

Abstract
PURPOSE
Evidence supports the immune system activity accompanying glaucomatous neurodegeneration. This study aimed to determine the in vitro effects of reactive oxygen species (ROS) on the phenotype and antigen-presenting function of the retina and optic nerve head glia.
METHODS
Cultures of rat retina and optic nerve head glia were treated with a mixture of ROS-generating compounds for 24 and 48 hours. Pretreated glial cells were then coincubated with syngeneic CD4+ T cells for 48 hours. ROS generation and cell viability were assessed with the use of dihydroethidium and calcein assays, respectively. Flow cytometry and immunocytochemistry were used to determine major histocompatibility complex (MHC) class II molecules. In addition, functional experiments were performed to determine the proliferation and cytokine secretion of T cells using [3H]-thymidine incorporation and TNF-α assays, respectively.
RESULTS
MHC class II molecules were upregulated on glial cells exposed to ROS. Compared with the control glia, glial cells in ROS-generating systems were found to be more potent inducers of T-cell activation in a cell density- and time-dependent manner, as assessed by increased T-cell proliferation (approximately threefold) and TNF-α secretion (approximately sixfold; P < 0.01). When an ROS scavenging treatment was applied, MHC class II upregulation on glial cells persisted, but antigen-mediated T-cell activation was significantly decreased (P < 0.01), indicating an additional costimulatory function of ROS during antigen presentation.
CONCLUSIONS
These in vitro findings support that ROS regulate the immune response by stimulating the antigen-presenting ability of glial cells and functioning as costimulatory molecules for antigen presentation.
Oxidative stress caused by increased generation of reactive oxygen species (ROS)1 and nitric oxide-induced damage2,3 has been implicated in retinal ganglion cell (RGC) death after axonal injury during glaucomatous neurodegeneration. Our recent in vitro studies using primary cultures of RGCs have also provided evidence that RGC death induced by different glaucomatous stimuli involves increased ROS generation and that antioxidant treatment provides additional protection to caspase inhibited RGCs.4 In addition, our more recent in vivo studies using a proteomic approach have revealed oxidative modification of many important retinal proteins during glaucomatous neurodegeneration in ocular hypertensive rat eyes.5
Growing evidence obtained from clinical and experimental studies over the past decade strongly suggests the involvement of the immune system in glaucoma.6–8 The association of the immune system to glaucoma has seemingly conflicting aspects as neuroprotective or neurodestructive. T-cell-mediated immune response may initially be beneficial to limit neurodegeneration.9–11 However, a failure to properly control aberrant, stress-induced immune response likely converts the protective immunity to an autoimmune neurodegenerative process that can facilitate the progression of neurodegeneration in some, if not all, glaucoma patients. Expansion and secondary recruitment of circulating T cells through an antigen-mediated process is supported by the evidence of abnormal T-cell subsets12 and increased production of serum autoantibodies to different optic nerve and retina antigens in many glaucoma patients.13–17 Furthermore, initial in vivo studies support the feasibility of eliciting an experimental autoimmune model of glaucomatous neurodegeneration in which RGCs progressively die in specific antigen-immunized animals by exhibiting a pattern of neuronal damage similar to that of human glaucoma (Wax MB, et al. IOVS 2006;47:ARVO E-Abstract 1828).
Activated immune response in glaucoma patients may partly be associated with the increased expression and exposure of neuronal antigens as a result of neuronal stress and injury. Several stress-associated factors are also known to be required for the activation of resting antigen-presenting cells.18–20 Microglia, which are derived from the monocyte/macrophage lineage, play a crucial role in the regulation of the immune response.21 In addition to microglia, considerable evidence indicates that astrocytes, the most numerous glial cells in the central nervous system (CNS), are also capable of regulating immune responses.22–24 Similarly, microglial and macroglial cells (including astrocytes and retinal Müller cells) are two important cell types with immunoregulatory functions in the optic nerve head and retina.25–27 Consistent with observations in other neurodegenerative injuries,28,29 chronic activation of glial cells in glaucomatous human eyes30,31 is accompanied by upregulation of major histocompatibility complex (MHC) class II molecules.32 Not only microglial cells33 but also glial fibrillary acidic protein (GFAP)-positive astrocytes exhibit increased immunolabeling for HLA-DR (a human MHC class II molecule) in glaucomatous human eyes.32 Thus, persistent activation of the retina and optic nerve head glia in glaucomatous eyes also involves the activation of their antigen-presenting ability, thereby facilitating the initiation of an autoimmune process through antigen presentation.
Based on the activated state of glial cells in glaucomatous eyes, along with the evidence of amplified ROS generation, we hypothesized that oxidative stress may be a major force that drives a resting immune system over the threshold of antigen-specific activation. To investigate the validity of this hypothesis, we aimed to determine the effects of ROS on the phenotype and function of glial cells in activating T cells. We therefore performed a series of in vitro experiments using syngeneic T cells, retinal macroglial and microglial cells, and optic nerve head astrocytes. Findings of these experiments demonstrated that glial cells exposed to ROS-generating compounds are more potent inducers of T-cell activation, as assessed by increased proliferation and cytokine secretion of T cells. Our findings also indicated that, in addition to an upregulation of MHC class II molecules on glial cells, ROS function as costimulatory molecules during antigen presentation. These findings provide evidence that in the presence of ROS, stimulated antigen-presenting ability of glial cells in the glaucomatous retina and optic nerve head can elicit an activated immune response.
MATERIALS AND METHODS
Primary Cell Cultures
Cell cultures were derived from Lewis rats (Harlan-Sprague-Dawley, Indianapolis, IN). All animals were handled according to the regulations of the Institutional Animal Care and Use Committee, and all procedures adhered to the tenets of the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
T-cell cultures were derived from Lewis rats immunized with myelin basic protein (MBP) according to previously described methods.34,35 T cells were isolated 10 days after immunization from lymph node and spleen cells by passage through a nylon wool column. Cells (1 × 107) were first stimulated with 10 µg/mL MBP 72 to 89 in the presence of 2 × 107 irradiated syngeneic spleen cells as antigen-presenting cells. After 2 days, activated T-cell blasts were isolated by gradient centrifugation (Lymphoprep; Robbins Scientific, Mountain View, CA) and cultured in RPMI-1640 medium (Mediatech, Herndon, VA) supplemented with 10% inactivated fetal calf serum, 5 × 10−5 M 2-mercaptoethanol, and 15% interleukin-2- containing medium (supernatant from Con A-stimulated rat spleen cells). T cells were maintained in a regular tissue culture incubator at 5% CO2 and 37°C by periodic restimulation for 48 hours (once every 10 days) with antigen in the presence of irradiated syngeneic antigen-presenting cells. T-cell lines were used after three or four stimulation/resting cycles.
Retinal glial cell cultures were prepared using retinal cells depleted of microglia and ganglion cells after the immunomagnetic selection process previously described. After loss of residual neuronal cells by two or three cycles of replating, these cultures contained glial cells, including astrocytes and Müller, as previously documented.36
Retinal microglial cells were isolated as previously described (Peng G, et al. IOVS 2002;42:ARVO E-Abstract 3383).37 Briefly, mixed retinal cells were cultured in Dulbecco modified Eagle medium (DMEM; Gibco, Grand Island, NY) supplemented with 10% serum,1 ng/mL granulocyte-macrophage colony-stimulating factor (Sigma-Aldrich, St. Louis, MO), and antibiotics. For purification of microglia, the cells were incubated with Ca2+-Mg2+ -free Hanks balanced salt solution (Sigma-Aldrich) containing 0.2% EDTA and 5% serum for 1 hour at 4°C and detached by vigorous pipetting. The resultant cell suspension was placed in 96-well plates. Previous studies confirmed the purity of these cultures by immunolabeling for specific cell markers CD11b and GFAP. Flow cytometry revealed that 96% of the cells were positive for CD11b, a marker of microglial cells. However, only 5% of the cell population was positive for GFAP, a marker of astrocytes (Peng G, et al. IOVS 2002;42:ARVO E-Abstract 3383).
Cultures of optic nerve head astrocytes were also prepared as previously described.32,38,39 Briefly, after removing the postlaminar myelinated nerve, the remaining optic nerve head tissue was bisected and the central vessels were removed under a dissecting microscope. Tissues were then cut into small pieces, and each explant was rinsed in growth medium (DMEM/F-12 supplemented with 10% serum and antibiotics), placed in a culture flask, and kept in the hood for 10 minutes until the explant adhered to the surface of the flask. Before placement in the incubator, 0.5 mL medium was added to each flask. When the first cells began to grow out of the explant, the medium was increased to 2 mL and then was changed twice a week. Flow cytometric analysis previously showed that more than 98% of these cells were GFAP-positive astrocytes.32
During the experimental period, glial cell cultures were incubated in a serum-free medium containing DMEM, 1.3% bovine albumin fraction V, 1 µL/mL culture supplement (ITS+ Premix; BD Biosciences, San Diego, CA), and antibiotics.
Coculture Experiments
Glial cells seeded on 96-well plates at two different concentrations (1 × 104 and 4 × 104 cells/well) were treated with ROS-generating compounds for 24 or 48 hours or were left untreated. Syngeneic CD4+ T cells (1 × 105 cells/well) were then added to these plates and coincubated with pretreated glial cells for an additional 48 hours in the presence of antigen. Additional wells without antigen were simultaneously processed as a control.
At the end of the experimental period, cells were immediately subjected to the experiments described, which included measurement of ROS generation and cell viability through specific assays and assessment of MHC class II molecules on glial cells through flow cytometry and immunocytochemistry. In addition, functional experiments were performed to determine the proliferation and cytokine secretion of T cells. For this purpose, at the end of the coincubation period, 50 µL coculture supernatant was collected from each well into a new plate to measure TNF-α levels by enzyme-linked immunosorbent assay (ELISA). The coculture plate was then subjected to T-cell proliferation assay.
To exclude the possibility that TNF-α production in cocultures was contributed by glial cells, we also treated glial cells with mitomycin C (100 µg/mL; Sigma-Aldrich) for 1 hour before mixing them with T cells. For induction of MHC class II expression, additional wells included interferon-gamma (IFN-γ) treatment (20 ng/mL; R&D Systems, Minneapolis, MN) as a positive control. In addition to treated or untreated coculture wells, control wells containing glial cells and T cells cultured alone in the absence and presence of treatments were included in the plates. All counts were conducted in a masked fashion. All experiments were performed in triplicate wells and repeated three times for each experimental condition. An integrated value was obtained for untreated and treated cultures after background subtraction using corresponding negative controls. Differences between treated and untreated cultures were statistically analyzed by the Wilcoxon signed-rank test using these normalized values. Differences between the cocultures of T cells with three glial cell types were tested by one-way analysis of variance (ANOVA) using the Tukey test, and the Mann-Whitney U test was used to test differences between the different treatments used. Data are presented as fold-change (mean ± SD), which was expressed by comparing the normalized values in treated cultures relative to untreated controls, followed by the calculation of mean ± SD.
ROS-Generating or -Scavenging Treatments
Standard systems were used to generate ROS in vitro. In addition to H2O2 (20 µM), paraquat (20 µM) was used to generate the superoxide anion O2−. Paraquat undergoes single electron reduction through the oxidation of NADPH to form paraquat free radical, which reacts with molecular oxygen to produce O2−. To generate nitric oxide, S-nitroso-N-acetyl penicillamine (SNAP; 200 µM) was used. The hydroxyl radical was generated through the Fenton reaction, which uses a transition metal to reduce H2O2 and form the radical. Therefore, a combination (CPA) of CuSO4 (1 µM), 1,10-phenanthroline (1 µM), and ascorbic acid (400 µM) was used (all from Sigma-Aldrich). Utilized concentrations were based on dose-response experiments and were chosen to yield glial cell survival rates greater than 70% of untreated cultures at 48 hours. Although we initially tested these ROS-generating compounds individually and in combination, which revealed insignificant differences in their effect on glial cells, they were used as a mixture in this study to better simulate in vivo conditions. In addition, a cell-permeable vitamin E analogue, 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox; 100 µM; Alexis, San Diego, CA) was used for nonspecific ROS scavenging.
Assessment of Cell Survival
Cell viability was determined with the use of a live/dead kit containing calcein AM (Molecular Probes, Eugene, OR), as previously described.4,36 The kit relies on the intracellular esterase activity within living cells, through which the calcein AM, a cell-permeable fluorogenic esterase substrate, hydrolyzes to a green fluorescent product, calcein. The green fluorescence of living cells was counted in at least 10 random fields of each well at ×200 magnification under a fluorescence microscope (Carl Zeiss, Thornwood, NY). Survival rate was expressed as the percentage of the total cell number in control wells (treated with no additives) at each time point.
Assessment of ROS Generation
To assess ROS generation, cultures were incubated with dihydroethidium (1 mM; Molecular Probes) for 30 minutes, as previously described.4 When reacted with intracellular ROS, this cell-permeable dye fluoresces in the rhodamine spectrum.40 After labeling, cells were washed and replenished with fresh medium, and fluorescence was measured with a microplate spectrofluorometer (Gemini EM; Molecular Devices, Sunnyvale, CA). ROS generation was reported as the percentage of measurement in control wells.
Flow Cytometry
Flow cytometric analysis of MHC class II molecules was performed as previously described.32,36 Briefly, cells were fixed with 2% paraformaldehyde solution for 20 minutes at room temperature. After centrifuge and resuspension, cells were permeabilized using 0.4% Triton X-100 solution (Sigma-Aldrich) for 30 minutes. Washed cells were then incubated with an FITC-conjugated monoclonal antibody against MHC class II (1:100; eBioscience, San Diego, CA) for 30 minutes. Replacing the antibody with serum served as the negative control. After washing and resuspension, cells were analyzed by flow cytometry (CellQuest software; FACSCalibur, Becton-Dickinson, San Jose, CA).
Immunocytochemistry
Double-immunofluorescence labeling was performed as previously described.4,36 Briefly, cells grown on coverslips were washed in phosphate-buffered saline solution and fixed with 4% paraformaldehyde solution for 30 minutes at room temperature. After washing, cells were permeabilized using a Triton X-100 solution (Sigma-Aldrich) for 4 minutes, followed by incubation in 3% bovine serum albumin for 30 minutes to block nonspecific binding sites. Cells were then incubated with a mixture of anti-mouse and anti-rabbit primary antibodies for 2 hours. Primary antibodies were against MHC class II molecules (1:100; eBioscience) or glia markers GFAP (1:200; Invitrogen, Carlsbad, CA) and CD11b (1:100; Chemicon, Temecula, CA). This incubation was followed by washing and then by incubation with a mixture of conjugated anti-mouse or anti-rabbit secondary antibodies (2 µg/mL; Alexa Fluor 488 or 568, respectively; Molecular Probes). Negative controls were performed by replacing the primary antibody with serum. In addition, incubation with each primary antibody was followed by the inappropriate secondary antibody to determine that each secondary antibody was specific to the species against which it was made. After washing, cells were examined under a fluorescence microscope (Carl Zeiss).
Lymphocyte Proliferation Assay
T-cell proliferation was measured as previously described.35,41 Briefly, at the end of incubation period, 0.5 µCi [3H]-thymidine/well was added to culture plates for 6 hours. Wells not incubated with [3H]-thymidine served as the negative control. Cells were harvested and assessed for isotope incorporation using a beta counter (Beckman, Fullerton, CA).
ELISA
A commercial kit was used to measure TNF-α levels in a fraction of culture supernatant by quantitative sandwich ELISA (R&D Systems), as previously described.36 The culture supernatant was incubated in microwells coated with a monoclonal antibody specific for rat TNF-α. After washing, a horseradish peroxidase- conjugated secondary antibody was added to the wells, followed by addition of a substrate solution containing hydrogen peroxide and tetramethylbenzidine. The enzyme reaction was terminated by the addition of hydrochloric acid solution, and absorbance was measured at 450 nm. Antibody-uncoated control wells and standard curve wells prepared from seven dilutions of recombinant rat TNF-α were simultaneously processed to calculate concentrations of TNF-α in the culture supernatant. ELISA sensitivity was lower than 5 pg/mL.
RESULTS
To determine immunologic consequences of amplified ROS generation during glaucomatous neurodegeneration, we performed a series of in vitro experiments examining glia-T-cell interactions in the absence and presence of ROS-generating treatment. Our initial experiments using primary cultures of retina and optic nerve head glia confirmed ROS generation after exposure of these cells to a mixture of ROS-generating compounds. As shown in Figure 1A, compared with the control untreated cells, ROS generation was amplified in glial cells treated with these compounds for up to 48 hours. Based on dose-response experiments, ROS-generating compounds were used at micromolar concentrations because higher concentrations caused cell death. As confirmed by survival rates presented in Figure 1B, the utilized concentrations of these compounds yielded survival rates greater than 70% of untreated cultures. However, cotreatment of glial cell cultures with ROS-generating compounds and IFN-γ (complementarily used as a positive control for the stimulation of the glial antigen-presenting ability) resulted in survival rates less than 70% of untreated cultures.
FIGURE 1.
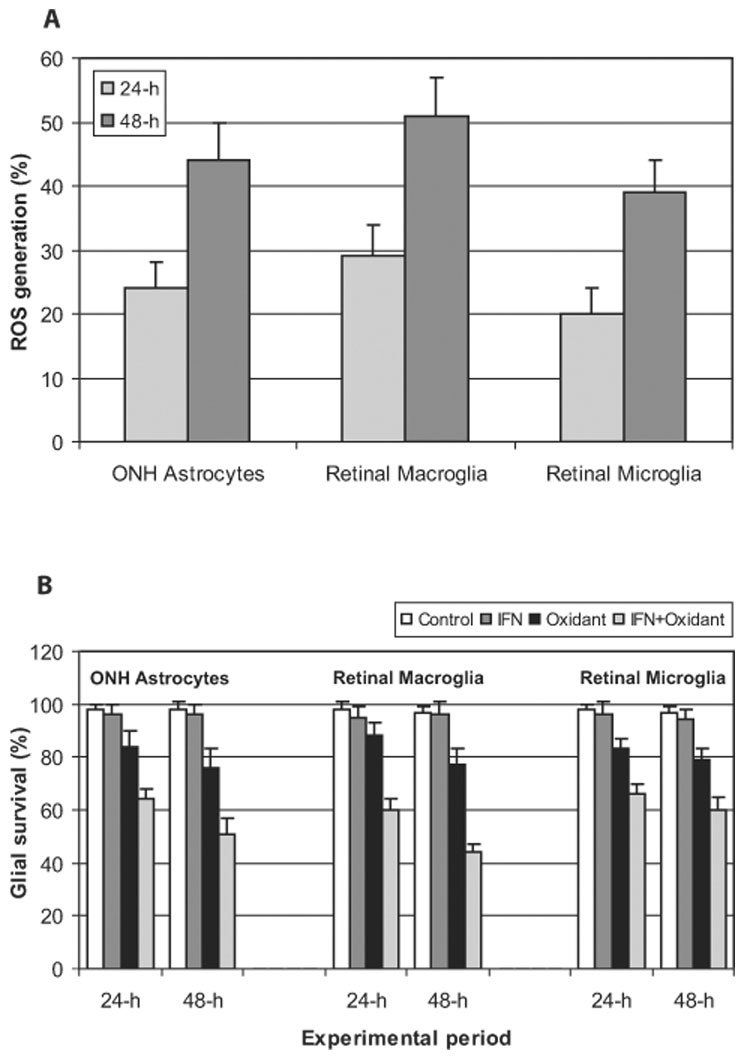
ROS generation (A) and survival (B) rates in glial cell cultures exposed to a mixture of ROS-generating compounds for 24 or 48 hours. ROS generation and cell survival, which were assessed with dihydroethidium and calcein assays, respectively, are expressed as percentages of controls. Despite ROS generation, the survival rate of glial cells after ROS-generating treatment was greater than 70% of untreated controls. All experiments were performed in triplicate and were repeated three times for each experimental condition. Data are presented as mean ± SD. ONH, optic nerve head.
We then examined phenotypic alterations of glial cells after their ROS-generating treatment. Despite cell debris and loosely attached cells, no prominent morphologic changes were detectable in surviving glial cells after ROS-generating treatment. As presented in Figure 2, no alteration in cell shape or in number or length of cellular processes possibly associated with cellular activity in glial immunoregulatory function could be detected. However, MHC class II molecules were significantly upregulated on glial cells after ROS-generating treatment (Wilcoxon signed-rank test; P < 0.01). As shown in Figure 3, based on flow cytometric analysis, MHC class II upregulation with ROS was predominant on microglial cells compared with optic nerve astrocytes or retinal macroglia. Treatment of microglia with chemical generators of ROS resulted in a sixfold upregulation of MHC class II molecules at 48 hours, whereas optic nerve head and retinal macroglial cells treated with these compounds exhibited an approximately threefold increase in MHC class II expression (one-way ANOVA; P < 0.001). Immunocytochemistry of cultured glial cells supported MHC class II upregulation after ROS exposure. As shown in Figure 4, MHC class II immunolabeling was virtually negative in control optic nerve head astrocytes and retinal macroglia and barely detectable in control cultures of retinal microglia. However, CD-11b-positive microglia and GFAP-positive optic nerve head and retinal macroglia exhibited prominent MHC immunolabeling after ROS-generating treatment. Thus, ROS induce the antigen-presenting ability of the optic nerve head and retinal glia.
FIGURE 2.
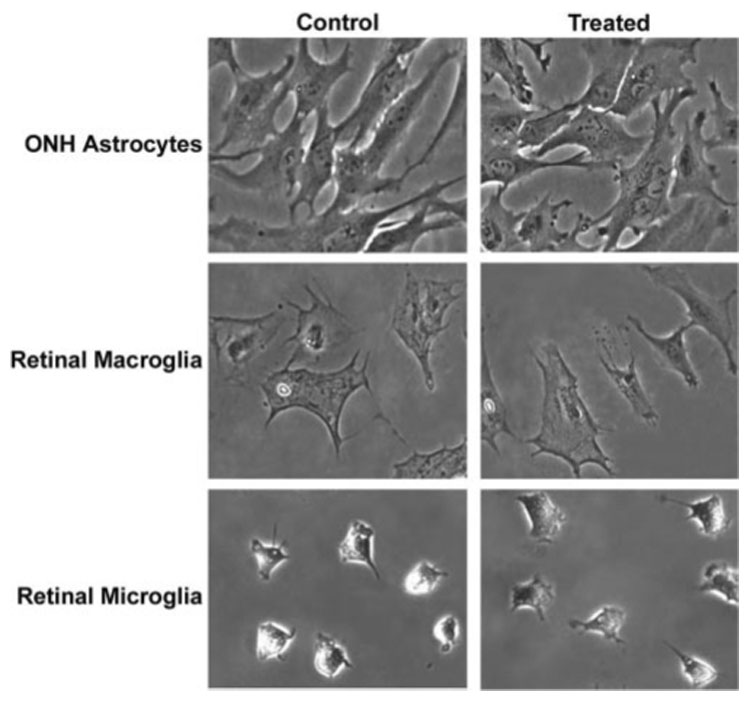
Morphologic assessment of glial cells after ROS-generating treatment. Phase-contrast images of glial cells incubated in the absence or presence of RO-generating treatment for 48 hours exhibit no morphologic changes. ONH, optic nerve head.
FIGURE 3.
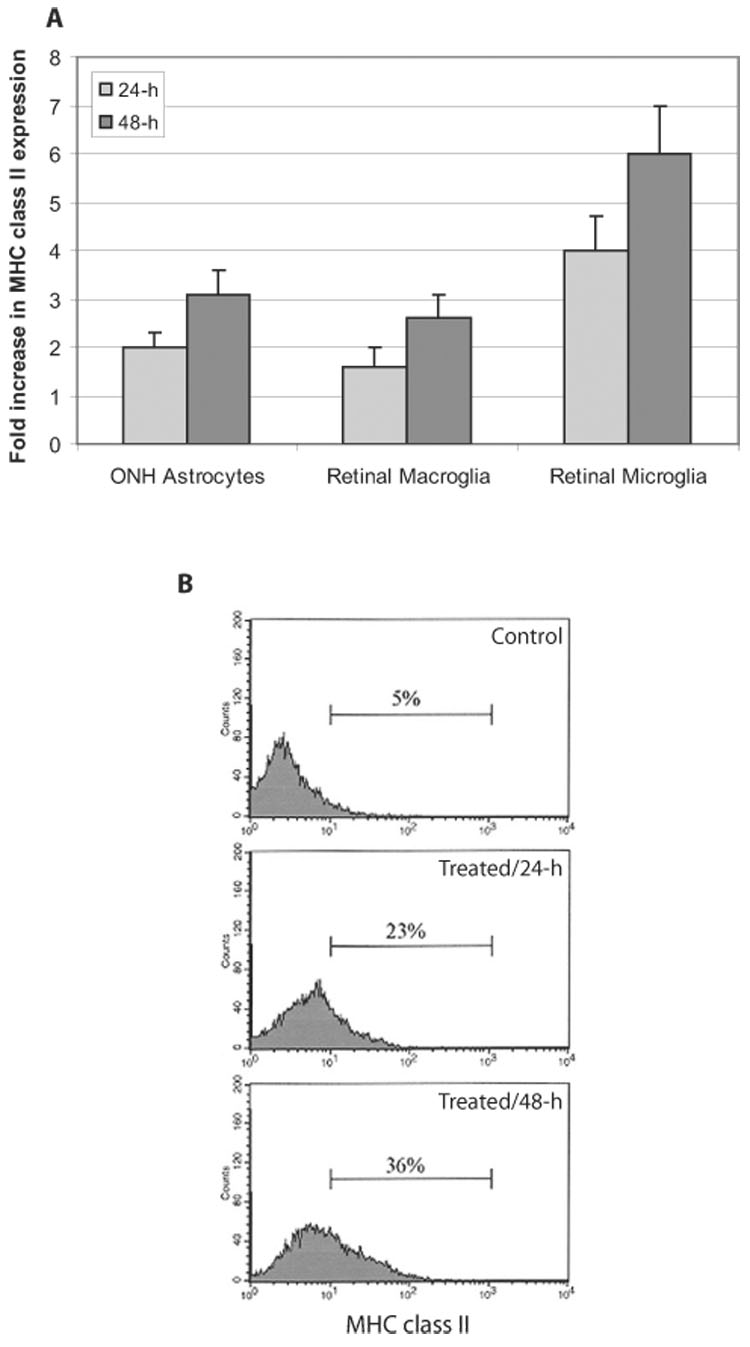
MHC class II upregulation on glial cells after ROS-generating treatment. (A) Fold increase in MHC class II molecules on glial cells was assessed by flow cytometric analysis. After ROS-generating treatment, MHC class II upregulation on glial cells was predominant for microglial cells. (B) Representative fluorescence histograms obtained from flow cytometric analysis of retinal microglia incubated in the absence or presence of ROS-generating treatment for 24 or 48 hours. All experiments were performed in triplicate and were repeated three times for each experimental condition. Data are presented as mean ± SD. ONH, optic nerve head.
FIGURE 4.

Immunocytochemical assessment of MHC class II molecules on glial cells. Fluorescence images show double immunolabeling of glial cells for specific cell markers (GFAP or CD11b) and MHC class II molecules in control cultures and cultures treated with ROS-generating compounds for 48 hours. Both CD-11b-positive microglia and GFAP-positive optic nerve head (ONH) and retinal macroglia exhibit prominent immunolabeling for MHC class II molecules after ROS-generating treatment.
Glia cultures exposed to ROS-generating compounds for 24 or 48 hours were then coincubated with syngeneic T cells for another 48 hours. These experiments revealed that glial cells pretreated with ROS-generating compounds are more potent inducers of T-cell activation, as assessed by increased proliferation and cytokine secretion of T cells. With the use of a T-cell proliferation assay, we observed that glial cells pretreated with ROS-generating compounds markedly stimulated (approximately threefold) the proliferation of coincubated T cells in a cell density- and time-dependent manner (Fig. 5). When the experiments were performed with different numbers of glial cells, stimulation of T-cell proliferation was greater (twofold vs fourfold) in plates with higher (4 × 104 cells/well) rather than lower (1 × 104 cells/well) glial cell density. The ability of ROS-exposed glial cells to stimulate T-cell proliferation was consistently higher than that of the control glia (Wilcoxon signed-rank test; P < 0.01), indicating that a functional effect had taken place but was only clearly manifested when high numbers of glial cells were used.
FIGURE 5.
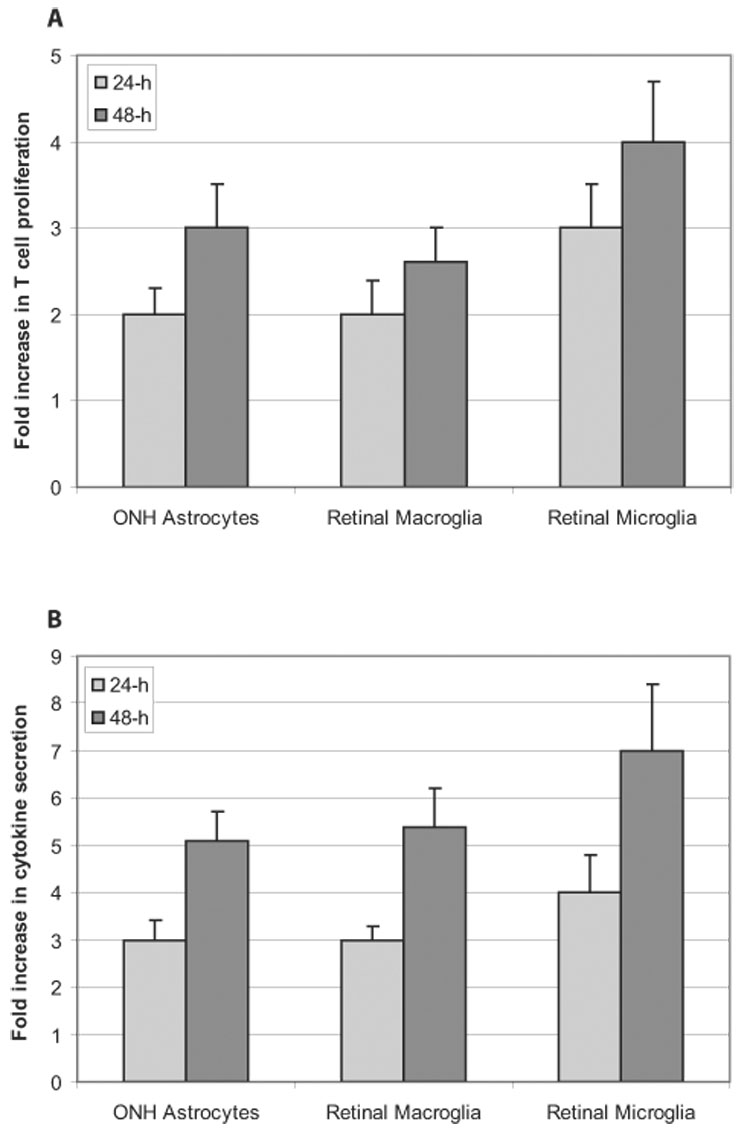
T-cell activation after ROS-generating treatment of antigen-presenting glia for 24 or 48 hours. Compared with the control glia, glial cells in ROS-generating systems were found to be more potent inducers of T-cell activation in a time-dependent manner, as assessed by increased proliferation (A) and increased TNF-α secretion (B) of T cells (Wilcoxon signed-rank test; P < 0.01). ROS-stimulated T-cell activation was predominant in cocultures of T cells with microglia compared with cocultures with optic nerve head (ONH) astrocytes or retinal macroglia (one-way ANOVA; P < 0.001). All experiments were performed in triplicate and were repeated three times for each experimental condition. Data are presented as fold-change (mean ± SD), which was expressed by comparing the normalized values in treated cultures compared with untreated controls.
TNF-α is the most abundantly produced cytokine generated by reactive T cells during glia-T-cell interactions.42,43 Therefore, we also determined whether the secretion of this cytokine was affected by antigen-presenting glia. Thus, in addition to T-cell proliferation, we also measured TNF-α secretion as an additional functional measure of T-cell activity. In parallel to the findings of the T-cell proliferation assay, ELISA of TNF-α levels in the coculture medium revealed that glial cells exposed to ROS-generating compounds significantly induced the secretion of this cytokine by T cells (Wilcoxon signed-rank test; P < 0.01). After coincubation of pretreated glia with T cells, TNF-α secretion was increased by approximately sixfold (Fig. 5). Compared with plates with lower glial cell density, stimulation of TNF-α secretion was greater in plates with higher glial cell density (fourfold vs eightfold). Increased secretion of this cytokine was greater in cocultures of T cells with microglial cells than in cocultures of T cells with optic nerve astrocytes or retinal macroglia (one-way ANOVA; P < 0.001).
After these observations, we determined whether the TNF-α detected in the coculture medium was preferentially produced by T cells or whether glial cells also produced this cytokine. When T cells and glia were cultured separately in the absence and presence of ROS, we detected that the relative TNF-α-secreting ability was increased in both cell types after their exposure to ROS-generating compounds. Indeed, T cells and glial cells are known to be able to release significant quantities of TNF-α, whereas microglia are more efficient in TNF-α secretion. However, TNF-α levels in the coculture medium were higher than when glial cells were cultured alone (without T cells) in the presence of identical ROS-generating treatments (approximately sixfold vs less than twofold). Moreover, to exclude the possibility that glial cell contributed to TNF-α production in coculture, after ROS-generating pretreatment, we also treated glia with mitomycin C before mixing them with T cells because mitomycin C was previously shown to block glial proliferation and function without interfering with MHC expression.42 Findings of this experiment demonstrated that despite mitomycin C treatment of glial cells, TNF-α levels were higher in the coculture medium (approximately sixfold vs fivefold; Mann-Whitney U test; P < 0.05), excluding the possibility that glial cells significantly contributed to TNF-α production in cocultures.
Thus, pretreatment of glia with ROS-generating compounds resulted in significant stimulation of the proliferation and TNF-α secretion of T cells. During our experiments, we also complementarily used IFN-γ treatment as a positive control for the stimulation of the glial antigen-presenting ability. As shown in Figures 6B–D, IFN-γ treatment of glial cells in additional control wells resulted in a significant increase in MHC class II expression, T-cell proliferation, and TNF-α secretion (Wilcoxon signed-rank test; P < 0.001). The average increase in MHC class II expression of glial cells with IFN-γ treatment was greater than sixfold, with an approximately fivefold increase in T-cell proliferation and an approximately 10-fold increase in TNF-α secretion. However, there was no synergistic effect of glial cotreatment with IFN-γ and ROS-generating compounds over IFN-γ treatment alone. T-cell proliferation (less than three-fold increase) and TNF-α secretion (fivefold increase) were lower in coculture wells containing pretreated glial cells with ROS-generating compounds and IFN-γ than in controls pretreated with ROS-generating compounds or IFN-γ alone, probably because IFN-γ synergizes ROS,44–46 thereby leading to glial cell death and interfering with their functional outcomes. In fact, counts of surviving cells (Fig. 1B) indicate that cotreatment of glial cell cultures with ROS-generating compounds and IFN-γ resulted in survival rates less than 70% of untreated cultures.
FIGURE 6.
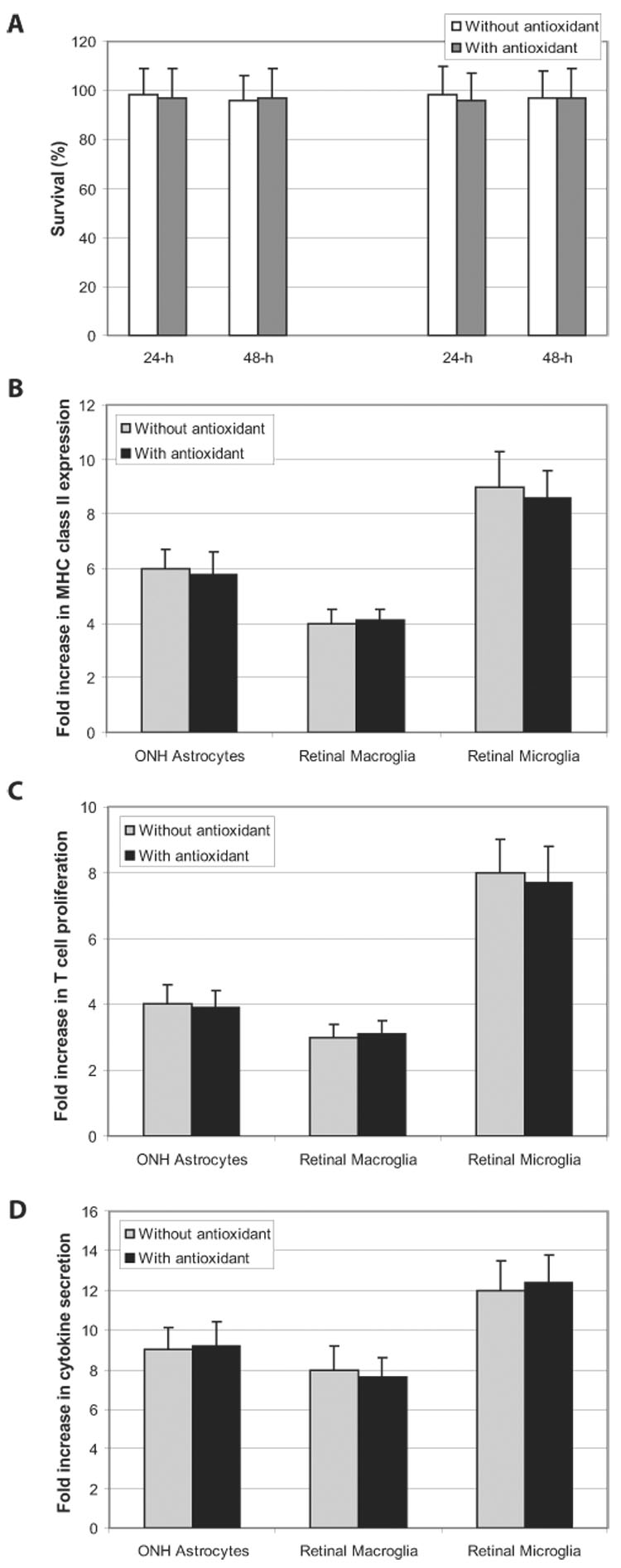
Effects of antioxidant treatment on cell survival and IFN-γ-stimulated antigen presentation. (A) Antioxidant treatment using a vitamin E analogue for 24 or 48 hours did not affect the survival of glial cells or T cells in control cocultures. Cell survival was assessed using a calcein assay and was expressed as a percentage of controls. In addition, the ROS-scavenging treatment did not affect the antigen presentation stimulated by IFN-γ. It also did not cause any significant alteration in glial MHC upregulation (B), T-cell proliferation (C), or TNF-α secretion (D) in cocultures treated with IFN-γ (Mann-Whitney U test; P > 0.05). All experiments were performed in triplicate and were repeated three times for each experimental condition. Data are presented as fold-change (mean ± SD), which was expressed by comparing the normalized values in treated cultures with untreated controls. ONH, optic nerve head.
As also supported by our findings, the stimulatory role of IFN-γ in antigen presentation is mainly associated with MHC upregulation.47,48 Based on a similar upregulation of MHC class II molecules on glial cells by ROS-generating treatment, the ROS-induced stimulation of glial antigen-presenting ability was thought to be similarly associated with MHC upregulation. To better determine whether ROS play additional roles in glial antigen presentation and T-cell activation, a cell-permeable vitamin E analogue (Trolox; Alexis) was used for nonspecific ROS scavenging. As shown in Figure 6A, this treatment alone did not exhibit any toxic effect on glial cells or T cells in control cocultures incubated in the absence of ROS-generating compounds. In addition, as shown in Figures 6B–D, this ROS-scavenging (Trolox; Alexis) treatment did not affect the antigen presentation stimulated by IFN-γ (Mann-Whitney U test P < 0.05). However, when the treatment was added to the ROS-containing medium of glia-T-cell cocultures, glial MHC upregulation persisted but T-cell activation was significantly decreased (Mann-Whitney U test; P < 0.01; Fig. 7). Thus, despite its having no effect on IFN-γ-stimulated antigen presentation, the ROS-scavenging treatment (Trolox; Alexis) resulted in a significant decrease in ROS-stimulated antigen presentation. This specific effect supports an important role of ROS in antigen presentation. These findings collectively indicate that stimulated antigen presentation by ROS was not solely the result of MHC class II upregulation on glial cells. Decreased stimulation of T-cell activation by ROS-scavenging treatment, despite the persistence of MHC upregulation on glial cells, points to an additional role of ROS during antigen presentation.
FIGURE 7.
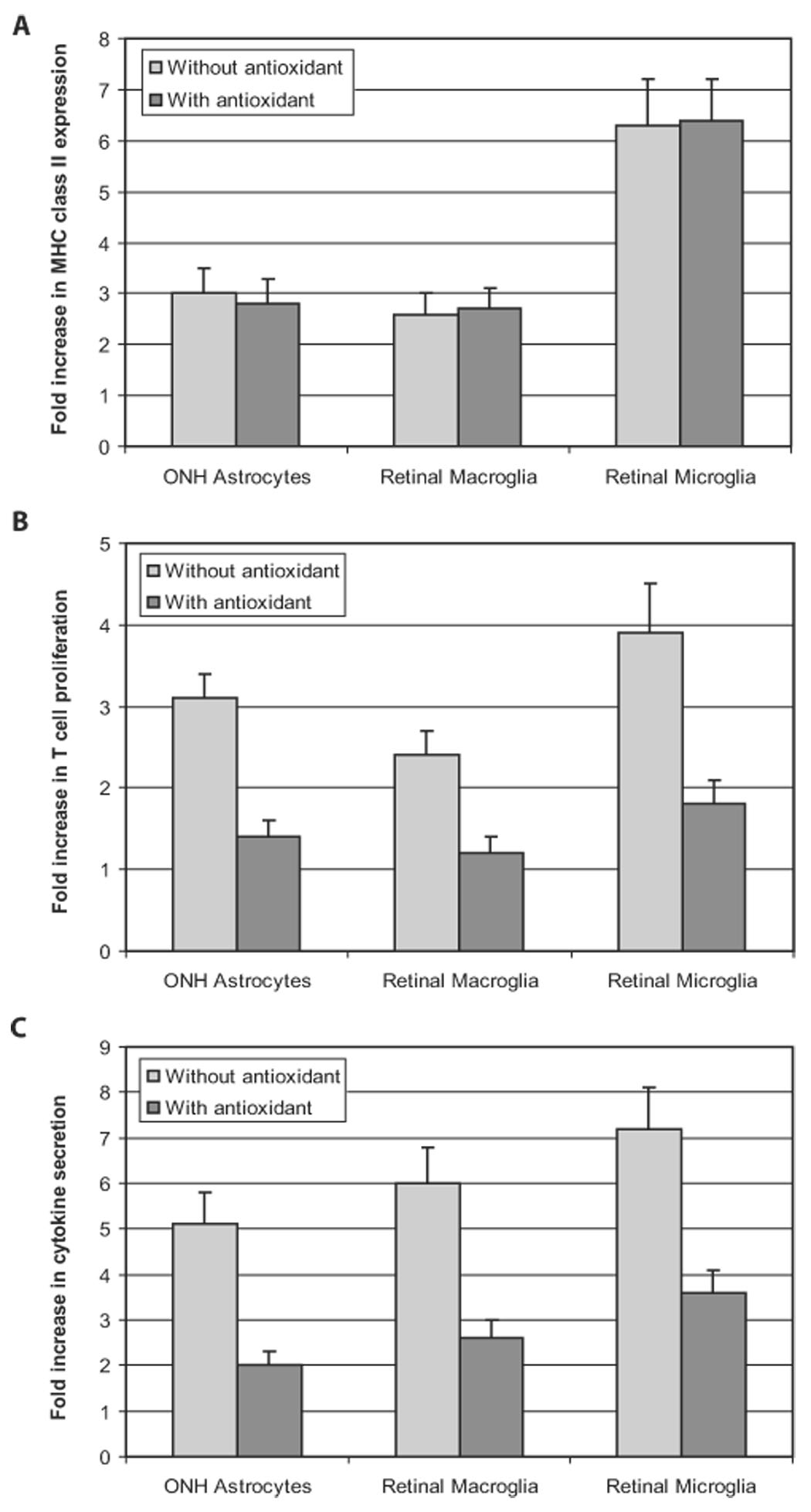
Costimulatory function of ROS in antigen presentation. When a ROS-scavenging treatment was applied, glial MHC class II upregulation persisted (A). However, T-cell proliferation (B) and cytokine secretion (C) were prominently decreased (Mann-Whitney U test; P < 0.01). All experiments were performed in triplicate and were repeated three times for each experimental condition. Data are presented as fold-change (mean ± SD), which was expressed by comparing the normalized values in treated cultures with untreated controls. ONH, optic nerve head.
DISCUSSION
Amplified generation of ROS during glaucomatous neurodegeneration, which leads to protein oxidation, depletion of the cellular redox balance, and cell death, has been associated with pathogenic mechanisms.4,5 In this study, we aimed to determine whether oxidative stress is also associated with immune system activity in glaucoma. Considering that the outcome of cellular interactions under specific disease conditions depends on the interplay of multiple factors, we established an in vitro system to determine interactions of glial cells with T cells in the absence and presence of ROS-generating treatment. Findings of this in vitro study demonstrated that the retina and the optic nerve head glia vigorously respond to ROS with alterations in their immunologically relevant functions, including stimulated antigen presentation to T cells. After pretreatment of glial cells with ROS-generating compounds, MHC class II molecules were upregulated. In the presence of ROS, glial cells more potently induced T-cell activation, as assessed by increased proliferation and TNF-α secretion of T cells. Findings of this study also supported that in addition to inducing MHC upregulation, ROS function as costimulatory molecules during antigen presentation. These important roles of ROS in antigen presentation by glial cells link oxidative stress to the initiation of an adaptive immune response during glaucomatous neuro-degeneration.
Immune privilege of the CNS makes it crucial that glial cells are capable of responding rapidly to any injurious condition or damage. In addition to their association with the innate immune response, by functioning as parenchymal antigen-presenting cells, these resident cells of the CNS also play a regulatory role in the initiation and modulation of the adaptive immune response, which is mediated by T cells (cellular response) with subsequent recruitment of B cells (humoral response).21–24,47 The fundamental basis of cellular immune recognition dictates that T cells that recognize antigens in the form of small peptides tightly bound to MHC class II molecules displayed on the surfaces of antigen-presenting cells.49 The MHC complex can then interact with antigen-specific receptors on histocompatible T cells to induce an antigen-specific reaction. MHC molecules thereby play an important role in autoimmune response, and a significant correlation has been reported between disease activity and MHC class II expression by resident cells of the organ involved.50 ROS-induced upregulation of glial MHC class II molecules in the present study was in accordance with previous findings supporting an upregulation of glial HLA-DR expression in glaucomatous human eyes.32,33 However, we found that the stimulated antigen presentation by ROS was not solely caused by glial MHC upregulation. Despite the persistence of upregulated MHC class II molecules on glial cells, ROS-scavenging treatment with a vitamin E analogue (Trolox; Alexis) resulted in a decrease in antigen-mediated T-cell activation by glial cells. This finding points to an additional role of ROS as costimulatory molecules. Thus, in addition to many other costimulatory molecules already identified, ROS can function as costimulatory molecules during antigen presentation by glia, which are also required for eliciting an activated immune response.
Although ROS have been regarded as toxic molecules because of their great reactivity and ability to damage cellular macromolecules, such as DNA, lipids, and proteins, controlled production of ROS and regulated redox modifications of transcription factors or enzymes are an essential part of signal transduction pathways.51,52 Our findings support that ROS also play an important role in the regulation of immune system activity. We observed that in the presence of ROS, glial cells markedly stimulated the proliferation of cocultured T cells. In addition to T-cell proliferation, we measured TNF-α secretion as a functional measure of T-cell activity. TNF-α is the most abundantly produced cytokine generated by reactive T cells,43,44 and TNF-α production has been associated with immune regulation and autoimmune pathogenesis of different neurodegenerative diseases of the CNS.41 RGCs are also susceptible targets of TNF-α-mediated death signaling.36,53,54 We therefore tested whether ROS affected TNF-α secretion of reactive T cells. In parallel with findings of the T-cell proliferation assay, we found that at higher cell concentrations, glial cells exposed to ROS-generating compounds induced the secretion of this cytokine by T cells. The density dependency we detected suggests that the context of synergy between activating signals requires a critical threshold number of cells before any influence can be demonstrated. There may also exist cascading responses by which multiple factors intensify or modify each other’s effects.55 For example, because TNF-α is known to induce ROS generation,4,56 mutual stimulation may lead to a vicious circle and to consequent deleterious overproduction of ROS and TNF-α.41 Dual stimulation of TNF-α by reactive T cells and glial cells could also be possible in our cocultures because TNF-α secretion by glial cells can also be upregulated by different stimuli.36,53 However, TNF-α levels in the coculture supernatant were higher than those detected when glial cells were cultured alone in the presence of identical treatments. Moreover, increased TNF-α secretion persisted even after mitomycin C treatment of glial cells before coincubation with T cells.
Constituents of tissue stress or damage may act as signals and potential activators of antigen-presenting cells,57 which evidently include ROS in glaucoma. This is supported by a recent study demonstrating that ROS can function as second messengers and modulators of the immune system.58 ROS have been reported to act as activators of antigen presentation by inducing phenotypic and functional changes in antigen-presenting cells. Alternatively, enhanced expression of heat shock proteins under oxidative stress may be an immunostimulatory signal because these stress proteins are also known to have the ability to elicit an activated immune response.59 Evidence suggests that in synergy with ROS themselves, advanced oxidation protein products generated under oxidative stress may also amplify the function of antigen-presenting cells.60 This may represent a route for the conversion of a short-term oxidant signal into a long-term one, thereby acting over a longer and possibly cumulative period. In addition, antigen-specific communication between antigen-presenting cells and T cells is blocked by interference with redox regulation pathways.61,62 Based on the identification of many redox-sensitive signal transduction pathways necessary for initiating the immune response, it has been suggested that the modulation of these oxidation-reduction reactions by antioxidants may have potential usefulness for the treatment of immunologic disease.62 Our findings provide evidence for a similar ROS-dependent regulation of glial antigen-presenting ability. With respect to the evidence of oxidative stress during glaucomatous neurodegeneration,4,5 such a ROS-dependent controlling pathway in immune system activation is potentially important for the immunogenic component of the neurodegenerative process.
Findings of this in vitro study provide important information relevant to glaucoma in humans. They indicate an important role of glial cells in the induction of an antigen-mediated immune process in response to oxidative stress in glaucoma. Enhanced ability of glial cells to function as antigen-presenting cells under oxidative stress facilitates their communication with T cells, thereby initiating the immune response to neurodegenerative injury and targeting T cells to the injury site. Early, moderate, and transient well-controlled activation of local microglia have been suggested to correlate with protective immunity in the injured CNS by providing tissue cleaning.63 However, given the widespread and persistent nature of glial activation in glaucomatous human eyes, which includes microglia and macroglia,31 stimulated antigen presentation and expanded T-cell activation seem more likely to lead to autoimmune injury. Among the additional evidence supporting this possibility is that antigenicity is also increased in the glaucomatous retina and optic nerve head because of glaucomatous tissue stress (upregulation of highly antigenic stress proteins64), increased protein expression, and increased exposure of retinal antigens because of neuronal injury. Most important, activation of an adaptive immune response, which requires antigen presentation, is evident in many glaucoma patients.8 The evidence of activated immune response in glaucoma patients include abnormal T-cell subsets12 and elevated titers of serum autoantibodies to many optic nerve head13,65 and retina antigens.14,16,17,66–68 Considering current in vitro findings, an activated immune response can be elicited by even a small number of autoreactive T cells on their entrance into the retina or optic nerve head, thereby leading to an autoimmune process. Despite immune privilege, autoreactive T cells are able to enter a normal, uninjured brain through an intact blood-brain barrier69 as part of the constitutive immune surveillance.70 Although there is no evidence of T-cell accumulation in the retina or optic nerve head tissues of glaucomatous eyes, possibly because of the transitory nature of sentinel T cells, episodic disruptions to the blood-eye barrier may facilitate the immune system contact. In addition, peripapillary chorioretinal atrophy, common in glaucomatous eyes71 and eyes in which the outer blood-retina barrier is disrupted,15 may serve as a site for the facilitated access of T cells into the retina. Aberrant activity of the immune system evidently leads to a number of neurotoxic consequences. Not only can T cells be directly toxic to RGCs through increased production of neurotoxic substances and death receptor binding (Wax MB, et al. IOVS 2006;47:ARVO E-Abstract 1828), serum antibodies can also facilitate RGC death during glaucomatous neurodegeneration.72 In addition, complement activation, a component of innate and adaptive immune responses, may exacerbate neuronal cell death in glaucomatous eyes.73,74
In conclusion, oxidative stress plays an important role in stimulating the antigen-presenting ability of glial cells and in activating the immune response in glaucoma. Mechanisms and consequences of ROS-mediated immune regulation warrant further investigation. An improved understanding of ROS-dependent regulation of glia-T-cell communication during antigen presentation can help to determine the therapeutic usefulness of antioxidants for the immunogenic component of neurodegeneration in glaucoma.
Acknowledgments
Supported in part by National Eye Institute Grants R01EY013813 (GT), R01EY014366 (DS), and R24EY015636 and by an unrestricted grant from Research to Prevent Blindness to the University of Louisville Department of Ophthalmology and Visual Sciences. In addition, GT is a recipient of the Research to Prevent Blindness Sybil B. Harrington Special Scholar Award.
Footnotes
Disclosure: G. Tezel, None; X. Yang, None; C. Luo, None; Y. Peng, None; S.L. Sun, None; D. Sun, None
References
- 1.Levin LA. Direct and indirect approaches to neuroprotective therapy of glaucomatous optic neuropathy. Surv Ophthalmol. 1999;43 suppl 1:S98–S101. doi: 10.1016/s0039-6257(99)00027-2. [DOI] [PubMed] [Google Scholar]
- 2.Neufeld AH, Sawada A, Becker B. Inhibition of nitric-oxide synthase 2 by aminoguanidine provides neuroprotection of retinal ganglion cells in a rat model of chronic glaucoma. Proc Natl Acad Sci USA. 1999;96:9944–9948. doi: 10.1073/pnas.96.17.9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu B, Neufeld AH. Expression of nitric oxide synthase-2 (NOS-2) in reactive astrocytes of the human glaucomatous optic nerve head. Glia. 2000;30:178–186. doi: 10.1002/(sici)1098-1136(200004)30:2<178::aid-glia7>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 4.Tezel G, Yang X. Caspase-independent component of retinal ganglion cell death, in vitro. Invest Ophthalmol Vis Sci. 2004;45:4049–4059. doi: 10.1167/iovs.04-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tezel G, Yang X, Cai J. Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Invest Ophthalmol Vis Sci. 2005;46:3177–3187. doi: 10.1167/iovs.05-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wax MB, Tezel G. Neurobiology of glaucomatous optic neuropathy: diverse cellular events in neurodegeneration and neuroprotection. Mol Neurobiol. 2002;26:45–55. doi: 10.1385/MN:26:1:045. [DOI] [PubMed] [Google Scholar]
- 7.Tezel G, Wax MB. Glial modulation of retinal ganglion cell death in glaucoma. J Glaucoma. 2003;12:63–68. doi: 10.1097/00061198-200302000-00014. [DOI] [PubMed] [Google Scholar]
- 8.Tezel G, Wax MB. The immune system and glaucoma. Curr Opin Ophthalmol. 2004;15:80–84. doi: 10.1097/00055735-200404000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz M, Kipnis J. Protective autoimmunity: regulation and prospects for vaccination after brain and spinal cord injuries. Trends Mol Med. 2001;7:252–258. doi: 10.1016/s1471-4914(01)01993-1. [DOI] [PubMed] [Google Scholar]
- 10.Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M. Neuroprotective autoimmunity: naturally occurring CD4+ CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc Natl Acad Sci USA. 2002;99:15620–15625. doi: 10.1073/pnas.232565399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwart M, Kipnis J. Autoimmunity on alert: naturally occurring regulatory CD4(+)CD25(+) T cells as part of the evolutionary compromise between a “need ” and a “risk. ”. Trends Immunol. 2002;23:530–534. doi: 10.1016/s1471-4906(02)02322-0. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Patil RV, Yu H, Gordon M, Wax MB. T cell subsets and sIL-2R/IL-2 levels in patients with glaucoma. Am J Ophthalmol. 2001;131:421–426. doi: 10.1016/s0002-9394(00)00862-x. [DOI] [PubMed] [Google Scholar]
- 13.Tezel G, Edward DP, Wax MB. Serum autoantibodies to optic nerve head glycosaminoglycans in patients with glaucoma. Arch Ophthalmol. 1999;117:917–924. doi: 10.1001/archopht.117.7.917. [DOI] [PubMed] [Google Scholar]
- 14.Wax MB, Tezel G, Saito I, et al. Anti-Ro/SS-A positivity and heat shock protein antibodies in patients with normal-pressure glaucoma [see comments] Am J Ophthalmol. 1998;125:145–157. doi: 10.1016/s0002-9394(99)80084-1. [DOI] [PubMed] [Google Scholar]
- 15.Wax MB, Tezel G, Edward PD. Clinical and ocular histopathological findings in a patient with normal-pressure glaucoma [see comments] Arch Ophthalmol. 1998;116:993–1001. doi: 10.1001/archopht.116.8.993. [DOI] [PubMed] [Google Scholar]
- 16.Tezel G, Seigel GM, Wax MB. Autoantibodies to small heat shock proteins in glaucoma. Invest Ophthalmol Vis Sci. 1998;39:2277–2287. [PubMed] [Google Scholar]
- 17.Yang J, Tezel G, Patil RV, Romano C, Wax MB. Serum autoantibody against glutathione S-transferase in patients with glaucoma. Invest Ophthalmol Vis Sci. 2001;42:1273–1276. [PubMed] [Google Scholar]
- 18.Young RA, Elliott TJ. Stress proteins, infection, and immune surveillance. Cell. 1989;59:5–8. doi: 10.1016/0092-8674(89)90861-1. [DOI] [PubMed] [Google Scholar]
- 19.van Eden W. Heat-shock proteins as immunogenic bacterial antigens with the potential to induce and regulate autoimmune arthritis. Immunol Rev. 1991;121:5–28. doi: 10.1111/j.1600-065x.1991.tb00821.x. [DOI] [PubMed] [Google Scholar]
- 20.van Noort JM. Multiple sclerosis: an altered immune response or an altered stress response? J Mol Med. 1996;74:285–296. doi: 10.1007/BF00207506. [DOI] [PubMed] [Google Scholar]
- 21.Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 22.Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol Today. 2000;21:141–147. doi: 10.1016/s0167-5699(99)01512-1. [DOI] [PubMed] [Google Scholar]
- 23.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 24.Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci. 2002;202:13–23. doi: 10.1016/s0022-510x(02)00207-1. [DOI] [PubMed] [Google Scholar]
- 25.Roberge FG, Caspi RR, Nussenblatt RB. Glial retinal Muller cells produce IL-1 activity and have a dual effect on autoimmune T helper lymphocytes: antigen presentation manifested after removal of suppressive activity. J Immunol. 1988;140:2193–2196. [PubMed] [Google Scholar]
- 26.Provis JM, Penfold PL, Edwards AJ, van Driel D. Human retinal microglia: expression of immune markers and relationship to the glia limitans. Glia. 1995;14:243–256. doi: 10.1002/glia.440140402. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Wu GS, Ishimoto S, Pararajasegaram G, Rao NA. Expression of major histocompatibility complex molecules in rodent retina: immunohistochemical study. Invest Ophthalmol Vis Sci. 1997;38:1848–1857. [PubMed] [Google Scholar]
- 28.Maehlen J, Olsson T, Zachau A, Klareskog L, Kristensson K. Local enhancement of major histocompatibility complex (MHC) class I and II expression and cell infiltration in experimental allergic encephalomyelitis around axotomized motor neurons. J Neuroimmunol. 1989;23:125–132. doi: 10.1016/0165-5728(89)90031-3. [DOI] [PubMed] [Google Scholar]
- 29.Molleston MC, Thomas ML, Hickey WF. Novel major histocompatibility complex expression by microglia and site-specific experimental allergic encephalomyelitis lesions in the rat central nervous system after optic nerve transection. Adv Neurol. 1993;59:337–348. [PubMed] [Google Scholar]
- 30.Hernandez MR. The optic nerve head in glaucoma: role of astrocytes in tissue remodeling. Prog Retin Eye Res. 2000;19:297–321. doi: 10.1016/s1350-9462(99)00017-8. [DOI] [PubMed] [Google Scholar]
- 31.Tezel G, Chauhan BC, LeBlanc RP, Wax MB. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Invest Ophthalmol Vis Sci. 2003;44:3025–3033. doi: 10.1167/iovs.02-1136. [DOI] [PubMed] [Google Scholar]
- 32.Yang J, Yang P, Tezel G, Patil RV, Hernandez MR, Wax MB. Induction of HLA-DR expression in human lamina cribrosa astrocytes by cytokines and simulated ischemia. Invest Ophthalmol Vis Sci. 2001;42:365–371. [PubMed] [Google Scholar]
- 33.Neufeld AH. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Arch Ophthalmol. 1999;117:1050–1056. doi: 10.1001/archopht.117.8.1050. [DOI] [PubMed] [Google Scholar]
- 34.Sun D, Hu XZ, Le J, Swanborg RH. Characterization of brain-isolated rat encephalitogenic T cell lines. Eur J Immunol. 1994;24:1359–1364. doi: 10.1002/eji.1830240618. [DOI] [PubMed] [Google Scholar]
- 35.Sun D, Hu XZ, Shah R, Coleclough C. The pattern of cytokine gene expression induced in rat T cells specific for myelin basic protein depends on the type and quality of antigenic stimulus. Cell Immunol. 1995;166:1–8. doi: 10.1006/cimm.1995.0001. [DOI] [PubMed] [Google Scholar]
- 36.Tezel G, Wax MB. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci. 2000;20:8693–8700. doi: 10.1523/JNEUROSCI.20-23-08693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsubara T, Pararajasegaram G, Wu GS, Rao NA. Retinal microglia differentially express phenotypic markers of antigen-presenting cells in vitro. Invest Ophthalmol Vis Sci. 1999;40:3186–3193. [PubMed] [Google Scholar]
- 38.Mi H, Barres BA. Purification and characterization of astrocyte precursor cells in the developing rat optic nerve. J Neurosci. 1999;19:1049–1061. doi: 10.1523/JNEUROSCI.19-03-01049.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang P, Hernandez MR. Purification of astrocytes from adult human optic nerve heads by immunopanning. Brain Res Brain Res Protoc. 2003;12:67–76. doi: 10.1016/s1385-299x(03)00073-4. [DOI] [PubMed] [Google Scholar]
- 40.Krohn AJ, Preis E, Prehn JH. Staurosporine-induced apoptosis of cultured rat hippocampal neurons involves caspase-1-like proteases as upstream initiators and increased production of superoxide as a main downstream effector. J Neurosci. 1998;18:8186–8197. doi: 10.1523/JNEUROSCI.18-20-08186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun D, Coleclough C, Cao L, Hu X, Sun S, Whitaker JN. Reciprocal stimulation between TNF-alpha and nitric oxide may exacerbate CNS inflammation in experimental autoimmune encephalomyelitis. J Neuroimmunol. 1998:122–130. doi: 10.1016/s0165-5728(98)00124-6. [DOI] [PubMed] [Google Scholar]
- 42.Sun D, Hu X, Shah R, Zhang L, Coleclough C. Production of tumor necrosis factor-alpha as a result of glia-T-cell interaction correlates with the pathogenic activity of myelin basic protein-reactive T cells in experimental autoimmune encephalomyelitis. J Neurosci Res. 1996;45:400–409. doi: 10.1002/(SICI)1097-4547(19960815)45:4<400::AID-JNR9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 43.Sun D, Coleclough C, Whitaker JN. Nonactivated astrocytes downregulate T cell receptor expression and reduce antigen-specific proliferation and cytokine production of myelin basic protein (MBP)-reactive T cells. J Neuroimmunol. 1997;78:69–78. doi: 10.1016/s0165-5728(97)00083-0. [DOI] [PubMed] [Google Scholar]
- 44.Lowenstein CJ, Alley EW, Raval P, et al. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc Natl Acad Sci USA. 1993;90:9730–9734. doi: 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin E, Nathan C, Xie QW. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J Exp Med. 1994;180:977–984. doi: 10.1084/jem.180.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferon-gamma. J Neurosci Res. 2004;77:540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- 47.Soos JM, Morrow J, Ashley TA, Szente BE, Bikoff EK, Zamvil SS. Astrocytes express elements of the class II endocytic pathway and process central nervous system autoantigen for presentation to encephalitogenic T cells. J Immunol. 1998;161:5959–5966. [PubMed] [Google Scholar]
- 48.Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- 49.Unanue ER, Beller DI, Lu CY, Allen PM. Antigen presentation: comments on its regulation and mechanism. J Immunol. 1984;132:1–5. [PubMed] [Google Scholar]
- 50.Feldmann M, Londei M, Kissonerghis M, et al. Regulation of HLA class II expression and the pathogenesis of autoimmunity. Concepts Immunopathol. 1988;5:44–56. [PubMed] [Google Scholar]
- 51.Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–253. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- 52.Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 53.Tezel G, Li LY, Patil RV, Wax MB. Tumor necrosis factor-alpha and its receptor-1 in the retina of normal and glaucomatous eyes. Invest Ophthalmol Vis Sci. 2001;42:1787–1794. [PubMed] [Google Scholar]
- 54.Tezel G, Yang X, Yang J, Wax MB. Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res. 2004;996:202–212. doi: 10.1016/j.brainres.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 55.Steinman L. A few autoreactive cells in an autoimmune infiltrate control a vast population of nonspecific cells: a tale of smart bombs and the infantry. Proc Natl Acad Sci USA. 1996;93:2253–2256. doi: 10.1073/pnas.93.6.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions: evidence for the involvement of mitochondrial radical generation. J Biol Chem. 1992;267:5317–5323. [PubMed] [Google Scholar]
- 57.Ibrahim MA, Chain BM, Katz DR. The injured cell: the role of the dendritic cell system as a sentinel receptor pathway. Immunol Today. 1995;16:181–186. doi: 10.1016/0167-5699(95)80118-9. [DOI] [PubMed] [Google Scholar]
- 58.Rutault K, Alderman C, Chain BM, Katz DR. Reactive oxygen species activate human peripheral blood dendritic cells. Free Radic Biol Med. 1999;26:232–238. doi: 10.1016/s0891-5849(98)00194-4. [DOI] [PubMed] [Google Scholar]
- 59.Young DB. Heat-shock proteins: immunity and autoimmunity. Curr Opin Immunol. 1992;4:396–400. doi: 10.1016/s0952-7915(06)80029-4. [DOI] [PubMed] [Google Scholar]
- 60.Alderman CJ, Shah S, Foreman JC, Chain BM, Katz DR. The role of advanced oxidation protein products in regulation of dendritic cell function. Free Radic Biol Med. 2002;32:377–385. doi: 10.1016/s0891-5849(01)00735-3. [DOI] [PubMed] [Google Scholar]
- 61.Matsue H, Edelbaum D, Shalhevet D, et al. Generation and function of reactive oxygen species in dendritic cells during antigen presentation. J Immunol. 2003;171:3010–3018. doi: 10.4049/jimmunol.171.6.3010. [DOI] [PubMed] [Google Scholar]
- 62.Tse HM, Milton MJ, Piganelli JD. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigenpresenting cells: implication for their use in targeting oxidationreduction reactions in innate immunity. Free Radic Biol Med. 2004;36:233–247. doi: 10.1016/j.freeradbiomed.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 63.Shaked I, Porat Z, Gersner R, Kipnis J, Schwartz M. Early activation of microglia as antigen-presenting cells correlates with T cellmediated protection and repair of the injured central nervous system. J Neuroimmunol. 2004;146:84–93. doi: 10.1016/j.jneuroim.2003.10.049. [DOI] [PubMed] [Google Scholar]
- 64.Tezel G, Hernandez MR, Wax MB. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Arch Ophthalmol. 2000;118:511–518. doi: 10.1001/archopht.118.4.511. [DOI] [PubMed] [Google Scholar]
- 65.Grus FH, Joachim SC, Bruns K, Lackner KJ, Pfeiffer N, Wax MB. Serum autoantibodies to alpha-fodrin are present in glaucoma patients from Germany and the United States. Invest Ophthalmol Vis Sci. 2006;47:968–976. doi: 10.1167/iovs.05-0685. [DOI] [PubMed] [Google Scholar]
- 66.Romano C, Barrett DA, Li Z, Pestronk A, Wax MB. Anti-rhodopsin antibodies in sera from patients with normal-pressure glaucoma. Invest Ophthalmol Vis Sci. 1995;36:1968–1975. [PubMed] [Google Scholar]
- 67.Maruyama I, Ohguro H, Ikeda Y. Retinal ganglion cells recognized by serum autoantibody against gamma-enolase found in glaucoma patients. Invest Ophthalmol Vis Sci. 2000;41:1657–1665. [PubMed] [Google Scholar]
- 68.Joachim SC, Pfeiffer N, Grus FH. Autoantibodies in patients with glaucoma: a comparison of IgG serum antibodies against retinal, optic nerve, and optic nerve head antigens. Graefes Arch Clin Exp Ophthalmol. 2005;243:817–823. doi: 10.1007/s00417-004-1094-5. [DOI] [PubMed] [Google Scholar]
- 69.Raivich G, Jones LL, Kloss CU, Werner A, Neumann H, Kreutzberg GW. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci. 1998;18:5804–5816. doi: 10.1523/JNEUROSCI.18-15-05804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 71.Tezel G, Kass MA, Kolker AE, Wax MB. Comparative optic disc analysis in normal pressure glaucoma, primary open-angle glaucoma, and ocular hypertension. Ophthalmology. 1996;103:2105–2113. doi: 10.1016/s0161-6420(96)30382-5. [DOI] [PubMed] [Google Scholar]
- 72.Tezel G, Wax MB. The mechanisms of hsp27 antibody-mediated apoptosis in retinal neuronal cells. J Neurosci. 2000;20:3552–3562. doi: 10.1523/JNEUROSCI.20-10-03552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stasi K, Nagel D, Yang X, et al. Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Invest Ophthalmol Vis Sci. 2006;47:1024–1029. doi: 10.1167/iovs.05-0830. [DOI] [PubMed] [Google Scholar]
- 74.Kuehn MH, Kim CY, Ostojic J, et al. Retinal synthesis and deposition of complement components induced by ocular hypertension. Exp Eye Res. 2006;83:620–628. doi: 10.1016/j.exer.2006.03.002. [DOI] [PubMed] [Google Scholar]