-His and 0.4 μm [Δ17]Arf1·GTP. D, 660 nm [1–415]Arf GAP1-His and 7 μm [Δ17]Arf1·GTP.
Previous reports proposed that coatomer contributed a catalytic residue to the Arf GAP·Arf1·GTP complex and therefore would affect the kcat and, consequently, Vmax, which is a product of the total amount of enzyme and the kcat. Our results were consistent with an effect on Km. To discriminate between these possibilities, we determined the Arf1·GTP dependence of the initial rate in the presence or absence of coatomer. We found an isolated effect on the Km value (Fig. 6B and Table 2). An effect of Km value could be the result of coatomer recruiting Arf GAP1 to the reaction surface. We performed two experiments to test this idea. First, we examined the effect of coatomer on Arf GAP1 binding to vesicles. We observed an ∼30% increase in Arf GAP1 binding, which was not sufficient to explain the increase in activity (Fig. 5). The second experiment we did was to exclude lipid vesicles from the reaction (Fig. 10), possibly because [Δ17]Arf1·GTP is soluble and does not require a lipid surface to be stabilized. Coatomer had a similar effect on GAP activity in the absence of vesicles as in the presence.
FIGURE 10.

Lipid dependence of coatomer activation of Arf GAP1. Coatomer was titrated into reactions containing 2.8 μm of [Δ17]Arf1·GTP, 1580 nm of [1–415]Arf GAP1-GFP, and as indicated either no LUVs or LUVs at a total phospholipid concentration of 500 μm and extruded through 1.0-μm pore membranes.
Inhibition of GAP activity by cargo binding to coatomer has been proposed to contribute to cargo selection during the formation of membrane traffic intermediates (15, 38, 39). A peptide derived from the cytoplasmic tail of p24β was found to be an inhibitor of Arf GAP1/coatomer-catalyzed hydrolysis of GTP (15). We determined the effect of p24β peptide on the conversion of myrArf1·GTP to myrArf1·GDP induced by [1–415]Arf GAP1-His with no coatomer present (Fig. 11). The peptide inhibited the reaction with a concentration dependence that was similar to that previously reported (15) (Fig. 11A); however, the peptide also inhibited activity of ASAP1 with a similar concentration dependence as for Arf GAP1 (Fig. 11B).
FIGURE 11.

Effect of p24 cargo peptide on Arf GAP1 activity. A, effect of p24 peptide on Arf GAP1. Activity was determined in a fixed time point assay using 0.6 μm myrArf1·[α-32P]GTP as a substrate in a reaction containing LUVs, 120 nm [1–415]Arf GAP1-His, and the indicated concentration of p24 cargo peptide. The rate was calculated as ln(S0/S)/t. B, effect of p24 peptide on ASAP1. The experiment is the same as that described in A but with 0.5 nm [325–724]ASAP1 as the enzyme in place of [1–415]Arf GAP1-His.
Lipid composition of membranes has also been found to regulate some Arf GAPs (7–9). We examined the effect of varying the concentration of four lipids previously identified either as regulators of Golgi, as products of Arf-regulated reactions, or as regulators of Arf GAPs. In these experiments, the C50 of Arf GAP1 was determined when assayed with LUVs containing the lipids indicated in Table 4. Phosphoinositides were examined because PI kinases are recruited to the Golgi and activated by Arfs, generating phosphatidylinositol 4-phosphate (PI4P) and phosphatidylinositol 4,5-bisphosphate (PIP2) (4). Neither reduced the C50. Phosphatidic acid is produced in a reaction catalyzed by phospholipase D, which is activated by Arf (4). We did not detect an effect of phosphatidic acid at concentrations up to 25 μm (5% of total lipid in the reaction) on the C50 for Arf GAP1. Activation of Arf GAP1 by diacylglycerol was the observation that led to the curvature sensor hypothesis (8). Under the conditions of our experiments, we did not observe a significant effect of diacylglycerol on GAP activity.
TABLE 4.
Effect of lipids on Arf GAP1 activity
The C50 of Arf GAP1 was determined in assays with LUVs extruded through 1-μm pore filters and containing the indicated lipids. Dioleoylglycerol (DAG) was the form of diacylglycerol used in these experiments. The total phospholipid concentration was 500 μm for all experiments. The data are the average ± range for two experiments. ND means not determined. PA means phosphatidic acid.
|
C50 (nm)
|
||||||
|---|---|---|---|---|---|---|
| Lipid (%) | 0 | 1 | 2.5 | 5 | 10 | 15 |
| PIP2 | 38 ± 2 | 32 ± 3 | 38 ± 3 | 25 ± 2 | ND | ND |
| PI4P | 46 ± 6 | 53 ± 4 | ND | 87 ± 8 | ND | ND |
| PA | 24 ± 3 | 39 ± 6 | ND | 32 ± 2 | ND | ND |
| DAG | 19 ± 4 | ND | ND | 34 ± 4 | 29 ± 4 | 36 ± 4 |
DISCUSSION
We have examined the enzymatic properties of Arf GAP1. In prevailing paradigms, the primary function of Arf GAP1 is regulation of Arf1 at the Golgi apparatus to control membrane traffic. Hydrolysis of GTP, converting Arf1·GTP to Arf1·GDP, is delayed until formation of a transport intermediate. In one model, the catalysis is positively regulated by membrane curvature. In another model, catalysis depends on a second protein, coatomer, and is negatively regulated by cargo. We found that catalytic rate of Arf GAP1 is slow compared with another Arf GAP (22) and compared with Ras GAP and Rho GAP (23–26) and that coatomer increased the affinity of Arf GAP1 for the substrate Arf1·GTP. Based on our results, we propose that interaction of Arf GAP1 with coatomer is critical to regulating Arf1·GTP concentrations. We speculate, based on the kinetic properties of the two characterized Arf GAPs, that Arf GAPs regulate the stability or turnover rate of specialized membrane surfaces.
Kinetic and mutational analysis of Arf GAP1 revealed differences from another Arf GAP, ASAP1, which has been characterized in a similar way. Previously it was speculated that Arf GAP1 and ASAP1 differed in that Arf GAP1 did not have the complete machinery to function as a GAP. Coatomer was thought to contribute a catalytic residue. ASAP1 was thought to contain all the catalytic machinery necessary to induce GTP hydrolysis. The catalytic constant of Arf GAP1 was about 1/250th the constant of ASAP1 and coatomer did stimulate activity. However, coatomer affected the Km and not the kcat values. Furthermore, mutation of an arginine highly conserved in Arf GAPs and suspected to have a catalytic role abrogated activity in both Arf GAP1 and ASAP1. On the basis of our data, we cannot exclude the idea that Arf GAP1 is an incomplete GAP. Such a conclusion would require more structural and kinetic information. On the other hand, the low catalytic efficiency of Arf GAP1 relative to other Arf GAPs in the cell could account for the cellular distribution of Arf1 and the residency time of Arf1 on the Golgi apparatus where Arf GAP1 functions.
Mutational analysis also revealed differences between ASAP1 and Arf GAP1. In particular a tryptophan within the zinc-binding motif (479 in ASAP1 and 32 in Arf GAP1) was critical for activity in ASAP1, but mutating the residue in Arf GAP1 affected activity by only 4-fold. Some structure/function differences within the Arf GAP domain are consistent with the important role of the pleckstrin homology domain in ASAP1 enzymatic activity. Arf GAP1 does not contain this domain.
As reported previously, we found that coatomer stimulated Arf GAP1 enzymatic activity. The effect of coatomer was complex to analyze. Previous work implicated switch 1 of Arf1 in binding both Arf GAP1 (32) and coatomer (40). Consequently, coatomer could potentially sequester Arf1·GTP from Arf GAP1. On the other hand, coatomer can directly bind to Arf GAP1 (41–43) and may allosterically modify activity. We found that coatomer both sequesters Arf1·GTP and allosterically modifies Arf GAP1 activity, reducing the Km. The effect of sequestration was observed with myrArf1. The affinity of [Δ17]Arf1 for coatomer may be less than the affinity of myrArf1. Allosteric modification of activity was observed using either myrArf1·GTP or [Δ17]Arf1·GTP as substrate. We analyzed the allosteric effect with [Δ17]Arf1 because the lack of detectable substrate sequestration simplified the analysis. The [Δ17]Arf1·GTP concentration dependence indicated an effect on Km but not kcat values. Consistent with this conclusion, the effect of coatomer on activity decreased with increasing substrate concentrations. These results explain the limited effect of coatomer on Arf GAP1 activity that was previously reported when using myrArf1 as a substrate (17). In those experiments, myrArf1·GTP was 4 μm, about 4-fold the Km value; therefore, coatomer could be predicted to have only a small effect on GTP hydrolysis observed in the in vitro assay. Depending on relative Km values for Arf1·GTP, the effect of coatomer on GTP hydrolysis in the in vitro assay may be greater for Arf GAP2 and Arf GAP3 than for Arf GAP1 because these proteins have a higher affinity for coatomer (44). Coatomer could be added at lower concentrations and would be less likely to sequester Arf1·GTP. Arf1·GTP could be used at lower concentration to maximize an effect. The effect of coatomer on Arf GAP1 could be significant in vivo because it would determine how effectively Arf GAP1 could compete for Arf1·GTP with coatomer and other effectors. An effect on affinity would also be significant if Arf GAP1 were functioning in part as an Arf effector as previously proposed for Arf GAP1 (43) and for yeast homologs of Arf GAP1 subtype Arf GAPs (45, 46). In this case, a change in affinity could result in relatively tight binding to the Golgi apparatus through Arf1. The relatively slow rate of GTP hydrolysis would be consistent with an effector function like sec23 in the COPII complex, which acts as an effector and GAP for Sar1p (47).
The basis of one proofreading model of membrane transport was the observation that peptides from cytoplasmic tails of cargo proteins inhibited Arf GAP1 activity (15, 41). The effect was assumed to be mediated by binding to coatomer, which was included in the GAP assays (15). Other studies found an effect of peptide that was independent of coatomer but still consistent with the proofreading model (39). We also observed inhibition by cargo peptide that was independent of coatomer; however, the effect was not specific for Arf GAP1. Peptide inhibited ASAP1 with similar potency and efficiency. These results do not exclude a role for cargo peptide interaction with Arf GAP for cargo sorting. A plausible explanation is that the cargo directly interferes with Arf binding to the catalytic site of an Arf GAP. Assuming that there is some conservation of the binding site among the Arf GAPs, the peptide might appear to be nonspecific in an in vitro assay but could be specific as a result of localization in a living cell. Further studies are necessary to determine the physiologic relevance of the effects of peptides from cargo cytoplasmic tails on Arf GAP activity.
We also examined the possible role of lipids in regulation of Arf GAP activity. We determined the effects of four lipids that have been implicated as regulators of Arf GAPs or are affected by Arf (7–9). Phosphatidylinositol 4,5-bisphosphate, which is an important regulator of ASAP1, did not affect Arf GAP1, as has been reported previously (18). Phosphatidylinositol 4-phosphate, a lipid that regulates the Golgi, also had little effect. Phosphatidic acid, produced in a reaction regulated by Arf, did not affect the reaction rate. Diacylglycerol, which can be derived from phosphatidic acid, has been found to increase Arf GAP activity, which was the observation that led to the curvature sensor hypothesis. We did not find an effect of diacylglycerol. The original observation (18) was compelling. Our inability to detect an effect of this lipid may be related to our inability to detect an effect on curvature.
Arf GAP1 has previously been found to function as a curvature sensor. The sensitivity to curvature is mediated by the two ALPS domains in the protein. Under the conditions we examined, we found a small effect of vesicle size on activity. Previous studies used very dilute solutions, for instance total lipids at 200 μm. We worked at higher concentrations (although still dilute compared with physiological conditions), which accounted for part of the difference. We also examined the effect of the method of protein preparation. We used recombinant Arf GAP1 purified from insect cells. In our purification scheme, the protein did not go through a denaturation/renaturation cycle. Most experiments in which differences in curvature were described used proteins that were solubilized by denaturation from inclusion bodies in a bacterial expression system and then renatured. We found that the bacterially expressed Arf GAP1 appeared to be more sensitive to membrane curvature than the protein expressed in insect cells. The sensitivity of Arf GAP1 expressed in insect cells could be increased by denaturing the protein in urea and renaturing it by dilution into the reaction. However, the effect of curvature was not as great as reported previously. Also, we did not find a large contribution of the ALPS (curvature sensing) domains of Arf GAP1 to GAP activity. Our results do not exclude the hypothesis that Arf GAP1 activity is regulated by membrane curvature. The hypothesis is well supported by results in the literature (19, 20, 48). Differences in the source of lipids, for instance, could account for differences in results (19). Differences in protein preparation could also account for the results. The ALPs domain could be partially unfolded depending on the method of protein preparation. The isolated ALPs domain from KES was a robust curvature sensor. The peptide, out of the context of a whole protein, would be anticipated to be in a random coil until association with a membrane as reported (21). We cannot exclude that a preparation of protein that may have the ALPs domain partially unfolded is the physiologically relevant form of the protein. However, our in vitro results are consistent with the dynamics of Arf GAP1 association with the Golgi in living cells (49). Arf GAP1 was found to associate with the Golgi apparatus under conditions in which transport vesicles were not formed. These in vitro results together with the data reported in this paper raise the possibility that Arf GAP1 is either regulated by a different mechanism, e.g. by coatomer, or is a constitutively active enzyme. Similarly, we cannot exclude a role for the ALPS domain in Arf GAP1 function; however, whatever the role of the ALPS domain, it would be specific for Arf GAP1; even closely related Arf GAP2 and Arf GAP3 were not identified in data base searches for proteins containing this motif (21).
The large difference in catalytic rate of Arf GAP1 and ASAP1 could be related to the structures regulated by these Arf GAPs. ASAP1 regulates podosomes and invadopodia, which turn over rapidly; Arf GAP1 regulates the Golgi apparatus, which, although dynamic, is a stable structure relative to podosomes. Each of these structures is a specialized membrane surface undergoing active remodeling. In the prevailing paradigm for the function of Arf1, it recruits vesicle coat proteins to the surface of the bilayer. The surface is then changed, deforming to form a transport intermediate. Another articulation of this model is that the role of Arf is maintenance of a specialized membrane surface, achieved by recruiting proteins and by activating enzymes such as phospholipid kinases and phosphodiesterases (4). The generation of the specialized membrane surfaces would be dependent on the activity of Arf guanine nucleotide exchange factors, but the turnover rates of the specialized membrane surfaces would be determined by the GAP activity. It is also possible that the Arf GAPs themselves, recruited to a surface by Arf, contribute directly to changing the surface, functioning as Arf effectors. For instance, Arf GAP1 has been proposed to function as a part of a basic coat subunit, while in complex with Arf and coatomer, which aggregates to form microdomains on the Golgi apparatus and drives the formation of transport intermediates (42, 43, 49). Arf GAPs homologous to Arf GAPs have also been identified as suppressors of Arf loss of function mutants in Saccharomyces cerevisiae (45, 46). Suppressor function is typical of downstream effectors.
In summary, we examined the kinetics and regulation of Arf GAP1. We found that Arf GAP1 had a similar affinity for Arf1·GTP as ASAP1 but a smaller kcat value. The vesicle coat protein coatomer affected the Km value, which could have a role in regulating Arf1·GTP concentration or function of an Arf GAP-coatomer complex.
Acknowledgments
We thank Drs. Alfred Wittinghofer, Jenny Hinshaw, Richard Kahn, and Bruno Antonny for insightful discussions and Drs. Peter Schuck and Xiaoying Jian for DLS analysis. We thank Drs. Ralph Hopkins and Dominic Esposito for expression of ArfGAP1 using baculovirus. We also thank the reviewers of this paper for insightful comments and for suggesting experiments.
APPENDIX
Kinetic Models—Using Scheme 1 in Fig. 8, in which coatomer does not sequester Arf1·GTP, we derived Equation 1 under rapid equilibrium assumptions (37). Vmax1 indicates maximum velocity when Arf GAP1 is not bound to coatomer. Vmax2 indicates maximum velocity when Arf GAP1 is bound to coatomer. Km1 indicates Michaelis constant for Arf GAP1 not bound to coatomer. Km2 indicates Michaelis constant for Arf GAP1 bound to coatomer. Kc indicates dissociation constant for coatomer-Arf GAP1 complex. Kcs indicates dissociation constant for the coatomer-Arf1·GTP complex. E indicates Arf GAP1 (enzyme). S indicates Arf1·GTP (substrate) and C indicates coatomer.
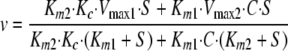 |
(Eq. 1) |
If Vmax1 = Vmax2, then Equation 2 is the result,
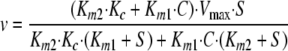 |
(Eq. 2) |
As S → ∞, v → Vmax and is independent of coatomer concentration. If Km1 = Km2, then we get Equation 3,
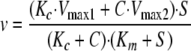 |
(Eq. 3) |
The proportional effect of coatomer is independent of Arf1·GTP concentration and, as S → ∞, the velocity approaches Equation 4,
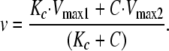 |
(Eq. 4) |
We also derived the equation assuming coatomer can sequester Arf1·GTP.
First, we assumed that Arf GAP1 << coatomer and Arf1·GTP but that Arf1·GTP<coatomer, so that a significant fraction of Arf1·GTP might be sequestered. If we assume sufficient coatomer-Arf1·GTP formation for the dimer to directly bind Arf GAP1 (Fig. 8, Scheme 2), the equation is complex. After collecting terms, the dependence of initial velocity on Arf1·GTP and coatomer has the form shown in Equation 5,
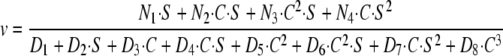 |
(Eq. 5) |
This equation predicts a biphasic concentration dependence on coatomer. The equation can be simplified by assuming either that there is not sufficient coatomer·Arf1·GTP complex to form significant amounts of a complex between Arf GAP1 and coatomer·Arf1·GTP or that coatomer·Arf1·GTP and coatomer associate with Arf GAP1 with the same kinetics and the same effect on activity (Fig. 8, Scheme 3). With these assumptions, the rapid equilibrium Equation 6 is shown.
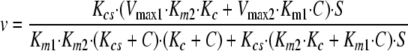 |
(Eq. 6) |
The dependence of velocity on coatomer is biphasic and on S is hyperbolic.
If we consider the case in which Arf1·GTP > coatomer, then Equation 7 has the following form,
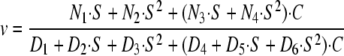 |
(Eq. 7) |
If we assume, as we did in deriving Equation 4, that coatomer·Arf1·GTP and coatomer association with Arf GAP1 with the same kinetics and the same effect on activity of the enzyme, then the dependence of velocity on coatomer and Arf1·GTP can be approximated by Equation 1, in which velocity is a hyperbolic function of both coatomer and Arf1·GTP. Furthermore, if Vmax1 = Vmax2, then the effect of coatomer diminishes with increasing Arf1·GTP concentration. If Km1 = Km2, the proportional effect of coatomer is constant, related to the difference between Vmax1 and Vmax2, and is independent of Arf1·GTP concentration.
This work was supported, in whole or in part, by a National Institutes of Health grant by the Intramural Research Program of the NCI, Department of Health and Human Services. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: GAP, GTPase-activating protein; NBD, nitrobenzoxadiazole; LUV, large unilamellar vesicle; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; PI, phosphatidylinositol; PI4P, phosphatidylinositol 4-phosphate; PIP2, phosphatidylinositol 4,5-bisphosphate; ALPS, Arf GAP lipid packing sensors.
References
- 1.Souza-Schorey, C., and Chavrier, P. (2006) Nat. Rev. Mol. Cell Biol. 7 347–358 [DOI] [PubMed] [Google Scholar]
- 2.Gillingham, A. K., and Munro, S. (2007) Annu. Rev. Cell Dev. Biol. 23 579–611 [DOI] [PubMed] [Google Scholar]
- 3.Rothman, J. E. (2002) Nat. Med. 8 1059–1062 [DOI] [PubMed] [Google Scholar]
- 4.Nie, Z. Z., Hirsch, D. S., and Randazzo, P. A. (2003) Curr. Opin. Cell Biol. 15 396–404 [DOI] [PubMed] [Google Scholar]
- 5.Bonifacino, J. S., and Glick, B. S. (2004) Cell 116 153–166 [DOI] [PubMed] [Google Scholar]
- 6.Bonifacino, J. S., and Lippincott-Schwartz, J. (2003) Nat. Rev. Mol. Cell Biol. 4 409–414 [DOI] [PubMed] [Google Scholar]
- 7.Randazzo, P. A., and Kahn, R. A. (1994) J. Biol. Chem. 269 10758–10763 [PubMed] [Google Scholar]
- 8.Nie, Z. Z., and Randazzo, P. A. (2006) J. Cell Sci. 119 1203–1211 [DOI] [PubMed] [Google Scholar]
- 9.Inoue, H., and Randazzo, P. A. (2007) Traffic 8 1465–1475 [DOI] [PubMed] [Google Scholar]
- 10.Randazzo, P. A., Inoue, H., and Bharti, S. (2007) Biol. Cell 99 583–600 [DOI] [PubMed] [Google Scholar]
- 11.Cukierman, E., Huber, I., Rotman, M., and Cassel, D. (1995) Science 270 1999–2002 [DOI] [PubMed] [Google Scholar]
- 12.Cassel, D. (2003) in Arf Family GTPases (Kahn, R. A., ed) pp. 157–158, Kluwer Academic Publishers Group, Dordrecht, Netherlands
- 13.Aoe, T., Cukierman, E., Lee, A., Cassel, D., Peters, P. J., and Hsu, V. W. (1997) EMBO J. 16 7305–7316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber, I., Cukierman, E., Rotman, M., Aoe, T., Hsu, V. W., and Cassel, D. (1998) J. Biol. Chem. 273 24786–24791 [DOI] [PubMed] [Google Scholar]
- 15.Goldberg, J. (2000) Cell 100 671–679 [DOI] [PubMed] [Google Scholar]
- 16.Goldberg, J. (1999) Cell 96 893–902 [DOI] [PubMed] [Google Scholar]
- 17.Szafer, E., Pick, E., Rotman, M., Zuck, S., Huber, I., and Cassel, D. (2000) J. Biol. Chem. 275 23615–23619 [DOI] [PubMed] [Google Scholar]
- 18.Antonny, B., Huber, I., Paris, S., Chabre, M., and Cassel, D. (1997) J. Biol. Chem. 272 30848–30851 [DOI] [PubMed] [Google Scholar]
- 19.Bigay, J., Casella, J. F., Drin, G., Mesmin, B., and Antonny, B. (2005) EMBO J. 24 2244–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bigay, J., Gounon, P., Robineau, S., and Antonny, B. (2003) Nature 426 563–566 [DOI] [PubMed] [Google Scholar]
- 21.Drin, G., Casella, J. F., Gautier, R., Boehmer, T., Schwartz, T. U., and Antonny, B. (2007) Nat. Struct. Mol. Biol. 14 138–146 [DOI] [PubMed] [Google Scholar]
- 22.Luo, R., Ahvazi, B., Amariei, D., Shroder, D., Burrola, B., Losert, W., and Randazzo, P. A. (2007) Biochem. J. 402 439–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nixon, A. E., Brune, M., Lowe, P. N., and Webb, M. R. (1995) Biochemistry 34 15592–15598 [DOI] [PubMed] [Google Scholar]
- 24.Ahmadian, M. R., Hoffmann, U., Goody, R. S., and Wittinghofer, A. (1997) Biochemistry 36 4535–4541 [DOI] [PubMed] [Google Scholar]
- 25.Gideon, P., John, J., Frech, M., Lautwein, A., Clark, R., Scheffler, J. E., and Wittinghofer, A. (1992) Mol. Cell. Biol. 12 2050–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham, D. L., Eccleston, J. F., and Lowe, P. N. (1999) Biochemistry 38 985–991 [DOI] [PubMed] [Google Scholar]
- 27.Yoon, H. Y., Jacques, K., Nealon, B., Stauffer, S., Premont, R. T., and Randazzo, P. A. (2004) Cell. Signal. 16 1033–1044 [DOI] [PubMed] [Google Scholar]
- 28.Randazzo, P. A., and Kahn, R. A. (1995) Methods Enzymol. 250 394–405 [DOI] [PubMed] [Google Scholar]
- 29.Ha, V. L., Thomas, G., Stauffer, S., and Randazzo, P. A. (2006) Methods Enzymol. 404 164–174 [DOI] [PubMed] [Google Scholar]
- 30.Huber, I., Rotman, M., Pick, E., Makler, V., Rothem, L., Cukierman, E., and Cassel, D. (2001) Methods Enzymol. 329 307–316 [DOI] [PubMed] [Google Scholar]
- 31.Che, M. M., Nie, Z., and Randazzo, P. A. (2006) Methods Enzymol. 404 147–163 [DOI] [PubMed] [Google Scholar]
- 32.Luo, R. B., Jacques, K., Ahvazi, B., Stauffer, S., Premont, R. T., and Randazzo, P. A. (2005) Curr. Biol. 15 2164–2169 [DOI] [PubMed] [Google Scholar]
- 33.Randazzo, P. A., Terui, T., Sturch, S., and Kahn, R. A. (1994) J. Biol. Chem. 269 29490–29494 [PubMed] [Google Scholar]
- 34.Nie, Z., Hirsch, D. S., Luo, R., Jian, X., Stauffer, S., Cremesti, A., Andrade, J., Lebowitz, J., Marino, M., Ahvazi, B., Hinshaw, J. E., and Randazzo, P. A. (2006) Curr. Biol. 16 130–139 [DOI] [PubMed] [Google Scholar]
- 35.Randazzo, P. A., Miura, K., and Jackson, T. R. (2001) Methods Enzymol. 329 343–354 [DOI] [PubMed] [Google Scholar]
- 36.Kam, J. L., Miura, K., Jackson, T. R., Gruschus, J., Roller, P., Stauffer, S., Clark, J., Aneja, R., and Randazzo, P. A. (2000) J. Biol. Chem. 275 9653–9663 [DOI] [PubMed] [Google Scholar]
- 37.Cha, S. (1968) J. Biol. Chem. 243 820–825 [PubMed] [Google Scholar]
- 38.Weiss, M., and Nilsson, T. (2003) Traffic 4 65–73 [DOI] [PubMed] [Google Scholar]
- 39.Lanoix, J., Ouwendijk, J., Stark, A., Szafer, S., Cassel, D., Dejgaard, K., Weiss, M., and Nilsson, T. (2001) J. Cell Biol. 155 1199–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao, L. Y., Helms, J. B., Brugger, B., Harter, C., Martoglio, B., Graf, R., Brunner, J., and Wieland, F. T. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 4418–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eugster, A., Frigerio, G., Dale, M., and Duden, R. (2000) EMBO J. 19 3905–3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee, S. Y., Yang, J. S., Hong, W. J., Premont, R. T., and Hsu, V. W. (2005) J. Cell Biol. 168 281–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang, J. S., Lee, S. Y., Gao, M. G., Bourgoin, S., Randazzo, P. A., Premont, R. T., and Hsu, V. W. (2002) J. Cell Biol. 159 69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frigerio, G., Grimsey, N., Dale, M., Majoul, I., and Duden, R. (2007) Traffic 8 1644–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang, C. J., Cavenagh, M. M., and Kahn, R. A. (1998) J. Biol. Chem. 273 19792–19796 [DOI] [PubMed] [Google Scholar]
- 46.Zhang, C. J., Bowzard, J. B., Anido, A., and Kahn, R. A. (2003) Yeast 20 315–330 [DOI] [PubMed] [Google Scholar]
- 47.Antonny, B., Madden, D., Hamamoto, S., Orci, L., and Schekman, R. (2001) Nat. Cell Biol. 3 531–537 [DOI] [PubMed] [Google Scholar]
- 48.Mesmin, B., Drin, G., Levi, S., Rawet, M., Cassel, D., Bigay, J., and Antonny, B. (2007) Biochemistry 46 1779–1790 [DOI] [PubMed] [Google Scholar]
- 49.Liu, W., Duden, R., Phair, R. D., and Lippincott-Schwartz, J. (2005) J. Cell Biol. 168 1053–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pavel, J., Harter, C., and Wieland, F. T. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 2140–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]