

Abstract
Peracetylation is a very common protection strategy that is widely implemented in carbohydrate synthesis. Here, a method for peracetylation of carbohydrates using catalytic In(OTf)3 in neat acetic anhydride is reported. In(OTf)3 has low toxicity and is mild and water tolerant, and the reactions are high yielding and efficient. Details regarding the scope and mechanism of the reaction are briefly discussed.
Keywords: Acetylation, Catalysis, Indium triflate, Acetic anhydride
Acetylation reactions are among the most common transformations in organic synthesis, and acetylation of monosaccharides is often the first step in the synthesis of complex carbohydrates. The peracetylation of sugars is most often performed using acetic anhydride and DMAP in pyridine.1 Although this reaction works well, long reaction times (12 h) are required and pyridine is high boiling and noxious.
Numerous ways to avoid the use of pyridine in acetylation reactions have been devised. Some notable examples include the use of catalytic TMSOTf in CH2Cl22 and of catalytic Bu3P in CH2Cl2.3 Use of acetic anhydride as both the solvent and the acylating reagent has been demonstrated using a wide variety of catalysts including sodium acetate,4 montmorillonite K-10,5 molecular sieves,6 iodine,7 FeCl3,8 3-nitrobenzeneboronic acid,9 LiClO4,10 HClO4-SiO2,11 CuSo4.5H2O,12 acetonyltriphenylphosphonium bromide,13 H14[NaP5W30O110],14 La(NO3)3.6H2O,15 and Mg(NTf 2)2.16 Metal triflate catalysis of acetylation reactions has been reported using Cu(OTf)2,17 Bi(OTf)3,18 V(O)(OTf)2,19 Sc(OTf)3,20 Ce(OTf)3,21 and In(OTf)3.22
Most of the conventional metal salts that are used as Lewis acids are water sensitive and are often used in stoichiometric amounts. Metal triflates are an attractive alternative because of their low toxicity and non-corrosive nature.23 Yamamoto's report of acetylation reactions using catalytic Sc(OTf)3 in CH3CN generated high interest in the area of metal triflate catalyzed acetylations.24 In(OTf)3 is a mild, water tolerant Lewis acid that can be used in catalytic quantities for a variety of organic transformations.25 Because In(OTf)3 is considerably less expensive than Sc(OTf)3, we are interested in its development as a catalyst for acetylation reactions. Frost reported the acetylation of alcohols and amines in acetonitrile using 0.1 mol % In(OTf)3, but a chromatographic purification step was required and only one carbohydrate was evaluated.22 Here, we report the In(OTf)3 catalyzed peracetylation of carbohydrates under solvent free conditions.
We found that 0.05 equiv of In(OTf)3 catalyzed the peracetylation of a number of sugars in acetic anhydride when the reactions were stirred for one hour at 0 °C. The results are summarized in Scheme 1. For reaction workup, ethyl acetate was added to the flask and a 10% aqueous solution of Na2CO3 was added. After stirring for 20 minutes to one hour to hydrolyze any unreacted acetic anhydride, the organic layer was collected and concentrated. The crude products were normally of sufficient purity to be used without further purification. The reactions of lactose and N-acetyl glucosamine were warmed to room temperature because of the low solubility of the substrates in acetic anhydride at low temperature (reactions 6 and 7 in Scheme 1).
Scheme 1.

In(OTf)3 catalyzed peracetylation of carbohydrates. Conditions: In(OTf)3 (0.05 equiv) in Ac2O (30 equiv), 1 h, 0 °C (reactions 1−5) or 0 °C to RT (reactions 6 and 7).
The compatibility of the In(OTf)3 catalyzed peracetylation reactions with a number of commonly used protecting groups was explored. The benzylidene acetal and the TBDPS ether were stable to the reaction conditions (Scheme 2), but TMS and TBS groups were readily hydrolyzed (and the corresponding acetylated sugars were obtained).
Scheme 2.

Protecting group compatibility studies.
Investigation into the mechanism of the indium-triflate catalyzed acetylation suggests that triflic acid is the active catalytic species. Acetylation of galactose occurred rapidly using 0.05 equiv. of triflic acid (Table 1, entry 1). However, the reaction does not proceed when 2,6-di-tert-butylpyridine (DTBP) is added with In(OTf)3, nor does acetylation occur when glucosamine is used as the substrate in a one hour reaction (Table 1, entries 2−4). Thus, our results suggest that indium triflate can be used to provide low levels of triflic acid. Dumeunier and Markó observed a similar triflic acid mediated acylation using scandium triflate in a benzoylation procedure,26 and free acid has also been proposed as the active catalyst in metal triflate mediated Friedel-Crafts reactions.27 Loh and co-workers capitalized on the lability of the triflate on indium when forming chiral indium(III)-pybox complexes.28 When we allowed our In(OTf)3 catalyzed reaction with DTBP to stir at room temperature for one day, an 11% yield of peracetylated product was obtained (Table 1, entry 2). This result indicates that an indium triflate mediated reaction pathway may also be possible but is much slower than the triflic acid-catalyzed process. A similar dual pathway process has been proposed by Dumeunier and Markó for Sc(OTf)3.26
Table 1.
Catalyst comparison.a
| entry | substrate | catalyst (equiv.) | time | yield |
|---|---|---|---|---|
| 1 | 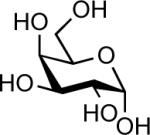 |
TfOH (0.05 equiv.) | 10 min | 84% |
| 2 | 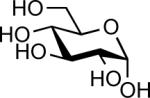 |
In(OTf)3 (0.05 equiv.) with DTBPb | 24 hc | 11% |
| 3 | 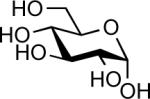 |
In(OTf)3 (0.05 equiv.) without DTBP | 1 h | 99% |
| 4 | 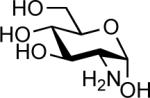 |
In(OTf)3 (0.05 equiv.) | 1 h | N.R.d |
| 5 |  |
InCl3 (0.05 equiv.) | 12 h | 71% |
| 6 |  |
InBr3 (0.05 equiv.) | 6 hc | 86% |
Reactions were performed in 30 equiv. Ac2O.
DTBP = di-tertbutylpyridine.
0 °C to RT; all other reactions 0 °C.
N.R. = no reaction.
To further explore the role of the catalyst in the acetylation reactions, we investigated the effect of the choice of ligand on the behavior of the catalyst in the reaction. We compared In(OTf)3 to InCl3 and InBr3. The acetylation of alcohols using indium chloride and ruthenium chloride catalysts has been reported previously.29 RuCl3 catalyzed reactions worked well without solvents for liquid substrates and was also successful for substrates compatible with acetonitrile (and slower in dichloromethane, chloroform, and tetrahydrofuran). Acetylations with carbohydrates were not explored.29a InCl3 catalyzed reactions were performed with only a small amount of catalyst and only one equivalent of Ac2O at room temperature, and peracetylation of glucose was reported.29b However, acetylation of glucose at 0 °C in our labs required 12 hours using 0.05 equiv. of InCl3 (Table 1, entry 5). InCl3 is a less reactive catalyst, and In(OTf)3 may be better suited for acetylations performed below room temperature. InBr3 catalyzed the acetylation of glucose at room temperature as shown in Scheme 1 (Table 1, entry 6). Thus, although InCl3 and InBr3 both effect the desired acetylation, the reactions are considerably slower with these catalysts than with In(OTf)3. Our results indicate that the strength of the respective acids that are generated from the catalysts (TfOH, HBr, and HCl) correlates well with the reaction rate.
Overall, we found that carbohydrates that were soluble in acetic anhydride could be peracetylated very effectively using the In(OTf)3-mediated reaction conditions described in this report. Acetylation of sugars such as sorbose and fructose that were insoluble in acetic anhydride could not be performed using this methodology. Carbohydrates are generally also insoluble in solvents such as acetonitrile that are commonly reported for In(OTf)3-mediated acetylation of simple alcohols. Although smaller amounts of acetic anhydride could be used for the peracetylation of carbohydrates, we found reactions with 30 equivalents of Ac2O to be most convenient, and excess anhydride is easily removed using the described workup procedure.
In addition to the peracetylation reaction, the use of In(OTf)3 to catalyze benzoylation reactions with carbohydrates was also briefly explored. Unfortunately, because benzoyl anhydride is a solid, solvent would be required for an analogous benzoylation reaction. Frost's work indicated that acetonitrile would be an appropriate solvent in which to invoke this transformation,22 but the low solubility of carbohydrate substrates in acetonitrile precluded product formation. In DMF, benzoyl protection of 1-methyl glucoside was performed using 0.05 equiv. of In(OTf)3 and 10 equiv. of benzoyl anhydride at room temperature for 12 h, affording a 50% yield of fully benzoyl-functionalized 1-methyl glucoside.
Microwave irradiation to improve carbohydrate solubility in the perbenzoylation reactions was also evaluated. The use of microwave irradiation in glycosylations and in protecting group manipulations for carbohydrates has recently been reviewed.30 We evaluated reactions using glucose and 10 equiv of benzoyl anhydride in acetonitrile with 0.05 equiv. of In(OTf)3. At temperatures ranging from 25 °C to 100 °C and with irradiations of 150 W to 300 W (all for five min), we were unable to achieve sufficient solubilization of substrates, and product formation was not observed.
We next tested microwave irradiation of the In(OTf)3-catalyzed peracetylation reactions. Microwave conditions have been successfully applied by Limousin and co-workers to peracetylations using ZnCl2, potassium acetate, or sodium acetate in acetic anhydride.31 An InCl3-catalyzed peracetylation reaction using microwave irradiation in acetonitrile was reported by Das and co-workers.32 We found that reactions that were performed according to our protocol described above (5mol% In(OTf)3, one hour, 0 °C, neat acetic anhydride) could also be performed under microwave irradiation. We irradiated mannose and N-acetyl glucosamine for 5 minutes at room temperature in a sealed tube with 150 watts to afford peracetylated products (Scheme 3). Although the yields we obtained were lower than those obtained without microwave irradiation, our results suggest that, with additional optimization, microwave irradiation could be used to successfully perform the transformations shown in Scheme 1. Reactions that were unsuccessful without microwave irradiation due to the insolubility of the substrates in acetic anhydride were equally unsuccessful when irradiated.
Scheme 3.

Peracetylation using microwave conditions.
In summary, the In(OTf)3-catalyzed peracetylation of carbohydrates in acetic anhydride works well for many carbohydrates. The new reaction conditions eliminate the need for unpleasant solvents such as pyridine, and In(OTf)3 has the advantage of being affordable, non-toxic, and non-corrosive.
1. Experimental
General peracetylation procedure
Carbohydrate was added to Ac2O (30 equiv.), and the solution was cooled to 0 °C. In(OTf)3 (0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 1 h. EtOAc and 10% aqueous Na2CO3 solution were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing twice with sat. aq. NaHCO3 solution, drying over MgSO4, and removal of solvent in vacuo afforded product.
General methods
Indium triflate was purchased from Aldrich Chemical Co. and was used without additional purification. Acetic anhydride was purchased from Acros Chemical Co. and was used without additional purification. NMR spectra were recorded on a Bruker 300 MHz spectrometer.
Experimental Protocols for In(OTf)3 Mediated Peracetylation Reactions
Preparation of 1,2,3,4,6-penta-O-acetyl-α-D-glucopyranoside
To Ac2O (3.3 mL, 30 equiv.) was added 200 mg (1.1 mmol) of α-D-glucose and the solution was cooled to 0 °C. In(OTf)3 (30 mg, 53 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 1 h. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), drying over MgSO4 followed by solvent removal in vacuo afforded 429 mg (99% yield) of product as a clear, colorless syrup. 1H NMR (CDCl3, 300 MHz, ppm) δ 6.32 (s, 1H), 5.32 (1H, t, J = 19.8 Hz, 1H), 5.11 (m, 2H), 4.25 (d, J = 9.0 Hz, 1H), 4.10 (s, 2H), 2.09 - 2.01 (m, 15H). These values agree with previously reported spectra.1
Preparation of 1,2,3,4,6-penta-O-acetyl-α-D-mannopyranoside
To Ac2O (3.1 mL, 30 equiv.) was added 186 mg (1.03 mmol) of α-D-mannose, and the solution was cooled to 0 °C. In(OTf)3 (29 mg, 51 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 1 h. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 386 mg of product (96% yield). 1H NMR (CDCl3, 300MHz, ppm) δ 6.08 (s, 1H), 5.34 (d, J = 6.0 Hz, 2H), 5.25 (s, 1H), 4.27 (dd, J = 12.3, 4.8 Hz, 1H), 4.15 - 4.14 (m, 2H), 2.21 (s, 1H), 2.17 (s, 3H) 2.09 (s, 3H), 2.04 (s, 3H), 2.0 (s, 3H). These values agree with previously reported spectra.1
Preparation of 1,2,3,4,6-penta-O-acetyl-α-D-galactopyranoside
To Ac2O (3.6 mL, 30 equiv.) was added 212 mg (1.17 mmol) of α-D-galactose, and the solution was cooled to 0 °C. In(OTf)3 (32 mg, 56 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 1 h. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 404 mg of product (88% yield). 1H NMR (CDCl3, 300 MHz, ppm) δ 6.36 (s, 1H), 5.48 (s, 1H), 5.38 (s, 2H), 4.32 (t, J = 12.8 Hz, 2H), 4.08 (d, J = 4.7 Hz, 2H), 2.14 (m, 15H). These values agree with previously reported spectra.1
Preparation of 1,2,3,4-tetra-O-acetyl-D-xylopyranoside
To Ac2O (2.02 mL, 30 equiv.) was added 116 mg (770 μmol) of D-xylose, and the solution was cooled to 0 °C. In(OTf)3 (24 mg, 42 μmol, 0.05 equiv.) was added, and the reaction was stirred for 1 h at 0 °C. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (35 mL) were added, and the mixture was stirred for 20 min at room temperature. Isolation of the organic layer, washing with water (10 mL) and brine (10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded crude product, which was chromatographed (3:1 hexane:EtOAc) to afford 155 mg of product (63 % yield). 1H NMR (CDCl3, 300 MHz, ppm) δ 6.24 (d, J = 3.6 Hz, 1H), 5.45 (ap t, J = 9.8 Hz, 1H), 5.02 (m, 2H), 3.92, (dd, J = 11.1, 5.9 Hz, 1H), 3.62 (ap t, J = 11.1 Hz, 1H), 2.16 (s, 3H), 2.03 (s, 6H), 2.00 (s, 3H). These values agree with previously reported spectra.2
Preparation of 1,2,3,4-tetra-O-acetyl-α-D-fucopyranoside
To Ac2O (1.98 mL, 30 equiv.) was added 110 mg (670 μmol) of α-D-fucose, and the solution was cooled to 0 °C. In(OTf)3 (20 mg, 35 μmol, 0.05 equiv.) was added, and the reaction was stirred for 1 h at 0 °C. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (35 mL) were added, and the mixture was stirred for 20 min at room temperature. Isolation of the organic layer, washing with water (10 mL) and brine (10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 214 mg of product (96% yield). 1H NMR (CDCl3, 300 MHz, ppm) δ 6.32 (s, 1H), 5.25 (s, 3H), 4.25 (q, J = 6.4 Hz, 1H), 2.16 (s, 3H), 2.13 (s, 3H), 1.99 (s, 6H), 1.14 (d, J = 6.4 Hz, 3H). These values agree with previously reported spectra.3
Preparation of 1,3,4,6-tetra-O-acetyl-2-N-acetyl-α-D-glucosamine
To Ac2O (2.7 mL, 30 equiv.) was added 176 mg (0.90 mmol) of N-acetyl α-D-glucosamine, and the solution was cooled to 0 °C. In(OTf)3 (30 mg, 55 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 1 h. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 287 mg of product (89% yield). 1H NMR (CDCl3, 300 MHz, ppm) δ 6.16 (d, J = 4.4 Hz, 1H), 5.28 (m, 2H), 5.23 (m, 1H), 4.51 (ap q, J = 9.1 Hz, 1H), 4.24 (m, 2H), 2.21 (s, 3H), 3.71 (bs, 1H), 2.18 (s, 3H), 2.10 (s, 3H), 2.08 (s, 3H), 1.91 (s, 3H). These values agree with previously reported spectra.4
Preparation of 2,3,4,6-tetra-O-acetyl-β-galactopyranose [1→4] 1,2,3,6-tetra-O-acetyl-β-glucopyranose
To Ac2O (7.9 mL, 30 equiv.) was added 1.0 g (2.76 mmol) of lactose, and the solution was cooled to 0 ° C. In(OTf)3 (79.5 mg, 141 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 1 h. EtOAc (100 mL) and 10% aqueous Na2CO3 solution (150 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with water (3 × 50 mL) and brine (3 × 50 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 1.88 g of product (97% yield). 1H NMR (CDCl3, 300 MHz, ppm) δ 1H NMR (CDCl3, 300 MHz, ppm) δ 6.24 (d, J = 3.7 Hz, 1H), 5.45 (t, J = 9.6 Hz, 1H), 5.34 (d, J = 3.0 Hz, 1H), 5.12 (m, 1H), 4.97 (m, 2H), 4.46 (m, 2H), 4.09 (m, 5H), 3.83 (m, 2H), 2.05 (m, 24H). These values agree with previously reported spectra.5
Preparation of 1-O-methyl-2,3,4-tri-O-acetyl-6-O-tertbutyldiphenylsilyl-α-D-glucopyranoside
To pyridine (16 mL) was added 1-O-methyl-α-D-glucopyranoside (345 mg, 1.61 mmol). The solution was cooled to 0 °C, tert-butyl-diphenyl-silyl chloride (443 mg, 1.61 mmol) was added, and and the reaction was stirred for 3.5 h.6 Pyridine was removed under reduced pressure. The resulting intermediate was cooled to 0°C, Ac2O (5.3 mL, 30 equiv.) and In(OTf)3 (49 mg, 88 μmol, 0.05 equiv.) were added, and the reaction was stirred for 1 h while warming to room temperature. EtOAc (20 mL) and 10% aq. Na2CO3 solution (20 mL) were added and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded crude product, which was chromatographed (9:1 hexane/EtOAc) to afford 853 mg of product (86% yield). 1H-NMR (CDCl3, 300 MHz, ppm) δ 7.66 (t, J = 5.0 Hz, 2H), 7.2 (ap d, J = 7.2 Hz, 3H), 5.32 (m, 2H), 5.19 (s, 1H), 4.71 (s, 1H), 3.74 (m, 3H), 3.37 (s, 3H), 2.09 (s, 3H), 1.95 (s, 3H). 1.85 (s, 3H) 1.04 (s, 9H). These values agree with previously reported spectra.7
Preparation of 4,6-O-benzylidene-2,3-O-diacetyl-1-O-methyl-α-D-glucopyranoside
To Ac2O (3.3 mL, 30 equiv.) was added 200 mg (1.11 mmol) of 4,6-O-benzylidene-1-O-methyl-α-D-glucopyranoside, and the solution was cooled to 0 °C. In(OTf)3 (30 mg, 55 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 1 h. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 228 mg of product as a syrup (88% yield). 1H NMR (300 MHz, CDCl3, ppm) δ 7.20-7.45 (m, 5H), 5.56 (t, J = 9.8 Hz, 1H), 5.52 (s, 1H), 4.91 (d, J = 3.8 Hz, 1H), 4.88 (dd, J = 3.8, 9.8, 1H), 4.30 (dd, J=3.8, 9.8, 1H), 3.92 (m, 1H), 3.39 (s, 3H), 2.05 (s, 3H), 2.00 (s, 3H). These values agree with previously reported spectra.8
Preparation of 1,2,3,4,6-penta-O-acetyl-α-D-glucopyranoside
To Ac2O (1.7 mL, 30 equiv.) was added 317 mg (0.58 mmol) of 1,2,3,4,6-O-penta(trimethylsilyl)-α-D-glucose,9 and the solution was cooled to 0 °C. In(OTf)3 (16 mg, 29 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 1 h. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 203 mg of 1-O-methyl-2,3,4,6-tetra-O-acetyl-α-D-glucopyranoside (89% yield, characterization data and reference are provided above). Products bearing trimethylsilyl groups were not obtained.
Preparation of 1-O-methyl-2,3,4,6-tetra-O-acetyl-α-D-glucopyranoside
To pyridine (26 mL) was added 1-O-methyl-α-D-glucopyranoside (512 mg, 2.63 mmol). The solution was cooled to 0°C, tert-butyl-dimethylsilyl chloride (395 mg, 2.63 mmol) was added, and the reaction was stirred for 3.5 h.10 Pyridine was removed under reduced pressure. The resulting intermediate was cooled to 0°C, Ac2O (7.9 mL, 30 equiv.) and In(OTf)3 (74 mg, 132 μmol, 0.05 equiv.) were added, and the reaction was stirred for 1 h while warming to room temperature. EtOAc (30 mL) and 10% aq. Na2CO3 solution (30 mL) were added and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 15 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 966 mg of 1-O-methyl-2,3,4,6-tetra-O-acetyl-α-D-glucopyranoside (87% yield).11 1-O-methyl-2,3,4-tri-O-acetyl-6-O-tertbutyldimethylsilyl-α-D-glucopyranoside was not obtained.
Experimental Protocol for TfOH Mediated Peracetylation Reactions
Preparation of 1,2,3,4,6-penta-O-acetyl-α-D-galactopyranoside
To Ac2O (2.7 mL, 30 equiv.) was added 103 mg (0.50 mmol) of α-D-galactose, and the solution was cooled to 0 °C. TfOH (2 μl, 28 μmol, 0.05 equiv.) was added, and the reaction was allowed to stir for 10 min. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 187 mg of product as a syrup (84% yield; see above for characterization data).1
Experimental Protocol for In(OTf)3 Mediated Peracetylation Reactions with Proton Scavenger
Preparation of 1,2,3,4,6-penta-O-acetyl-α-D-glucopyranoside
To Ac2O (1.6 mL, 30 equiv.) was added 97 mg (0.53 mmol) of α-D-glucose and the solution was cooled to 0 °C. In(OTf)3 (30 mg, 55 μmol, 0.05 equiv.) and 2,6-di-tert-butyl pyridine (0.226 mL, 1.06 mmol, 2.2 equiv.) were added, and the reaction was allowed to warm to room temperature while stirring for 24 h. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 23 mg of product (11% yield; see above for characterization data).1
Experimental Protocols for Indium Trihalide Mediated Peracetylation Reactions
Preparation of 1,2,3,4,6-penta-O-acetyl-α-D-glucopyranoside
To Ac2O (3.1 mL, 30 equiv.) was added 196 mg (1.03 mmol) of α-D-glucose and the solution was cooled to 0 °C. InCl3 (12 mg, 54 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 12 h. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 301 mg (71% yield) of product as a syrup (see above for characterization data).1
Preparation of 1,2,3,4,6-penta-O-acetyl-α-D-glucopyranoside
To Ac2O (3.5 mL, 30 equiv.) was added 213 mg (1.18 mmol) of α-D-glucose and the solution was cooled to 0 °C. InBr3 (21 mg, 59 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 5 h. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. Isolation of the organic layer, washing with sat. aq. NaHCO3 solution (2 × 10 mL), and drying over MgSO4 followed by solvent removal in vacuo afforded 396 mg (86% yield) of product as a syrup (see above for characterization data).1
Experimental Protocols for In(OTf)3 Mediated Perbenzoylation Reactions
Preparation of 1-O-methyl-2,3,4,6-penta-O-benzoyl-α-D-glucopyranoside
Procedure 1
To CH3CN (6.6 mL) was added 121 mg (0.66 mmol) of 1-O-methyl-α-D-glucose and benzyol anhydride (1.5 g, 6.6 mmol, 10 equiv.) and the solution was cooled to 0 °C. In(OTf)3 (18 mg, 30 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 24 h and refluxed for 5 h. CH3CN was removed in vacuo. EtOAc (20 mL) and 10% aqueous Na2CO3 solution (20 mL) were added, and the mixture was stirred for 1 h. The organic layer was isolated, washed with sat. aq. NaHCO3 solution (2 × 10 mL), and dried over MgSO4 followed by solvent removal in vacuo, but no product was obtained.
Procedure 2
To DMF (9.0 mL) was added 173 mg (0.89 mmol) of 1-O-methyl-α-D-glucose and benzyol anhydride (2.01 g, 8.9 mmol, 10 equiv.) and the solution was cooled to 0 °C. In(OTf)3 (25 mg, 45 μmol, 0.05 equiv.) was added, and the reaction was allowed to warm to room temperature while stirring for 12 h. The reaction was quenched with 20 mL H2O and washed with EtOAc (10 mL × 3). The organic layer was isolated, washed with sat. aq. NaHCO3 solution (2 × 10 mL), and dried over MgSO4 followed by solvent removal in vacuo to afford 293 mg of product (50% yield). 1H NMR (500 MHz, CDCl3, ppm) δ 7.96 (d, J = 7.43 Hz, 7H), 7.94 (d, J = 8.0 Hz, 2H), 7.51 (q, J = 10.2 Hz, 1H), 7.41 (m, 7H), 7.37 (t, J = 7.4 Hz, 3H), 6.24 (t, J = 9.4 Hz, 1H), 5.52 (t, J=9.4 Hz, 1H), 5.28 (m, 2H), 4.03 (d, J = 9.76 Hz, 1H), 3.8 (q, J = 3.4 Hz, 2H), 3.45 (s, 3H), 2.05 (d, J = 1.5 Hz, 2H). These values agree with previously reported spectra.12
Protocols for Microwave Assisted Reactions
General
A CEM Discover BenchMate microwave was used for all of the reported microwave experiments.
Attempted Preparation of 1-O-methyl-2,3,4,6-penta-O-benzoyl-α-D-glucopyranoside
1-O-methyl-α-D-glucose (50 mg, 0.257 mmol) was weighed into a 10 mL tube equipped with a stir bar. Benzoic anhydride (0.583 g, 2.57 mmol) and acetonitrile (2 mL) were added to the tube. The tube was sealed and placed in a microwave. The reaction was carried out under the following conditions:
Preparation of 1,2,3,4,6-penta-O-acetyl-α-D-mannopyranoside
α-D-Mannose (100 mg, 0.555 mmol) was weighed into a 10 mL tube equipped with a stir bar. Indium triflate (0.016 g, 0.028 mmol) and acetic anhydride (1.56 mL, 16.7 mmol) were added to the tube. The tube was sealed and placed in a microwave. The reaction was carried out at 25° C (150 W) for 5 minutes. Ethyl acetate (1.56 mL) and a solution of 10% aq. Na2CO3 soln (1.56 mL) were added, and the resulting solution was allowed to stir for 1 h. The organics were extracted with ethyl acetate (30 mL) and washed with 10% aq. Na2CO3 soln (3 × 30 mL). The organic layer was dried over MgSO4, and the solvent was removed in vacuo to yield 0.183 g (84%) of the peracetylated sugar (characterization data is given above).1
Preparation of 1,3,4,6-tetra-O-acetyl-2-N-acetyl-α-D-glucosamine
N-acetyl α-D-glucosamine (100 mg, 0.452 mmol) was weighed into a 10 mL tube equipped with a stir bar. Indium triflate (0.0127 g, 0.026 mmol) and acetic anhydride (1.27 mL, 13.6 mmol) were added to the tube. The tube was sealed and placed in a microwave. The reaction was carried out at 25° C (150 W) for 5 minutes. Ethyl acetate (1.27 mL) and a solution of 10% Na2CO3 (1.27 mL) were added, and the resulting solution was allowed to stir for 1 hour. The organics were extracted with ethyl acetate (30 mL) and washed with 10% Na2CO3 (3 × 30 mL). The organic layer was dried over MgSO4, and the solvent was removed in vacuo to yield 0.082 g (46%) of the per-acetlyated sugar (characterization data is given above).4
Attempted Preparation of 1,3,4,6-tetra-O-acetyl-2-N-acetyl-α-D-glucosamine
Procedure 1
α-D-Glucosamine HCl (100 mg, 0.464 mmol) was weighed into a 10 mL tube equipped with a stir bar. Indium triflate (0.013 g, 0.023 mmol) and acetic anhydride (1.3 mL, 13.91 mmol) were added to the tube. The tube was sealed and placed in a microwave. The reaction was carried out under the following conditions:
Procedure 2
α-D-Glucosamine HCl (130 mg) was weighed out and stirred in a 0.1 M NaOMe/MeOH solution (6.5 mL) for 10 minutes. The resulting solution was filtered, and the filtrate was collected. The solvent was removed to leave 107 mg of the free amine in the tube. Indium triflate (0.0168 g, 0.03 mmol) and acetic anhydride (1.67 mL, 17.9 mmol) were added to the tube. The tube was sealed and placed in a microwave. The reaction was carried out at 25° C (150 W) for 5 minutes. Ethyl acetate (1.67 mL) and a solution of 10% aq. Na2CO3 soln (1.67 mL) were added, and the resulting solution was allowed to stir for 1 hour. The organics were extracted with ethyl acetate (30 mL) and washed with 10% Na2CO3 (3 × 30 mL). The organic layer was dried over MgSO4, and the solvent was removed in vacuo.
References
1. Lemieux, R. U.; Stevens, J. D. Can. J. Chem. 1965, 43, 2059-2070.
2. Durette, P. L.; Horton, D. J. Org. Chem. 1971, 36, 2658-2669.
3. Pozsgay, V.; Brisson, J.-R.; Jennings, H. J. Carbohydr. Res. 1990, 205, 133-146.
4. Kartha, K. P. R.; Field, R. A. Tetrahedron 1997, 53, 11753-11766.
5. Ross, A. J.; Ivanova, I. A.; Ferguson, M. A. J.; Nikoleav, A. V. J. Chem. Soc. Perkin Trans. 1 2001, 72-81.
6. Komiotis, D.; Agelis G.; Manta, S.; Tzioumaki, N.; Tsoukala, E.; Antonakis, K. J. Carbohydr. Chem. 2007, 25, 441-450.
7. Spencer, R. P.; Cavallaro, C. L.; Schwartz, J. J. Org. Chem. 1999, 64, 3987–3995.
8. Adinolfi, M.; De Napoli, L.; Di Fabio, G.; Iadonisi, A.; Montesarchio, D.; Piccialli, G.; Tetrahedron, 2002, 58, 6697-2704.
9. Sweeley, C. C.; Bentley, R.; Makita, M.; Wells, W. W. J. Am. Chem. Soc. 1963, 85, 2497-2507.
10. Komiotis, D.; Agelis G.; Manta, S.; Tzioumaki, N.; Tsoukala, E.; Antonakis, K. J. Carbohydr. Chem. 2007, 25, 441-450.
11. Spencer, R. P.; Cavallaro, C. L.; Schwartz, J. J. Org. Chem. 1999, 64, 3987–3995.
12. Zerrouki, R.; Roy, V.; Hadj-Bouazza, A.; Krausz, P. J. Carbohydr. Chem. 2004, 23, 299-303.
Supplementary Material
Acknowledgments
Support from NIGMS RO1 62444 and from NSF REU 0552959 (JRT) is gratefully acknowledged. CEM Corporation loaned the microwave to us.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.a Steglich W, Holfe G. Angew. Chem. Int. Ed. Engl. 1969;8:981–981. [Google Scholar]; b Hofle G, Steglich W, Vorbruggen H. Angew. Chem. Int. Ed. Engl. 1978;17:569–583. [Google Scholar]
- 2.Procopiou PA, Baugh SPD, Flack SS, Inglis GGA. J. Org. Chem. 1998;63:2342–2347. [Google Scholar]
- 3.a Vedejs E, Diver ST. J. Am. Chem. Soc. 1993;115:3358–3359. [Google Scholar]; b Vedejs E, Bennett NS, Conn LM, Diver ST, Gingras M, Lin S, Oliver PA, Peterson MJ. J. Org. Chem. 1993;58:7286–7288. [Google Scholar]
- 4.Wolfrom ML, Thompson A. Meth. Carbohydr. Chem. 1963:211–215. [Google Scholar]
- 5.Bhaskar PM, Loganathan D. Tetrahedron Lett. 1998;39:2215–2218. [Google Scholar]
- 6.Adinolfi M, Barone G, Iadonisi A, Schiattarella M. Tetrahedron Lett. 2003;44:4661–4663. [Google Scholar]
- 7.Phukan P. Tetrahedron Lett. 2004;45:4785–4787. [Google Scholar]
- 8.Dasgupta F, Singh PP, Srivastava HC. Carbohydr. Res. 1980;80:346–349. [Google Scholar]
- 9.Tale RH, Adude RN. Tetrahedron Lett. 2006;47:7263–7265. [Google Scholar]
- 10.Lu K-C, Hsieh S-Y, Patkar LN, Chen C-T, Lin C-C. Tetrahedron. 2004;60:8967–8973. [Google Scholar]
- 11.Misra AK, Tiwari P, Madhusudan SK. Carbohydr. Res. 2005;340:325–329. doi: 10.1016/j.carres.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 12.Heravi MM, Behbahani FK, Zadsirjan V, Oskooie HA. J. Braz. Chem. Soc. 2006;17:1045–1047. [Google Scholar]
- 13.Khan AT, Choudhury LH, Ghosh S. Eur. J. Org. Chem. 2005:2782–2787. [Google Scholar]
- 14.Heravi MM, Behbahani FK, Bamoharram FF. J. Mol. Cat. A: Chem. 2006;253:16–19. [Google Scholar]
- 15.Reddy TS, Narasimhulu M, Suryakiran N, Mahesh KC, Ashalatha K, Venkateswarlu Y. Tetrahedron Lett. 2006;47:6825–6829. [Google Scholar]
- 16.Chakraborti AK, Shivani J. Org. Chem. 2006;71:5785–5788. doi: 10.1021/jo0605142. [DOI] [PubMed] [Google Scholar]
- 17.a Chandra KL, Saravanan P, Singh RK, Singh VK. Tetrahedron. 2002;58:1369–1374. [Google Scholar]; b Tai C-A, Kulkarni SS, Hung S-C. J. Org. Chem. 2003;68:8719–8722. doi: 10.1021/jo030073b. [DOI] [PubMed] [Google Scholar]
- 18.a Orita A, Tanahashi C, Kakudo A, Otera J. Angew. Chem. Int. Ed. 2000;39:2877–2879. doi: 10.1002/1521-3773(20000818)39:16<2877::aid-anie2877>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]; b Orita A, Tanahashi C, Kakuda A, Otera J. J. Org. Chem. 2001;66:8926–8934. doi: 10.1021/jo0107453. [DOI] [PubMed] [Google Scholar]
- 19.Chen C-T, Kuo J-H, Li C-H, Barhate NB, Hon S-W, Li T-W, Chao S-D, Liu CC, Li Y-C, Chang I-H, Lin J-S, Liu C-J, Chou Y-C. Org. Lett. 2001;3:3729–3732. doi: 10.1021/ol016684c. [DOI] [PubMed] [Google Scholar]
- 20.a Ke W, Whitfield DM. Carbohydr. Res. 2004;339:2841–2850. doi: 10.1016/j.carres.2004.10.006. [DOI] [PubMed] [Google Scholar]; b Lee J-C, Tai CA, Hung S-C. Tetrahedron Lett. 2002;43:851–855. [Google Scholar]
- 21.Bartoli G, Dalposso R, De Nino A, Maiuolo L, Nardi M, Procopio A, Tagarelli A. Green Chem. 2004;6:191–192. [Google Scholar]
- 22.Chauhan KK, Frost CG, Love I, Waite D. Synlett. 1999;11:1743–1744. [Google Scholar]
- 23.Kulkarni SS, Hung S-C. Lett. Org. Chem. 2005;2:670–677. [Google Scholar]
- 24.a Ishihara K, Kubota M, Kurihara H, Yamamoto H. J. Am. Chem. Soc. 1995;117:4413–4414. [Google Scholar]; b Ishihara K, Kubota M, Kurihara H, Yamamoto H. J. Org. Chem. 1996;61:4560–4567. doi: 10.1021/jo952237x. [DOI] [PubMed] [Google Scholar]
- 25.a Ghosh R, Maiti S. J. Mol. Cat. A: Chem. 2007;264:1–8. [Google Scholar]; b Chauhan KK, Frost CG. J. Chem. Soc. Perkin Trans. 1. 2000:3015–3019. [Google Scholar]; c Thakur AJ. Synlett. 2003:899–900. [Google Scholar]; d Auge J, Lubin-Germain N, Uziel J. Synthesis. 2007:1739–1764. [Google Scholar]
- 26.Dumeunier R, Markó IE. Tetrahedron Lett. 2004;45:825–829. [Google Scholar]
- 27.Goodrich P, Hardacre C, Mehdi H, Nancarrow P, Rooney DW, Thompson JM. Ind. Eng. Chem. Res. 2006;45:6640–6647. [Google Scholar]
- 28.Fu F, Teo Y-C, Loh T-P. Tetrahedron Lett. 2006;47:4267–4269. [Google Scholar]
- 29.a De SK. Tetrahdron Lett. 2004;45:2919–2922. [Google Scholar]; b Chakraborti AK, Gulhane R. Tetrahedron Lett. 2003;44:6749–6753. [Google Scholar]
- 30.Das SK. Synlett. 2004:915–932. [Google Scholar]
- 31.Limousin C, Cleophax J, Petit A, Loupy A, Lukacs G. J. Carbohydr. Chem. 1997;16:327–342. [Google Scholar]
- 32.Das SK, Reddy KA, Krovvidi VLNR, Mukkanti K. Carbohydr. Res. 2005;340:1387–1392. doi: 10.1016/j.carres.2005.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.